
Abstract
The advancement of synthetic biology requires the ability to create new DNA sequences to produce unique behaviors in biological systems. Automation is increasingly employed to carry out well-established assembly methods of DNA fragments in a multiplexed, high-throughput fashion, allowing many different configurations to be tested simultaneously. However, metrics are required to determine when automation is warranted based on factors such as assembly methodology, protocol details, and number of samples. The goal of our synthetic biology automation work is to develop and test protocols, hardware, and software to investigate and optimize DNA assembly through quantifiable metrics. We performed a parameter analysis of DNA assembly to develop a standardized, highly efficient, and reproducible MoClo protocol, suitable to be used both manually and with liquid-handling robots. We created a key DNA assembly metric (Q-metric) to characterize a given automation method’s advantages over conventional manual manipulations with regard to researchers’ highest-priority parameters: output, cost, and time. A software tool called Puppeteer was developed to formally capture these metrics, help define the assembly design, and provide human and robotic liquid-handling instructions. Altogether, we contribute to a growing foundation of standardizing practices, metrics, and protocols for automating DNA assembly.
Introduction
DNA assembly technology is central to synthetic biology research. While there have been many powerful advancements in DNA assembly,1–6 large-scale experimental efforts would benefit from establishing more standard engineering practices and metrics. These practices and metrics would enable and evaluate, respectively, the automation capabilities of the processes involved. There are often comparisons made of DNA assembly to electronic circuit manufacturing.7–9 However, the types of metrics that apply to the latter do not necessarily translate to the former. The methodologies of assembling electronic parts have well-defined metrics for commonly accepted practices. Such metrics have yet to become well established for DNA assembly methods, which are often impacted by the vast sequence space possible for DNA-based designs. The many permutations of DNA sequence constructs make biological systems particularly well suited for performing advanced biological functions and control that can be exploited in many applications.10,11 However, this feature, along with varying construct-specific sensitivity to assembly method parameters, can make standard practices elusive for the assembling of DNA-based devices.
As the field of synthetic biology grows, there is a need in industry and academia for best practices, metrics, and protocols for DNA assembly to enable automation, reproducibility, and meaningful sharing of reliable results. 12 While many methodologies exist for DNA assembly, virtually none have risen as the standard for automation or been thoroughly evaluated using quantitative metrics. Academic labs require the ability for their results to be replicated by others, and variations between DNA assembly protocols can cause challenges for other groups trying to replicate tricky assemblies. Industry and academia would benefit from standardization and automation to develop high-fidelity and high-throughput DNA assemblies in a cost-effective manner. Adoption of these standards cannot be made compulsory, but the field would be well served to take up a unified set of methods and measurements to facilitate communication between researchers and compare results across separate experiments and institutions.
Incorporating standard practices into DNA assembly workflows will also allow for the facilitation of experimentation including the choice of automated processes. 13 The use of integrated automation to design, build, and test systems is well established in traditional rapid prototyping and can be applied with equal benefit to DNA assembly. Hillson et al. developed the j5 DNA Assembly Design Software to enable design of multipart DNA assemblies in silico, 14 and Linshiz et al. developed automated methods for generating human and machine-readable liquid-handling robot instructions for the construction of DNA. 15 However, there is a need for the next step to develop metrics that facilitate comparisons across both hardware platforms and assembly methods. Rapid, multiplexed, and high-throughput automated methods can accelerate the pace of development of experimental and industrial production. But researchers should understand these processes’ tolerances and bounds, and assess whether the affordability, efficiency, and reproducibility of automation compares favorably with manual methods for a particular experiment. 16 In many cases, an experiment’s scale is the primary reason for using automation instead of manual methods.
Along with the automation of any process comes the necessity of computer-aided handling of the process, as well as the tracking of material traversing the possible routes through the system. 17 Automation of DNA assembly requires the conversion of protocols to a language that both a human and a machine can understand, which is nontrivial and often requires redundant systems to achieve both. In addition, the flexibility to track samples through multiple possible methods based on measurements throughout the process can require an adaptable system that relies on consistent performance and well-established metrics that indicate a clear course of action.
