
Abstract
Glycoscience has been recognized as an important area in biomedical research. Currently, a major obstacle for glycoscience study is the lack of diverse, biomedically relevant, and complex glycans in quantities sufficient for exploring their structural and functional aspects. Complementary to chemoenzymatic synthesis, natural glycans could serve as a great source of biomedically relevant glycans if they are available in sufficient quantities. We have recently developed oxidative release of natural glycans (ORNG) for large-scale release of N-glycans as free reducing glycans. While free reducing glycans can be readily derivatized with ultraviolet or fluorescent tags for high-performance liquid chromatography (HPLC) and mass spectrometry (MS) analysis, it is difficult to remove tags for the regeneration of free reducing glycans without affecting the structural integrity of glycans. To address this inconvenience, we explored the use of a cleavable tag, O-benzylhydroxylamine (BHA). Free reducing glycans are easily and efficiently labeled with BHA under mild conditions, enabling UV detection during HPLC purification. Individual glycan–BHA conjugates can then be separated using multidimensional HPLC and characterized by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) and MS/MS. The BHA tag can then be easily removed by palladium-on-carbon (Pd/C)-catalyzed hydrogenation to efficiently regenerate free reducing glycans with little effect on glycan structures. This procedure provides a simple and straightforward way to tag free reducing glycans for purification at a preparative scale using multidimensional HPLC and subsequently recover purified free reducing glycans.
Keywords
Introduction
As one of the most important posttranslational modifications, glycosylation plays important roles in many biological processes including protein folding and stability, cell differentiation and adhesion, signal recognition and transmission, viral entry and bacterial infection, and immune response and regulation.1–4 However, glycoscience lags far behind the study of other major biomolecules such as proteins/peptides and nucleic acids, due to the high structural complexity of glycans. Unlike nucleic acids and proteins, the biosynthesis of glycans is not template driven and often involves the combined operation of multiple glycosyltransferases. The resulted glycans are often branched structures defined by monosaccharide composition and sequence, linkage position, and anomeric stereochemistry, making detailed structural characterization much more challenging than that of nucleic acids and proteins/peptides.
Owing to the great technological advancement in high-performance liquid chromatography (HPLC), mass spectrometry (MS), and online LC-MS, glycomics analysis has seen great improvement recently.5–8 While many challenges still exist, one immediate impediment is the lack of many diverse complex glycans with defined structures serving as standards, which would greatly facilitate the development of an MS database, fragmentation pattern analysis, and sequencing algorithm development and eventually lead to high-throughput analytical glycomics similar to current practice in proteomics. On the other hand, the functional study of glycans is also dependent on access to more biomedically relevant glycans. In the last decades, glycan microarray has become an efficient tool to study protein–glycan interactions by analyzing the binding specificity of glycan binding proteins (GBPs) on microarray slides immobilized with a library of glycan structures.9–11 The utility of a glycan microarray is highly dependent on the size, diversity, and biological relevance of the underlying glycan library, which is always challenging to establish. Therefore, for both analytical and functional glycomics, access to more relevant glycan structures has become a pivotal point for glycoscience development.
Due to their structural complexity, it is not surprising that glycans are among the most challenging target for chemoenzymatic synthesis. Synthetic methodologies for glycans have improved greatly in the past decades, and many elegant syntheses of extremely complex glycans have been achieved.12–18 However, glycan synthesis remains a laborious and costly practice that can only be carried out in a limited number of specialized laboratories. It has yet to meet the urgent need for expansion of a biomedically relevant glycan library for glycoscience. An alternative and complementary route is to isolate glycans from natural sources, which is an unlimited reservoir for diverse and biologically relevant complex glycans. One particular challenge for this route is the low natural abundance of glycans in natural materials except some common polysaccharides, making a large amount of starting material a necessity. Gram-scale glycopeptides and glycans can be isolated from a large amount of egg yolk. 19 As a more general approach, we have developed the oxidative release of natural glycans (ORNG), which uses household bleach to directly release gram-scale glycans from kilogram-scale natural materials such as animal and plant tissues. 20 Another challenge is the separation of individual glycans from a complex mixture that occurs in nature by chromatography. Free reducing glycans are versatile reagents for both analytical and functional glycomics. Due to the special reactivity of the reducing end, they can be easily derivatized for HPLC and MS analysis or microarray preparation if a bifunctional linker is used.21,22 Most commercial glycans are available as free reducing glycans, and many previous glycomics studies have been based on free reducing glycans, making them an ideal form as structural standards. While most natural glycans either occur as or can be released as free reducing glycans, the separation of these glycans is not an easy task due to the lack of chromophore for detection. The most common practice has been the fluorescent or chromogenic tagging at the reducing end to aid detection in chromatography. 23 These tags, however, are often permanent and difficult to remove without affecting structural integrity. Tags installed by reductive amination can be removed by hydrogen peroxide at low yields. 24 We also developed a method to remove these tags using N-bromosuccinimide (NBS); however, a significant amount of the reducing glycans generated lost one carbon. 25 Hydrazide beads have been used to enrich free reducing glycans, but the removal of free reducing glycans from these beads requires a significantly acidic condition, 26 posing potential problems for acid-labile groups such as sialic acids. Also, the use of beads is not compatible with the HPLC separation of individual glycans.
