
Abstract
Traditional microbiological methods enable genus-level identification of Streptococcus spp. isolates. However, as the species of this genus show broad phenotypic variation, species-level identification or even differentiation within the genus is difficult. Herein we report the evaluation of protein spectra cluster analysis for the identification of Streptococcus species associated with disease in swine by means of matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS). A total of 250 S. suis–like isolates obtained from pigs with clinical signs of encephalitis, arthritis, pneumonia, metritis, and urinary or septicemic infection were studied. The isolates came from pigs in different Brazilian states from 2001 to 2014. The MALDI-TOF MS analysis identified 86% (215 of 250) as S. suis and 14% (35 of 250) as S. alactolyticus, S. dysgalactiae, S. gallinaceus, S. gallolyticus, S. gordonii, S. henryi, S. hyointestinalis, S. hyovaginalis, S. mitis, S. oralis, S. pluranimalium, and S. sanguinis. The MALDI-TOF MS identification was confirmed in 99.2% of the isolates by 16S rDNA sequencing, with MALDI-TOF MS misidentifying 2 S. pluranimalium as S. hyovaginalis. Isolates were also tested by a biochemical automated system that correctly identified all isolates of 8 of the 10 species in the database. Neither the isolates of the 3 species not in the database (S. gallinaceus, S. henryi, and S. hyovaginalis) nor the isolates of 2 species that were in the database (S. oralis and S. pluranimalium) could be identified. The topology of the protein spectra cluster analysis appears to sustain the species phylogenetic similarities, further supporting identification by MALDI-TOF MS examination as a rapid and accurate alternative to 16S rDNA sequencing.
The Streptococcus genus, which is composed of gram-positive cocci that grow in variable-length strings, consists of non-motile, catalase-negative, facultative anaerobes. 5 The genus comprises several pathogens that pose a serious threat to human and animal health. 10 Streptococcus suis is the most important species for the worldwide swine industry, as it is both a part of the upper respiratory tract microbiota, especially the tonsils and nasal cavity, and a significant cause of disease in 3–12-wk-old piglets. 5 The infection can cause meningitis, pneumonia, arthritis, endocarditis, septicemia, abscesses, and sudden death in peracute cases. However, several other Streptococcus species can infect swine and cause similar clinical manifestations. 5
Traditional microbiological methods, such as biochemical and serologic characterization, enable the identification of isolates of Streptococcus to the genus level. However, the species in the genus show broad phenotypic variation, making species-level identification difficult. 8 Molecular methods, especially 16S ribosomal (r)DNA sequencing, have revolutionized the taxonomy and nomenclature of the genus Streptococcus with >50 species and 6 species groups recognized to date. 4
Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) has become an important laboratory tool, providing rapid and accurate identification of bacterial species. 9 In our study, we assessed the application of the MALDI-TOF MS technique, as well as spectra cluster analysis, for the identification of diverse Streptococcus species, all of which are associated with disease in swine.
A total of 250 presumptive Streptococcus isolates were studied. The Streptococcus-like isolates were obtained from diseased pigs across different Brazilian states between 2001 and 2014. The conditions observed in these pigs were encephalitis, arthritis, pneumonia, metritis, urinary tract infection, and septicemia. The isolates were grown on Columbia blood agar plates a (5% defibrinated sheep blood) incubated aerobically for 24 h at 37°C. Single colonies were then inoculated into brain–heart infusion broth a supplemented with 5% fetal calf serum and incubated for 24 h at 37°C, for further bacterial protein and DNA extraction.
For MALDI-TOF MS sample preparation, bacterial proteins were extracted using an ethanol–formic acid protocol. 11 The protein suspension (1 µL) was transferred to a polished steel MALDI target plate b and overlaid with 1 µL of matrix (10 mg/mL α-cyano-4-hydroxy-cinnamic acid in 50 % acetonitrile–2.5 % trifluoroacetic acid). Bacterial mass spectra in the 2–20 kDa range were acquired using a commercial mass spectrometer. c Spectra replicates were obtained by loading each sample into multiple spots and examining each spot twice. Initially, for MALDI-TOF MS identification, the spectra were loaded and compared with the manufacturer’s library. d Standard interpretive criteria b were applied: scores ≥2.0 were accepted for species assignment and scores ≥1.7 but <2.0 for genus identification. For further analysis, spectra replicates were used to generate a main spectrum e for each isolate. Cluster analysis e was performed using the number of different peaks detected and the unweighted pair group method with arithmetic mean method. Principal component analysis (PCA) e was also applied to analyze spectra homogeneity.