Developing metrics to quantitatively evaluate the benefits of DNA assembly automation has proven challenging as there are a wide variety of DNA assembly methods and hardware available. 18 However, since the evaluation of all these protocols is similar, we developed a new type of metric, “Q-metrics,” to quantitate the benefit of automation. Q-metrics originated from chemistry to describe the energy released or required for a chemical reaction, and were later used to evaluate the ratio of energy output to energy input of nuclear fusion reactors enabling a comparative metric to determine if a specific reactor will break even (Q = 1). 19 Q-values, or Q-metrics, have been used extensively in determining the economic viability of fusion to provide a quantitative measurement for when a nuclear fusion reactor is economically viable. 20 Instead of comparing energy output, our Q-metrics compare factors representing researchers’ resources: cost and time (eqs 1 and 2):
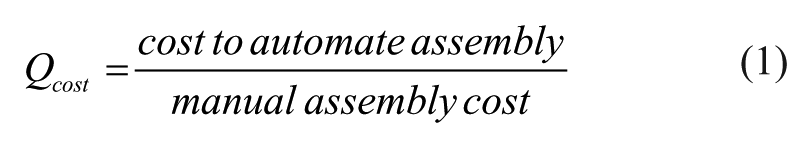
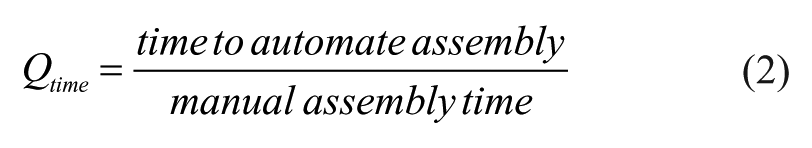
Q-metrics are automation method dependent, and a set of Q-metrics are made for each available liquid-handling robot. An example calculation can be seen in
Here we apply all these factors, proposed standard practices and metrics, automation, and computer-aided processing, to achieve efficient and high-throughput DNA assembly. We explore the effects on efficiency of automation, including the incorporation of a computer-aided workflow we call Puppeteer.
Materials and Methods
Evaluating DNA Assembly Efficiency
Using the Modular Cloning (MoClo) DNA assembly methodology,
6
we tested changes in DNA assembly outcomes (i.e., blue/white colony screening results) across a variety of parameters, including the total number of DNA parts in a reaction (two parts, five parts, and eight parts), the final concentration of the individual DNA parts (1, 2, and 4 nM), and the plating volume of recovered transformation reactions (5% or 50% of recovery volume). A summary of the DNA parts used in this experiment is in
Head to Head: Automated versus Manual
We next sought to directly compare DNA assembly metrics of five-part MoClo reactions prepared either manually (at the bench, by the researcher) or using a Freedom EVO 150 automated liquid-handling platform (Tecan, Männedorf, Switzerland), with an emphasis on capturing data that would later be fed into our Q-metrics calculations. We performed two different experiments, the first using only a single combination of promoter, ribosome binding site (RBS), gene, terminator, and backbone, and the second that explored 42 different combinations of similar five-part DNA assemblies. A detailed list of the DNA parts used in these experiments can be found in
Results and Discussion
Evaluating DNA Assembly Metrics
DNA assembly can be evaluated by many methods. Our analysis focused on the common blue-white colony-forming unit (CFU) screening assay where assembly reactions were transformed into highly competent bacterial cells and plated on media supplemented with X-Gal and IPTG. In this screen, colonies formed from cells containing empty destination vectors yield blue CFUs, while those containing properly assembled DNA produce white CFUs. While identifying a desired clone typically requires only a few white CFUs, much can be learned from the total number of white CFUs, as well as the percent of white CFUs present. As our proof of concept using MoClo, we wanted to understand how different factors (number of DNA parts, total assembly size, and concentration of DNA parts) affect the overall success of the process. Our goal of this parameter sweep was to understand critical variables when performing MoClo DNA assembly. We used the original MoClo protocol suggested by Weber and Fussenegger 22 as a starting point. The full protocol for our experiment can be read in Supplemental Protocol 1.