In this paper, we seek to develop a cleavable linker that can be used to tag free reducing glycans to facilitate HPLC separation and that can be removed to recover free reducing glycans efficiently under mild conditions.27,28 O-Benzylhydroxylamine (BHA) is commercially available at a cheap price. We envisioned that the conjugation of BHA with free reducing glycan through oxime formation will install a benzyl group as a hydrophobic tag for C18 solid-phase extraction and a chromogenic tag for HPLC separation. Once a glycan–BHA conjugate is purified, palladium-on-carbon (Pd/C)-catalyzed hydrogenation can be used to remove the benzyl group to initiate the hydrolysis of hydroxylamine to generate free reducing glycans, 28 as illustrated in Figure 1 . This simple strategy would facilitate access to more natural glycan structures in free reducing form.
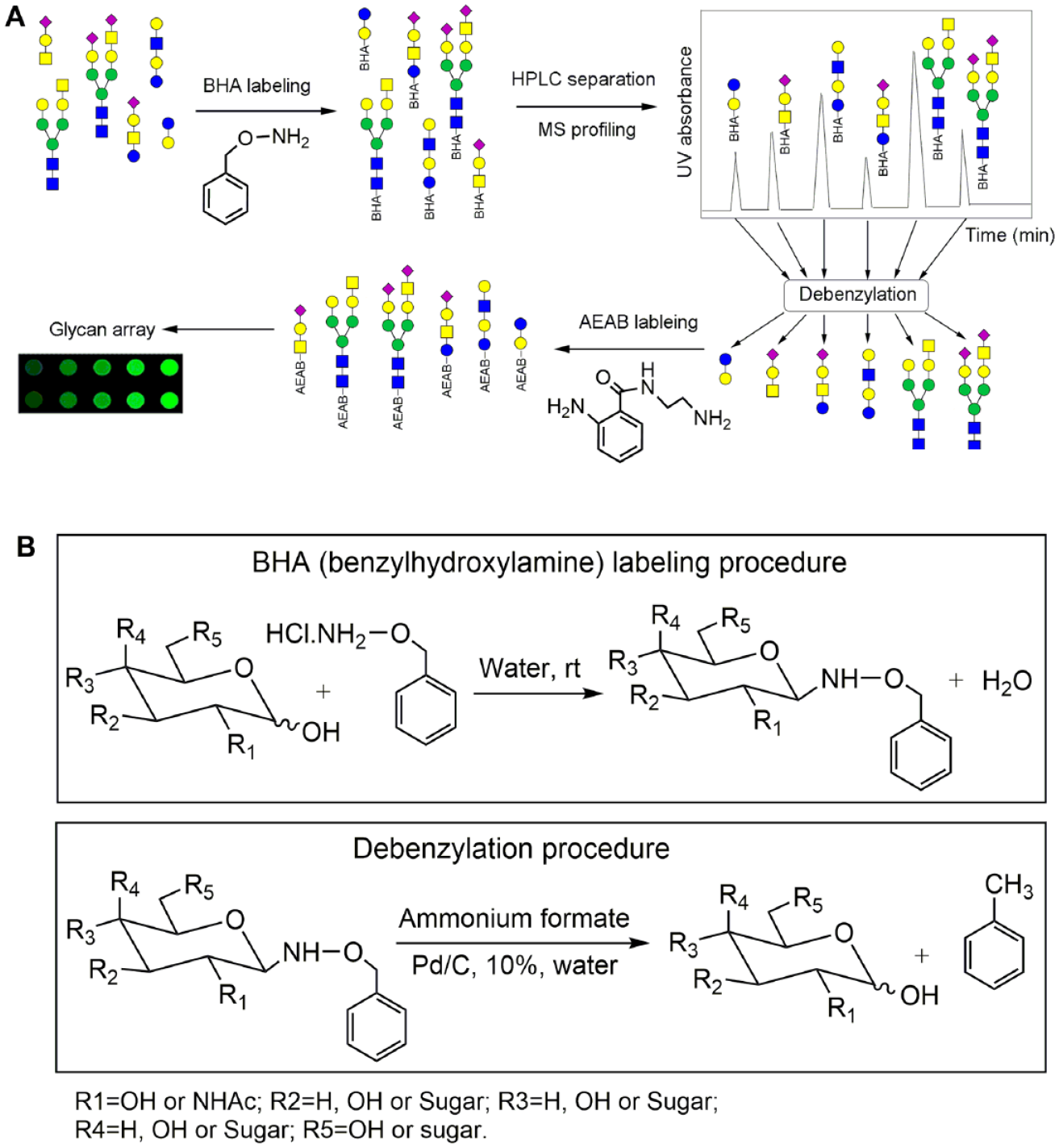
(
Materials and Methods
Chemicals
BHA, 2-anthranilic acid (2-AA), sodium cyanoborohydride (NaBH3CN), 2-amino-N-(2-aminoethyl) benzamide (AEAB), 10% Pd/C, ammonium formate, 2,5-dihydroxybenzoic acid (DHB), and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). HPLC solvents were obtained from Thermo Fisher Scientific (Waltham, MA). The water used in the experiment was prepared by a Milli-Q system (Millipore, Bedford, MA). Human milk neutral glycans (HMGs) were prepared in our lab. 29
HPLC Separation Conditions
HPLC profiles were acquired using a Shimadzu HPLC CBM-20A system equipped with a UV detector SPD-20A (Shimadzu Co., Kyoto, Japan). UV absorption at 254 nm or fluorescence at 330 nm excitation and 420 nm emission was used for the detection of BHA tag or AEAB. Phenomenex amino columns were used for normal-phase HPLC. For normal-phase HPLC, the mobile phases were acetonitrile, water, and aqueous ammonium acetate buffer at pH 4.5. A linear gradient from 20 mM ammonium acetate in 80% acetonitrile to 20 mM ammonium acetate in 10% acetonitrile for 45 min was used.
MALDI-TOF-MS Conditions
A Bruker Daltonics Ultraflex-II matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF)/TOF system and an anchor-chip target plate were used for MS analysis (Bruker Daltonic Billerica, MA, USA). A reflective positive mode was used for glycans. DHB (10 mg/mL in 50% acetonitrile containing 0.1% trifluoroacetic acid) was used as the matrix.