To confirm species identification, the biochemical profile of each isolate was characterized using an automated system, f and16S rDNA sequencing was also performed. DNA extraction was performed as described previously 2 with prior enzymatic treatment with lysozyme (100 mg) g and proteinase K (20 mg) g at 37°C for 60 min. Partial gene amplification was performed as described previously. 12 The polymerase chain reactions (PCRs: 50 μL) consisted of 5 μL of genomic DNA, ultrapure water, 10× PCR buffer, 1.5 mM MgCl2, 200 μM of dNTPs, h 200 μM of each primer, and 1.25 U of Taq polymerase. h The amplicons were purified, i and sequencing was performed by the Human Genome Research Center (University of São Paulo, Brazil). A phylogenetic tree was constructed using the maximum-likelihood method. e The DNA sequences from our study were deposited in GenBank under accessions KR819485, KR819487–KR819494, KR819496, KR819498, KR819500, KR819502, KX485315, KX485314, KX500124, KX500127–KX500131, KX500136, KX500138, KX500139, KX500149–KX500156, and KX586713–KX586715.
The isolation sites (n = 250) of the Streptococcus strains were as follows: 117 (46.8%) were isolated from the central nervous system, 82 (32.8%) from the respiratory tract, 25 (10%) from joints, 13 (5.2%) from the genitourinary system, and 13 (5.2%) from the peritoneum, heart, blood, or a skin lesion (Table 1).
Distribution of Streptococcus spp. isolates according to isolation site and identified species.*
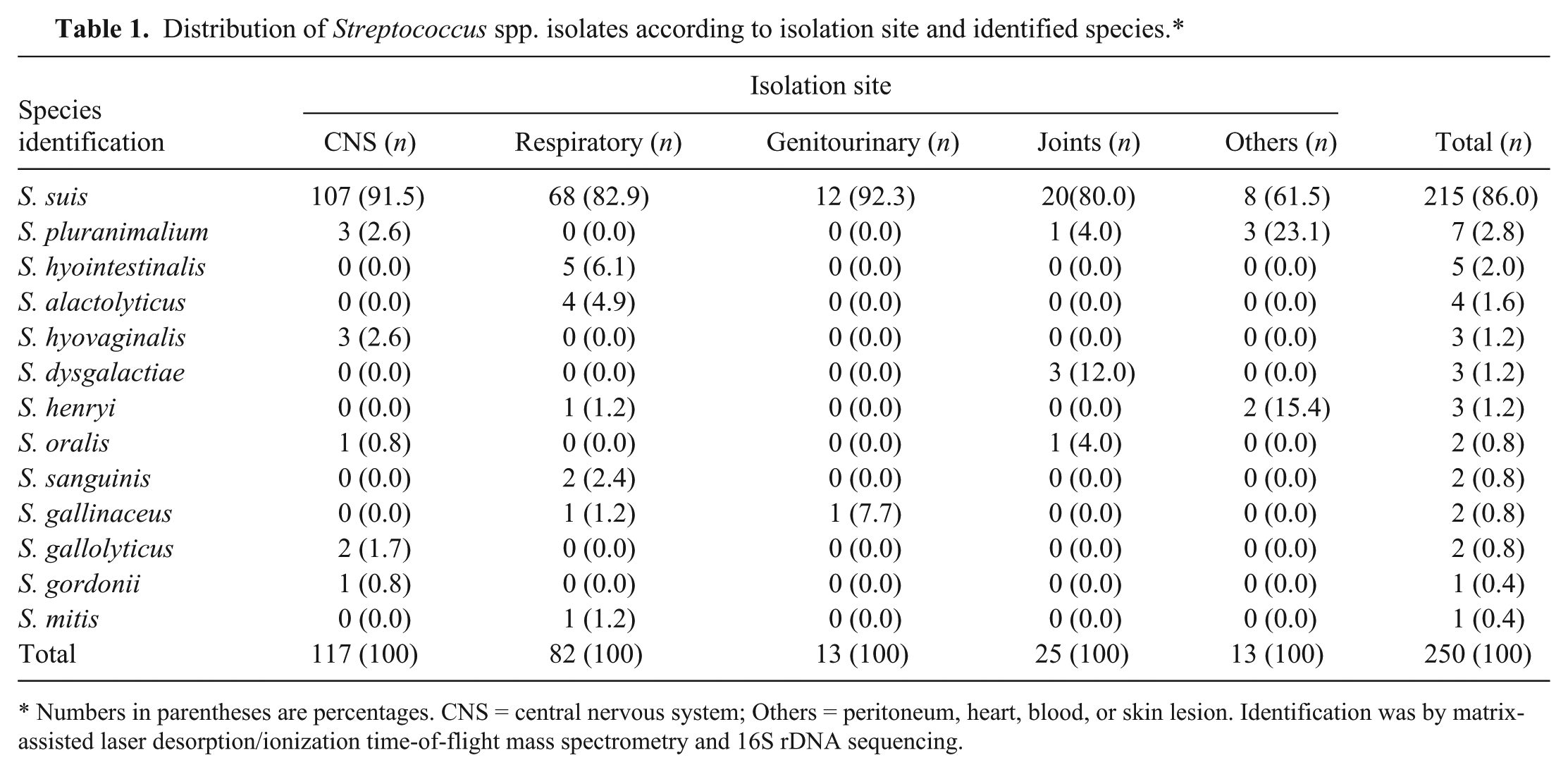
Numbers in parentheses are percentages. CNS = central nervous system; Others = peritoneum, heart, blood, or skin lesion. Identification was by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry and 16S rDNA sequencing.