We first wanted to assess the effect the number of DNA parts would have on assemblies of a comparable final size. Briefly, we assembled three similarly sized assemblies (2828, 3152, and 3285 bp), composed of two, five, or eight parts, respectively, using identical protocols. Regardless of part concentration, as the number of parts in a given assembly increased, white CFUs decreased (
Fig. 1A
). While the lowest number of undesired blue CFUs resulted from the five-part assembly (
Fig. 1B
), the highest percentage of white CFUs was observed in reactions having the fewest parts, which decreased as the number of parts increased (
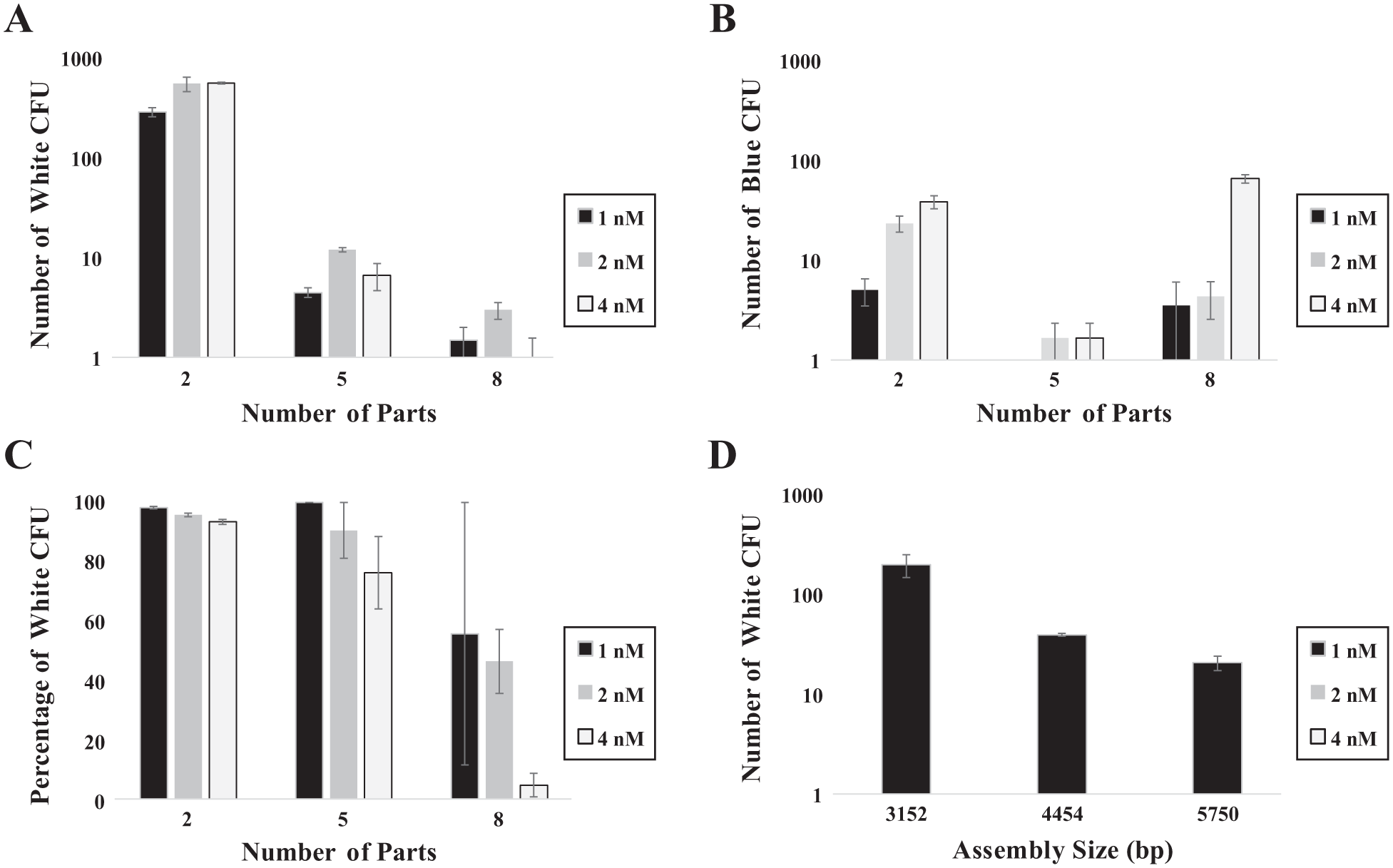
Parameter sweep performed to analyze, optimize, and standardize MoClo DNA assembly protocol for differing numbers of parts (
Next, we investigated the process of building three differently sized five-part assemblies using identical assembly protocols. As expected, we saw the most white CFUs from the smallest assembly (3152 bp) and the fewest from the largest assembly (5750 bp) ( Fig. 1D ). These data suggest that there may be a limit to the size of DNA construct that can be assembled or transformed with this method, potentially necessitating alternative methods for larger constructs. This finding agrees with previous studies showing that larger circuits are more difficult to transform, 25 and more parts in an assembly reaction decrease the probability of a complete assembly coming together. 26 Junction fidelity of MoClo overhangs has also been shown in the literature to impact CFUs. 27 To mitigate the effects of different junction fidelities, all our test parts used the same five MoClo overhangs for all assemblies.
We then tested our two-, five-, and eight-part assemblies with different final DNA part concentrations (1, 2, and 4 nM), with a particular focus on identifying concentrations that would yield a reasonable number of white CFUs for reactions with varying numbers of DNA parts. In general, as DNA part concentration increased, white CFU percentage decreased ( Fig. 1C ). As the number of parts increased, the negative impact of higher part concentration was exacerbated. We hypothesize that more total DNA going into a reaction, with either more parts or higher part concentration, may overwhelm the processing power of the available enzymes, limiting the overall reaction efficiency. These deficiencies might be addressed through increasing enzyme concentration, altering the balance of ligase versus endonuclease, or adjusting reaction temperatures or times. We also note that due to lower total CFU counts for five- and eight-part assemblies, error bars are larger.