BHA Labeling of Reducing Glycans
Glycans were dissolved in water. BHA hydrochloride was prepared at 20 mg/mL in water as labeling buffer. To complete the reaction, a sufficient amount of BHA (fivefold over the glycan) was used. Briefly, glycans were reacted with BHA labeling solution at room temperature for 6 h. After the labeling reaction was complete, the product could be submitted to the Bruker Daltonics Ultraflex-II MALDI-TOF-MS (Bruker Daltonic) or Shimadzu HPLC CBM-20A (Shimadzu Co.) system directly. Alternatively, the products can be subjected to a Sephadex G25 column to remove excess BHA tags. 19
Reductive Amination of Sugar with AEAB
The procedure was conducted according to Song et al. 20 Glycans were reacted with AEAB (about 10-fold) and NaBH3CN (about 10-fold excess) in a solvent mixture (DMSO–glacial acetic acid [7:3]), and the mixture was mixed in a 2 mL polyethylene vial. The tightly capped vial was heated at 60 °C for 2 h. After cooling to ambient temperature, a 10-fold volume of acetonitrile was added to precipitate the glycan–AEAB derivatives. After centrifugation, the pellet was dissolved in water and then submitted to HPLC or MALDI-TOF-MS analysis after dilution with water.
Debenzylation of BHA Derivatives
The procedure was conducted according to Sajiki et al. 28 with minor modifications. Debenzylation of 2 mg of glycan–BHA conjugates was catalyzed by 2 mg of 10% Pd/C and 50 mg of ammonium formate in 0.2 mL of water at room temperature for 12 h. After the reaction was complete, the residue was submitted to a Sephadex G25 column to remove excess BHA tags. 19
Permethylation of Glycan–BHA Derivatives for the MS Profile
Permethylation of glycan–BHA derivatives was carried out according to reported procedures. 20 Briefly, the lyophilized glycan–BHA derivatives were treated with 200 µL of a DMSO–NaOH slurry and 50 µL of methyl iodide at room temperature for 30 min. After the reaction was complete, 500 µL of water and 500 µL of chloroform were added to the reaction mixture, vortexing for 1 min. After centrifugation, the supernatant was then partitioned between 500 µL of water and 500 µL of chloroform. The organic layer was washed with 500 µL of water twice, dried, and redissolved in 50% methanol for MS analysis.