The MALDI-TOF MS identified all isolates with log (score) values >2.0. Of the 250 isolates studied, 215 (86%) were identified as S. suis; the remaining 35 (14%) belonged to S. alactolyticus, S. dysgalactiae, S. gallinaceus, S. gallolyticus, S. gordonii, S. henryi, S. hyointestinalis, S. hyovaginalis, S. mitis, S. oralis, S. pluranimalium, and S. sanguinis (Table 1; Fig. 1). The protein spectra cluster analysis enabled the distinction of the Streptococcus isolates according to the identified species. The S. gordonii, S. mitis, and S. oralis isolates tended to group together comprising a nonuniform cluster with higher proximity to the S. suis group (group highlighted by a red circle in Fig. 2). The clustering tendency of protein spectra per species was also evidenced by PCA (Fig. 2). The S. suis spectra comprise the largest group, as expected, and were separated from the spectra of the remaining Streptococcus species.
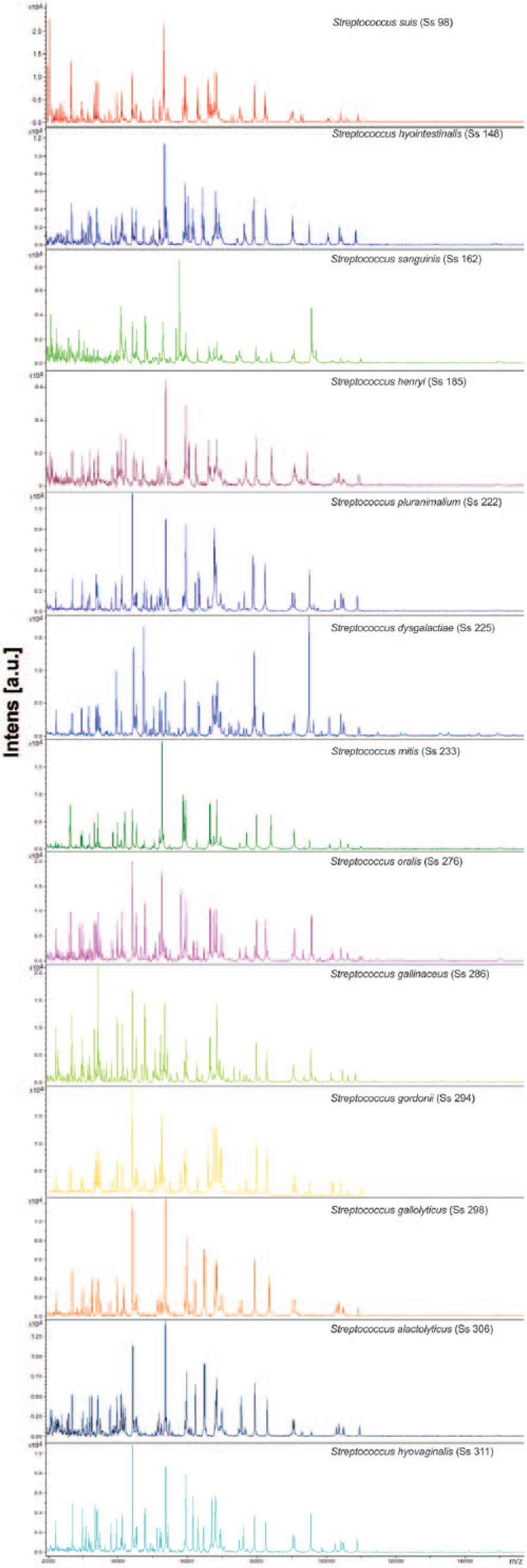
Comparison of the protein spectra of the Streptococcus species examined in the current study.