While the focus of this study was on total CFU number and blue-white percentage, we also examined common molecular biology metrics such as transformation efficiency. Molecular and synthetic biology literature has reported transformation efficiency variously as CFU/µL reaction,
28
CFU/fmol DNA, and CFU/pg DNA,
29
depending on the context. From a biochemical standpoint, we should favor CFU/fmol DNA as a more useful metric than CFU/pg DNA, since the size of the DNA construct is factored in. The unit CFU/µL reaction may be more meaningful to industry as it gives the user information about how much assembly reaction mix is required to obtain the target number of CFUs while minimizing reagent cost. However, we calculated these values (
DNA Assembly Using Liquid-Handling Robots
Once the assembly protocol standardization was finalized, we uploaded our five-part assembly GenBank files into our Puppeteer software. The software generates both human-readable manual and Tecan robotic liquid-handling instructions. A demo of the Puppeteer software is available via GitHub, and instructions can be found in Supplemental Protocol 4. Puppeteer pulled DNA part sequence information from our SynBioHub in silico library to define a total of 42 unique assemblies that were all composed of five parts, had properly matching MoClo DNA overhangs, and followed the defined assembly organization of Promoter: Ribosome Binding Site (RBS): Gene: Terminator: Destination Vector. Using these instructions, we performed a head-to-head study to compare hands-on time, cost, and assembly efficiency between assemblies performed either on a Tecan robot or manually by a graduate student at Boston University and by an assistant staff member at MIT Lincoln Laboratory. We manually performed transfers to a thermocycler, as well as subsequent transformation steps, for all methods. We also executed DNA dilutions manually, and DNA source plate layout was summarized in a human-readable experimental summary file generated by Puppeteer.