Peracetylation of Glycan–BHA Derivatives for the MS Profile
Peracetylation of glycan–BHA derivatives was conducted by a previously reported procedure. 30 Briefly, the lyophilized sample was dissolved in 300 µL of pyridine and 300 µL of acetic anhydride and held at 60 °C for 2 h. After the reaction was complete, 500 µL of water and 500 µL of chloroform were added to the reaction mixture, as described above. The organic layer was washed with 500 µL of water twice, dried, and redissolved in 50% methanol for MS analysis.
Results and Discussion
Overall Evaluation of the Whole Procedure
For method development, lacto-N-neotetraose (LNnT) was employed as the model glycan to evaluate the feasibility of the whole strategy. After reaction with BHA, the LNnT–BHA conjugate gained stronger hydrophobicity and ultraviolet absorption at 254 nm, which enabled the subsequent HPLC separation with a UV detector, as shown in
Figure 2A
(top). MALDI-TOF-MS analysis (
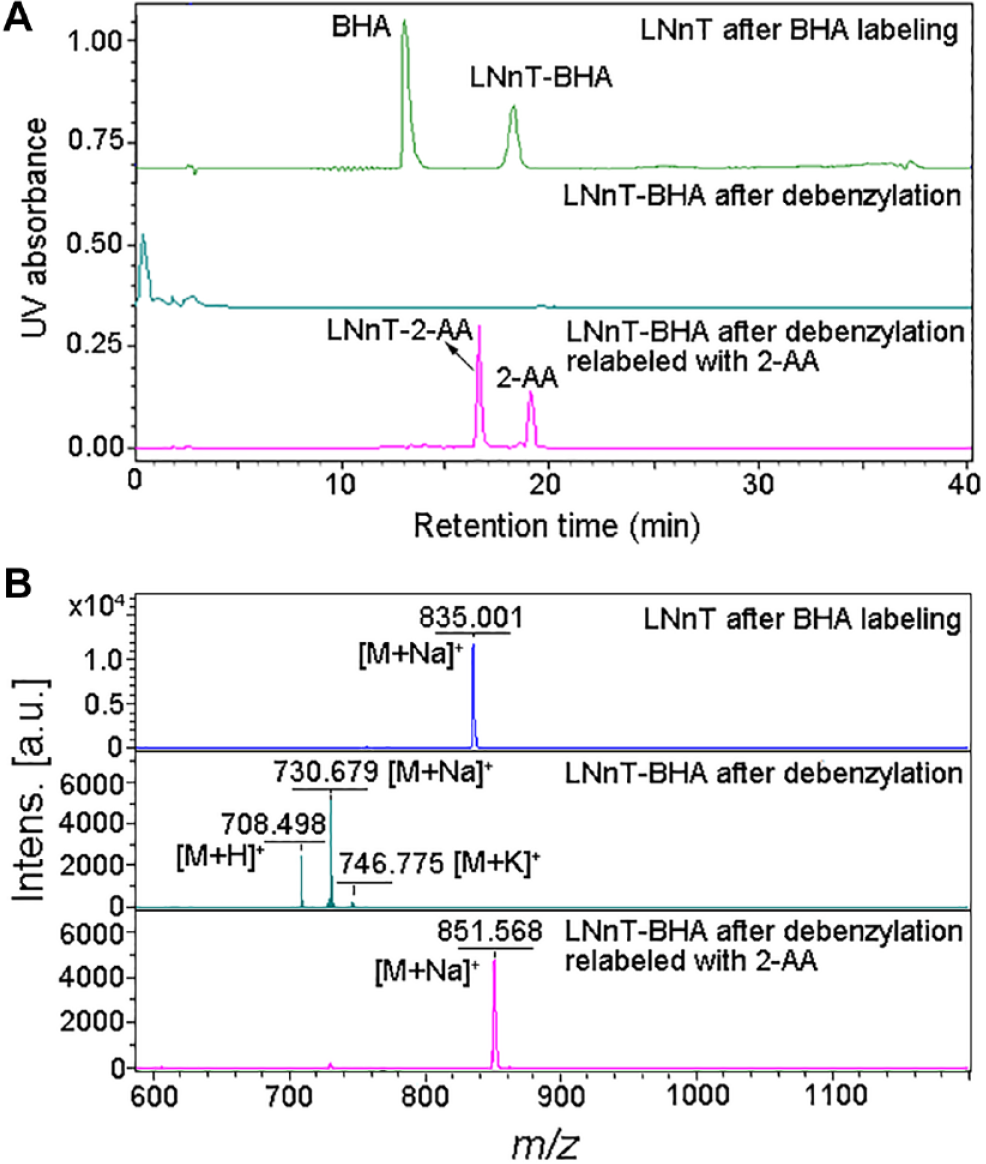
Overall procedure of LNnT after BHA tagging, HPLC separation, and debenzylation (BHD strategy). (
Pd/C-catalyzed hydrogenation is commonly used to remove the benzyl-protecting group.28,31 We investigated this mild debenzylation of LNnT–BHA in both water and methanol, as shown in
Supplemental Figure S1
and
Figure 2A