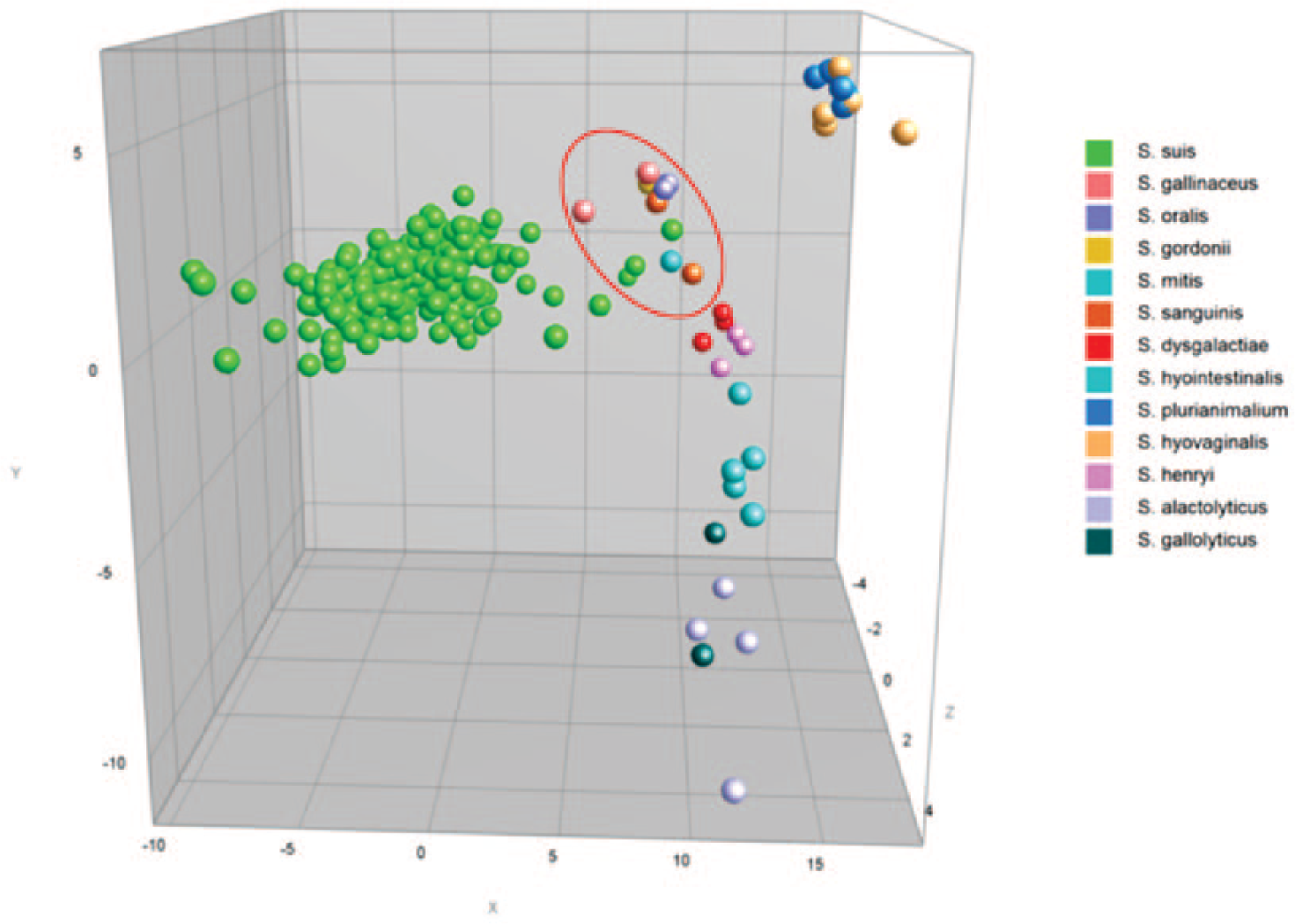
Principal component analysis of the spectra profiles of the Streptococcus spp. isolates. The color indicates the species. The “mitis-sanguinis” group species is within the red circle.