One challenge of using our Tecan robot was the minimum allowable volume for reproducible fluid transfer, which was 2 µL for our hardware setup. We standardized all reactions to have this as a minimum transfer volume, giving us a total reaction volume of 20 µL for both manual and automated assembly. Another challenge was dead volume in the source DNA plate. Depending on the liquid-handling hardware used, varying amounts of additional liquid are required in each well that is accessed. The current version of Puppeteer does not account for this; however, future versions of the software will adjust reagent volume needs based on the platform used and its respective dead volume requirements. An additional risk is the evaporation of reagents over the course of pipetting tasks being executed for an assembly job. Use of an alternative liquid-handling system such as an acoustic dispenser with reproducible fluid transfers in the nanoliter regime would drastically improve both timing (using acoustics as opposed to a robotic arm and disposable tips to move fluid) and cost (lower reagent volume required), 15 and would address many of these issues. An advantage of using the Tecan liquid-handling robot paired with the Puppeteer assembly software was that after the sample was transferred to the source plate, the Tecan instructions generated by Puppeteer allowed the researcher to simply run the machine and perform other tasks instead of entering pipette steps manually into the robot control software (EvoWare), which is very time-consuming for combinatorial assembly. The benefit here is that automating tedious pipetting tasks would free up valuable researcher time.
Our head-to-head study between manual and automated assembly showed no notable difference in number of CFUs or percent white CFUs (data not shown). Sanger sequencing of plasmid insert regions of three white colonies from 10 randomly chosen plates confirmed that the assemblies were correct for all DNA parts and overhangs as well. Our Q-metrics ( Fig. 2 ) confirmed the straightforward expectation that single assemblies are preferably done manually, whereas performing a set of 42 assemblies is better suited to automated methods.
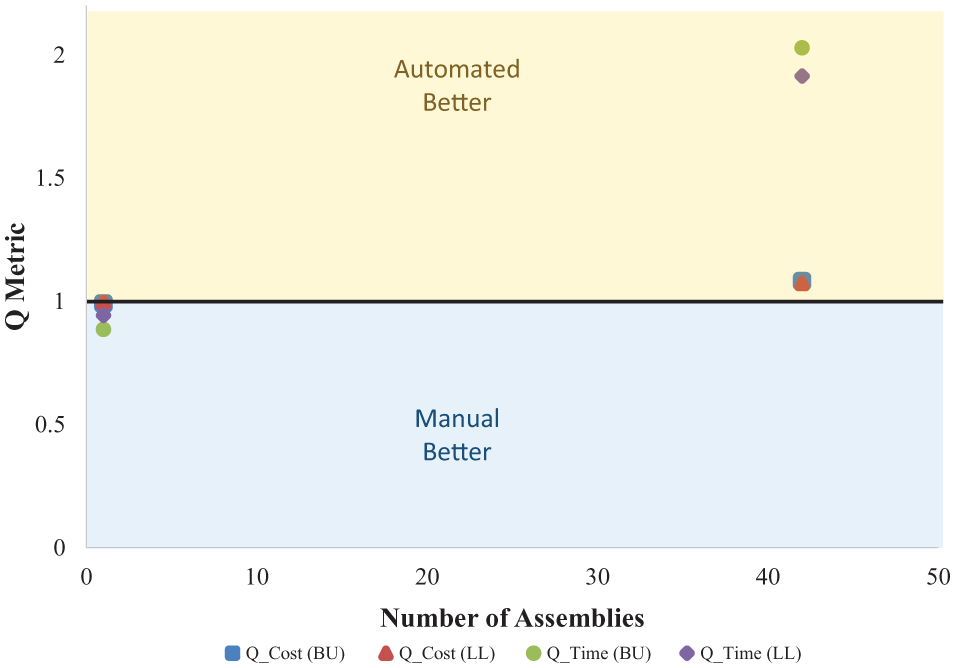
Graph showing results of head-to-head study comparing manual and automated DNA assembly methods using a liquid-handling robot. Q-metrics are defined in eqs 1 and 2. A Q-metric higher than 1 infers cost savings using robotics, whereas the opposite is true for Q-metrics less than 1. The labels BU and LL refer to where the manual assembly took place (Boston University or Lincoln Laboratory), and both are compared to the automated Tecan liquid-handling robot located at BU.