(middle). As expected, the BHA tag could be easily removed from LNnT–BHA conjugates by Pd/C-catalyzed hydrogenation to afford the reducing glycan product in either solvents, while reaction in water was more complete, presumably due to better solubility. No signal of the LNnT–BHA conjugate and other side products was observed by HPLC analysis (
To further confirm the structural integrity of the recovered LNnT after Pd/C debenzylation, we relabeled the product with 2-AA and analyzed it by HPLC and MS. A single peak besides 2-AA was observed in the HPLC profile ( Fig. 2A , bottom), which was confirmed to be LNnT–2-AA conjugates (m/z 851 [M+Na]+) by MS spectrometry ( Fig. 2B , bottom). The HPLC and MS data demonstrated the presence as well as structural integrity of the reducing ends of generated glycan through the BHD strategy, as shown in Figure 2 (both lower layers). Together, the data demonstrated the potential feasibility of the strategy to access pure reducing glycans through BHA tagging, HPLC separation, and debenzylation (BHD strategy) ( Figure 1 ).
We then investigated the efficiency as well as the recovery yield of glycan after the BHD strategy using several mono- and oligosaccharides. The recovery yields of four glycans, LNnT, lactose (Lac), N-acetylgalactosamine (GalNAc), and N-acetyl-
MS and MS/MS Assignation of LNnT–BHA Derivatives after Permethylation and Peracetylation