The 16S rDNA sequencing showed 99.2% agreement with MALDI-TOF MS for species identification; 2 S. pluranimalium isolates were misidentified as S. hyovaginalis by MALDI-TOF MS (SS224 and SS296). The phylogenetic tree (Supplemental Fig. 1, available at http://vdi.sagepub.com/content/by/supplemental-data) presents the field isolates and the Streptococcus type strains, and demonstrates the close relationship of the clusters comprised of the following pairs of species: S. hyovaginalis and S. pluranimalium; S. alactolyticus and S. gallolyticus; and S. mitis and S. sanguinis, as expected for these non–beta-hemolytic streptococci. 3 The high genetic similarity expected for the “mitis-sanguinis” group species (S. gordonii, S. mitis, S. oralis, and S. sanguinis) 8 was also observed and thus corroborated the clustering results of spectra analysis.
Among the 13 identified Streptococcus species, the automated biochemical systemg correctly identified all isolates of S. alactolyticus, S. dysgalactiae, S. gallolyticus, S. gordonii, S. hyointestinalis, S. mitis, S sanguinis, and S. suis. The isolates of S. gallinaceus, S. henryi, and S. hyovaginalis could not be identified, as these species are not included in the manufacturer’s database. The S. oralis and S. pluranimalium isolates were also not identified. Given that these species are in the database, it appears that the isolates of these species had biochemical profiles different from those contained in the manufacturer’s database.
The identification of Streptococcus species based on biochemical characterization is challenging because of their broad variation. 8 Even with the advent of commercial kits and automated systems, complete biochemical characterization of the species is still difficult. As an example, it has been reported that the Streptococcus-specific commercial identification kits achieve only an overall 50% correct identification level, with that level showing marked variation between species. 7 The automated system g used in our study appears to present a higher rate of correct identification. However, the system still has problems with closely related species, such as the “mitis-sanguinis” group, where misidentification can occur. 6 As shown in our study, the automated system g also has the disadvantage of a limited database, with some Streptococcus species absent, and a failure to recognize all possible biochemical variations for the species in the database.
The MALDI-TOF MS approach has been applied previously to a few Streptococcus species–specific studies1,13 and proved to be a valuable tool for genus and species identification. Our results corroborate the relevance of MALDI-TOF MS for Streptococcus species identification. Furthermore, the topology of spectra cluster analysis appears to sustain the species phylogenetic similarities, further supporting MALDI-TOF MS analysis as a rapid and accurate alternative to 16S rDNA sequencing.
Even though S. suis was the most frequently isolated species, corroborating the importance of the organism for the swine industry, the remainder of the identified species may represent unrecognized pathogens that may have significance to swine health. Therefore, MALDI-TOF MS is not only a rapid and accurate tool for microbiologic diagnosis but it also enables the identification of species not yet recognized for the role that they play in animal health.
Footnotes
Authors’ contributions
CEC Matajira, LZ Moreno, and AM Moreno contributed to conception and design of the study and to analysis and interpretation of data. LZ Moreno and AM Moreno critically revised the manuscript. VTM Gomes, APS Silva, DS Doto, FF Calderaro, and FN de Souza contributed to acquisition of data. RE Mesquita contributed to acquisition and analysis of data. APG Christ and MIZ Sato contributed to acquisition and interpretation of data. All authors drafted the manuscript, gave final approval, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
a.
BD Difco, Becton, Dickinson, Franklin Lakes, NJ.
b.
Bruker Daltonik, Bremen, Germany.
c.
Microflex mass spectrometer, Bruker Daltonik, Bremen, Germany.
d.
MALDI-Biotyper 3.1 software, Bruker Daltonik, Bremen, Germany.
e.
BioNumerics 7.5 software, Applied Maths, Sint-Martens-Latem, Belgium.
f.
VITEK 2 System-GP ID Card, bioMérieux, Hazelwood, MO.
g.
US Biological, Swampscott, MA.
h.
Fermentas, Glen Burnie, MD.
i.
Illustra GFXTM PCR DNA and gel band purification kit, GE Healthcare, Piscataway, NJ.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação de Amparo à Pesquisa do Estado de São Paulo, and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior. CEC Matajira, VTM Gomes, and LZ Moreno are recipients of PhD fellowships from FAPESP (2015/26159-1, 2013/16946-0, and 2013/17136-2). AM Moreno is a CNPq fellow.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.