The primary reason for our Q-metrics favoring automation at high numbers of assemblies is the savings in staff time. Both Q-cost and Q-time use staff time for salary and manual assembly calculations, respectively (
In this study, the transition phase from “prefer manual” to “prefer automated” does not require high numbers of assemblies to benefit from automation. For other laboratories and conditions, different reagent costs and staff salaries will yield different results, potentially shifting the transition point in either direction. In our case, Q-time considered only hands-on time done by an experienced user; however, new users would have to be treated differently. And Q-cost only considered costs from the assembly itself and did not include up-front capital equipment costs or maintenance costs for the liquid-handling robot. We intend to incorporate these additional cost elements into our Q-metrics to enable quick cost/benefit analyses to groups interested in acquiring new hardware. Also, while our setup times were similar for a single assembly and 42 assemblies, moving beyond a single well plate will impact time and cost. Finally, other robotic liquid-handling systems will require a similar study to be performed to benchmark speed and optimize the protocol for a new robot. The use of Q-metrics to evaluate the specific needs of a given context is easily adaptable to include additional factors such as amortization of equipment costs or employee training times.
Puppeteer Gene Assembly Wizard
Developing a pipeline for automated DNA assembly requires both hardware and software. The Puppeteer software platform manages the automation planning and scheduling aspects of the pipeline, taking a user-defined assembly composed of available DNA parts (e.g., promoters, ribosome binding sequences, coding sequences, terminators, and vectors) in GenBank format from an in silico library as the input. Puppeteer uses this information to create an assembly plan that takes into account possible successful combinatorial permutations of parts that meet the user’s specification. This plan is then transformed into a series of protocols given the target assembly format and the capabilities of the lab. Currently, Puppeteer is available as a proof of concept that can provide up to 96 possible design combinations of promoters, RBSs, genes, terminators, and vector backbones. While the position of each DNA component is fixed in the demo, the full version of Puppeteer we are developing now will allow the user to define the part “category” and assembly order. Future Puppeteer will generate full factorial designs for all compatible series of parts within the user-defined order, but can be downselected afterward by the user if not all combinations are desired. The current output of Puppeteer is threefold: (1) final assembled composite DNA sequence files (GenBank format) that feed back into the user’s SynBioHub in silico library, (2) liquid-handling instructions for performing the DNA assembly with the user-selected liquid-handling format (robots, or manually by hand), and (3) assembly evaluation Q-metrics that determine the relative cost and time savings of each liquid-handling option ( Fig. 3 ). SynBioHub is an open-source design repository for synthetic biology, built in Synthetic Biology Open Language (SBOL), and it serves as a standard for genetic designs enabling the sharing of design parts. The adoption and use of SynBioHub, a community-driven effort, has the potential to overcome the reproducibility challenge across laboratories by helping to address the current lack of information on published designs. 30 Puppeteer imports part libraries from SynBioHub and exports selected assemblies made using the software.