Structural characterization is essential for purified natural glycans, especially if they are to be used as MS or HPLC standards. Permethylation and peracetylation greatly improve ionization efficiency during MS analysis and have been widely used to aid in the structural characterization of glycans.
Figure 3
shows the MADLI-TOF-MS spectra and MS/MS fragmentation patterns of permethylated (
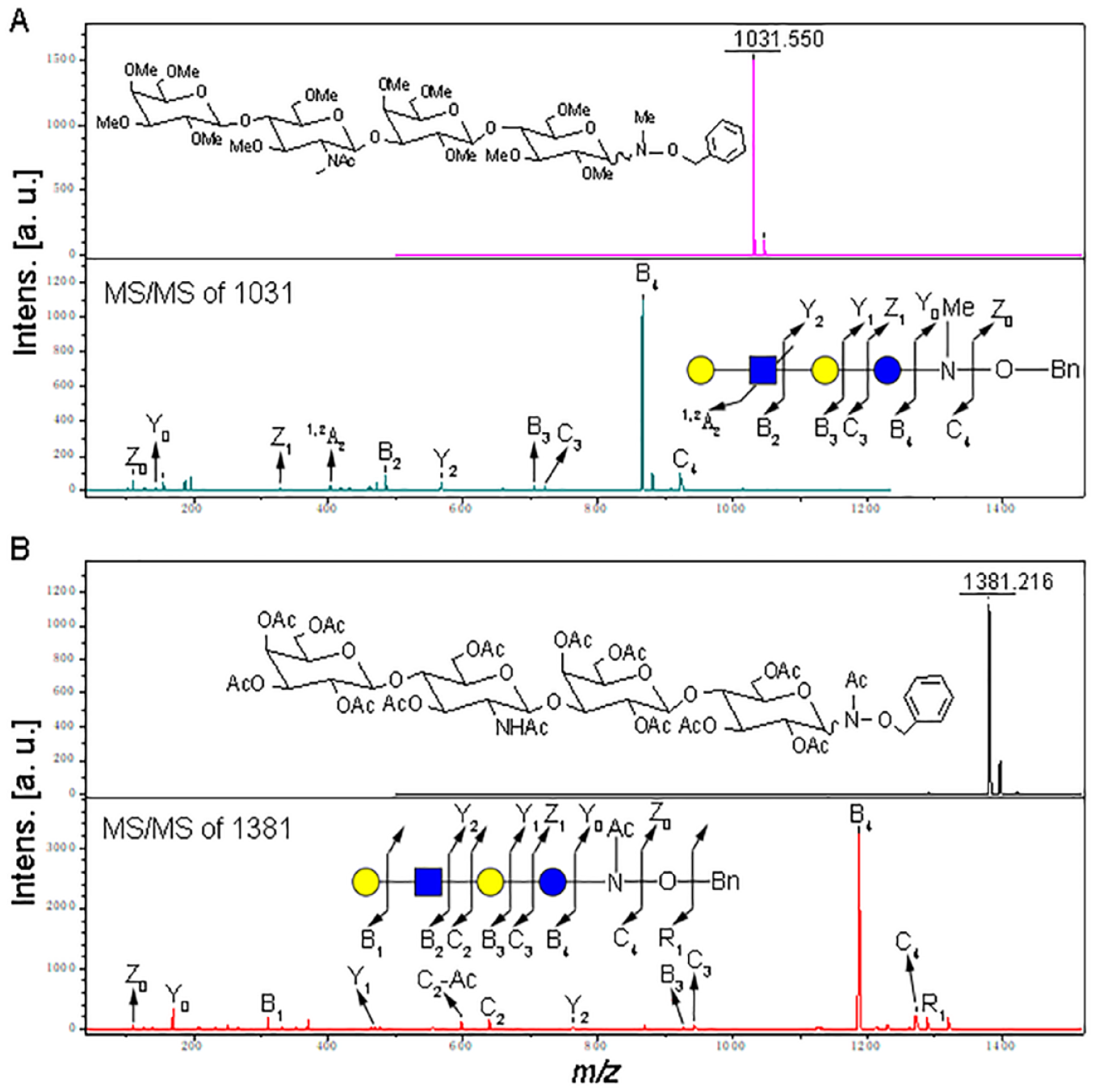
(
Under MALDI-TOF-MS/MS fragmentation conditions, two sets of fragmentation ions, B and Y ions, were observed in both the permethylation and peractylation products of LNnT–BHA conjugates, as presented in
Application of the Assay to the Preparation of Single Reducing Glycan from HMGs
We then applied the BHD strategy to the purification of human milk glycans. These HMGs occur in free reducing form under physiological conditions. However, to study their functions, it is of great interest to separate individual components at the reducing form. We applied BHA conjugation to a neutral fraction of HMG.
Figure 4A
shows the HPLC profile using the hydrophilic interaction chromatography (HILIC) mode on the amine column of HMG after BHA labeling, which afforded nine separated peaks (numbered P1–9). Each peak was collected to afford nine fractions named P1–9 and reprofiled by HPLC to check the purities of each fraction (P1–9), as presented in
Figure 4B
. Then the collected peaks were characterized individually by MALDI-TOF-MS, as presented in
Figure 4C
. Mass signals of nine separated peaks (P1–9) at m/z 470, 615, 762, 835, 981, 1127, 1346, 1492, and 1639 were assigned to nine glycan–BHA conjugates (H2–, H2F1–, H2F2–, H3N1–, H3N1F1–, H3N1F2–, H3N2F1–, H3N2F2–, and H3N2F3–BHA derivatives), respectively, as clearly presented in
Figure 4C
. Further details on the structure of the glycan–BHA conjugates identified were obtained through MS/MS fragmentation of their corresponding permethylated products, as shown in
Figure 5
and the Supplementary Information file (
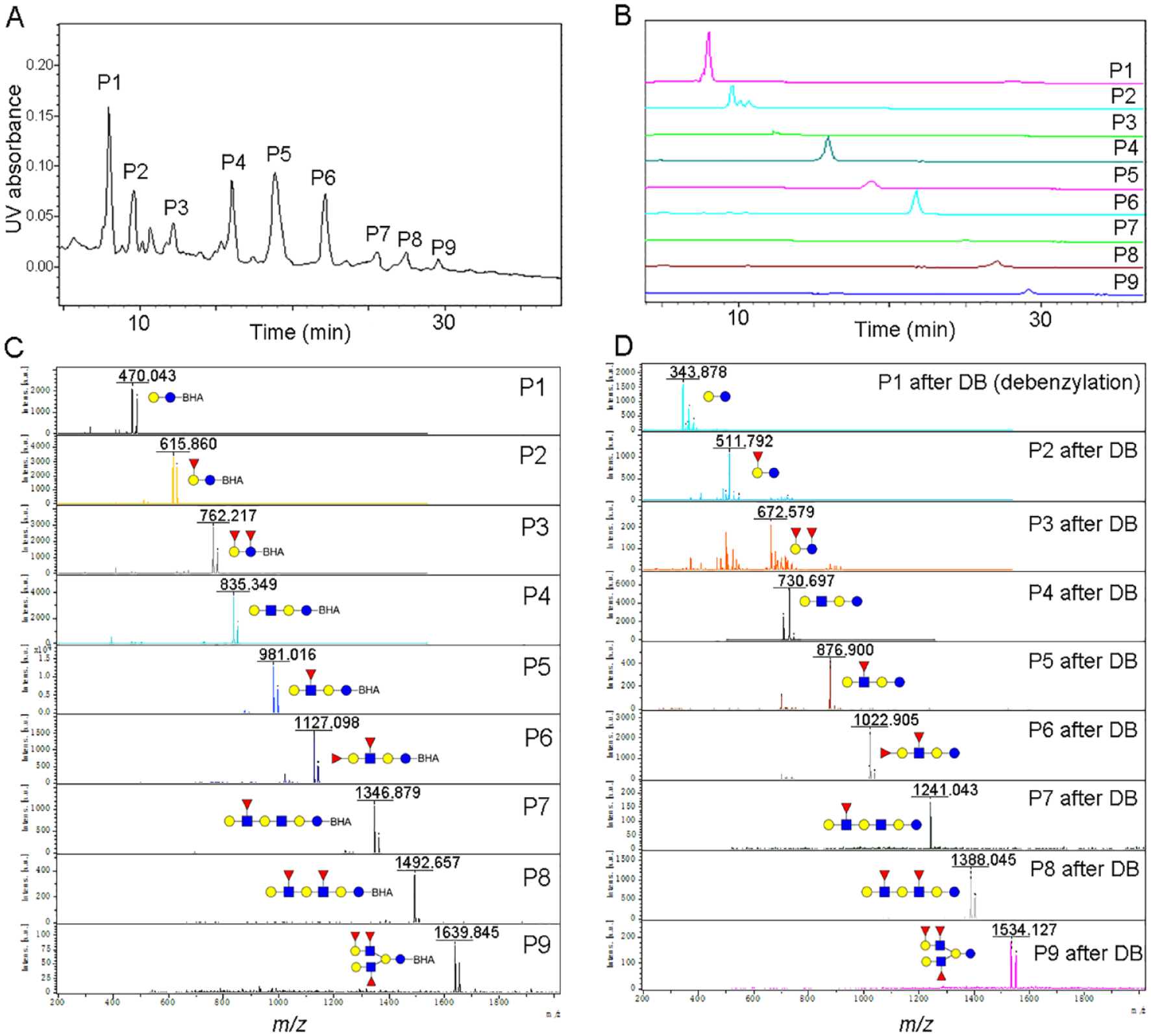
(
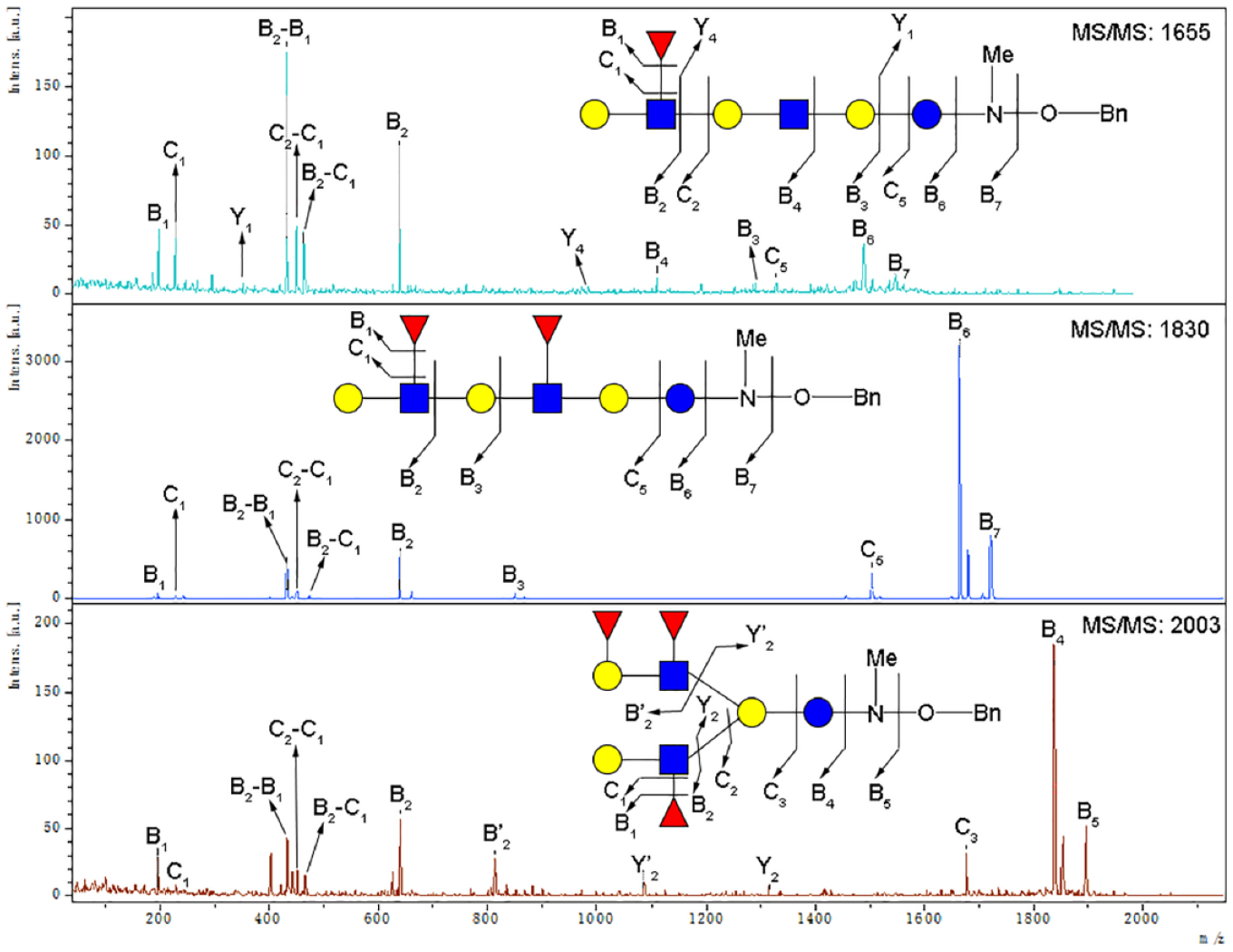
MALDI-TOF-MS/MS fragmentation of three collected peaks (P7–9) after permethylation.
Nine peaks, named P1–9, were submitted to Pd/C-catalyzed dehydrogenation (debenzylation) reaction to afford nine products determined by MS with signals at m/z 343, 511, 672, 730, 876, 1022, 1241, 1388, and 1534 ([M+H]+ or [M+Na]+), which were assigned to the nine corresponding reducing glycans, H2, H2F1, H2F2, H3N1, H3N1F1, H3N1F2, H3N2F1, H3N2F2, and H3N2F3 by MS (
Conclusions
In this study, BHA was employed as a cleavable tag for the labeling of reducing glycans, which allows the isolation and purification of reducing glycans by HPLC. Free reducing glycans are easily and efficiently labeled with BHA using water as a solvent at room temperature, and show significant improvement in HPLC separation compared with unlabeled material. Purified glycan–BHA conjugates can be permethylated or peracetylated