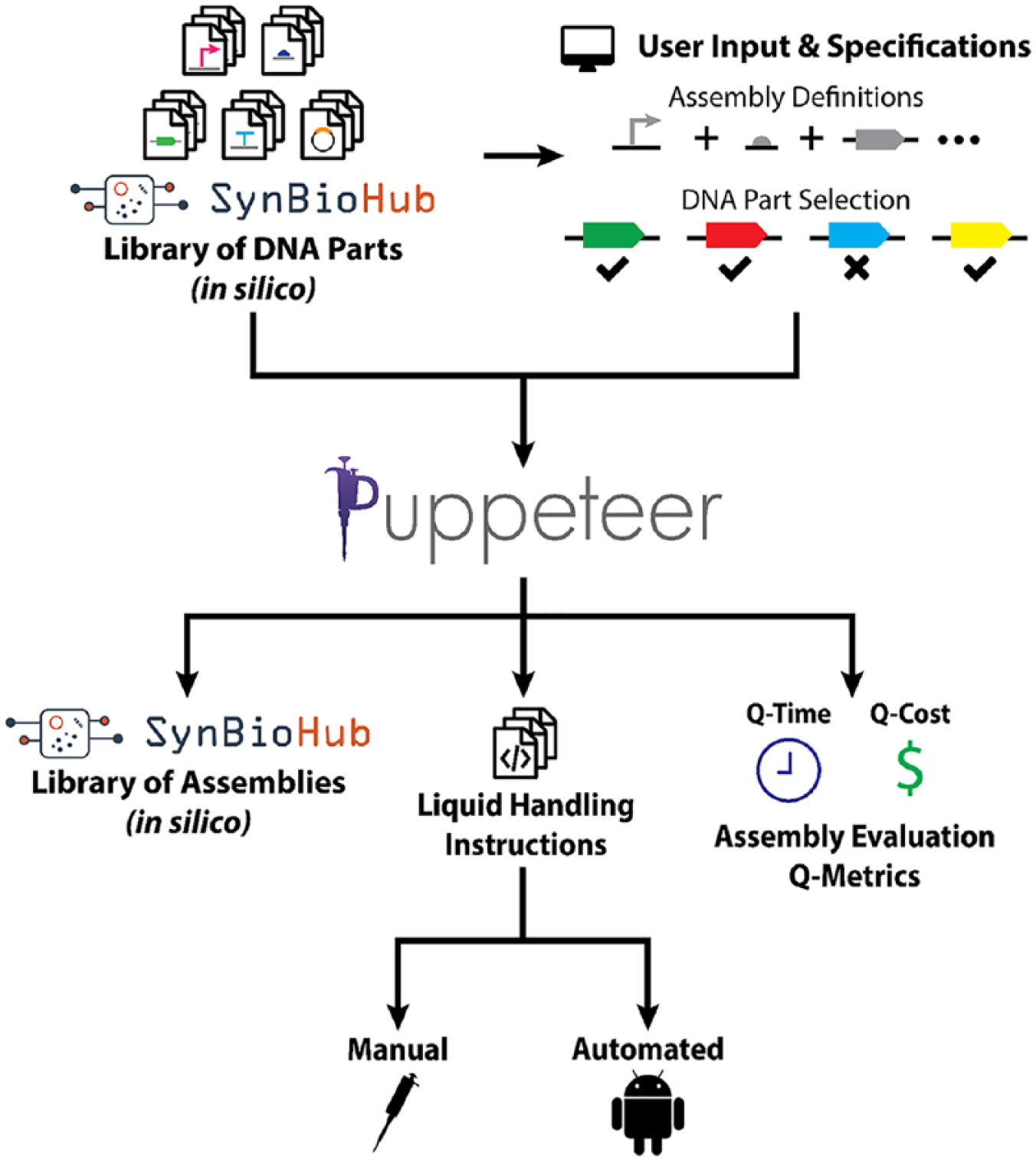
Schematic for DNA assembly pipeline automated by Puppeteer Gene Assembly Wizard software. The left portion of the figure illustrates that Puppeteer can connect to remote repositories of DNA parts (e.g., SynBioHub) as well as take input regarding required final DNA assemblies. The output of the process (right) is a protocol description for humans or liquid-handling robotics that meets the Q-metrics reported.