for MS analysis without generating complex side products. Permethylated glycan–BHA conjugates give clear MS/MS fragmentation patterns, facilitating detailed structural elucidation. Specially, the BHA tag can be easily removed by Pd/C-catalyzed hydrogenation to efficiently regenerate free reducing glycans with no effect on glycan structures. This procedure provides a simple and straightforward way to tag free reducing glycans for purification at a preparative scale using multidimensional preparative HPLC and subsequently recover the purified free reducing glycans from the purified BHA conjugate for direct functional glycomics studies or other derivatization procedures.
Supplemental Material
Supporting_information_manucript_TECH-19-0066 – Supplemental material for O-Benzylhydroxylamine (BHA) as a Cleavable Tag for Isolation and Purification of Reducing Glycans
Supplemental material, Supporting_information_manucript_TECH-19-0066 for O-Benzylhydroxylamine (BHA) as a Cleavable Tag for Isolation and Purification of Reducing Glycans by Ying Zhang, Yuyang Zhu, Yi Lasanajak, David F. Smith and Xuezheng Song in SLAS Technology
Footnotes
Acknowledgements
This work was supported by NIH Common Fund Glycoscience grant U01GM116254 and partially supported by STTR grant R41GM122139 and SBIR grant R43GM133252. We also acknowledge Emory Comprehensive Glycomics Core (ECGC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.