The Q-metrics output by the Puppeteer demo are currently hard-coded to output the values corresponding to a 42-part assembly, but the full Puppeteer software in development now will generate Q-values at run-time based on the assembly job submitted, and the liquid-handling hardware chosen, in a future release. The liquid-handling instructions are particularly helpful as programming the Tecan in EvoWare took 2 h, rather than the minutes it took to generate the same pipetting commands using Puppeteer. Generating the pipetting commands for DNA assembly tasks where the source and destination wells are a function of the job submitted is particularly problematic and time-consuming if done manually, since reagent plate layouts will likely change between jobs, requiring new commands to be generated for each job submitted. Manual generation also increases the possibility of human-based errors, necessitating a software tool that can procedurally generate pipetting commands based on unique job submissions. Overall, Puppeteer guides the user from DNA parts to final assemblies (both in vitro and in silico) and provides quantitative metrics to assess when automated platforms will save both money and time. The instructions to run our Puppeteer demo can be found in Supplemental Protocol 4.
Automation and standardization of synthetic biology processes require new tools and metrics that can support rapid, reproducible, systematic DNA assembly and screening. Our work demonstrates a useful methodology (Q-metrics) to analyze pipelines for DNA assembly, enabling better sharing, automation, and evaluation of these protocols to quantify their worth.
To guarantee the generation and collection of robust, repeatable DNA assembly data, we tested a MoClo methodology-based protocol in two different laboratories (Boston University and MIT Lincoln Laboratory). Protocols were standardized between the two laboratories and assembly parameters were optimized through a parameter sweep, studying the impact of the number of DNA parts, the DNA part concentration, and the total size of DNA assembly products. We used our optimized protocol to then execute the manual and automated assembly of 42 DNA circuits via an off-the-shelf liquid-handling robot. While the nonlabor costs and cloning efficiencies remained similar, there was a significant decrease in researcher hands-on time when using a liquid-handling robot.
We augmented DNA assembly automation using our design-and-build software tool, Puppeteer, to plan both physical and in silico assembly of DNA parts. Using only GenBank files as inputs, Puppeteer provides a platform that integrates the steps of genetic circuit design, planning, and building, while providing useful metrics (Q-metrics) to evaluate the automated assembly process. Our Q-metrics provide a quantitative approach to tackle the question of when to automate, and their flexibility in calculation can allow users to explore and identify which factors contribute to their costs and time in automation. While our work described here focuses on a relatively simple subset of the processes necessary to automate DNA assembly, namely, the preparation of DNA assembly reactions, we are working to incorporate downstream bacterial transformation and plating, as well as colony picking and plasmid DNA isolation, into the automated workflow. These additional processes will be used to update our Q-metrics to present a more holistic and informative picture of the entire DNA assembly process. Altogether, our work provides a proof-of-concept design of an automated DNA assembly pipeline suitable for testing and evaluation to enable scale-up of assembly construction.
Supplemental Material
TECH825335_Supplemental_Material – Supplemental material for Standardizing Automated DNA Assembly: Best Practices, Metrics, and Protocols Using Robots
Supplemental material, TECH825335_Supplemental_Material for Standardizing Automated DNA Assembly: Best Practices, Metrics, and Protocols Using Robots by David I. Walsh, Marilene Pavan, Luis Ortiz, Scott Wick, Johanna Bobrow, Nicholas J. Guido, Sarah Leinicke, Dany Fu, Shreya Pandit, Lucy Qin, Peter A. Carr and Douglas Densmore in SLAS Technology
Footnotes
Acknowledgements
We also acknowledge the software engineering effort and insights of the Software & Application Innovation Lab (SAIL) within the Hariri Institute for Computing at Boston University as well as the efforts of Dr. Swapnil Bhatia, now with Catalog DNA.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Douglas Densmore declares his involvement with the companies Lattice Automation (Boston, MA) and Asimov (Cambridge, MA); however, all authors declare no conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors wish to acknowledge the following sources of financial support. Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award number R01CA173712, the National Institute of General Medical Sciences of the National Institutes of Health grant P50 GM098792, and the National Science Foundation’s Expeditions in Computing Program under grants 1522074, 1521925, and 1521759. Distribution statement: approved for public release; distribution unlimited. This material is based upon work supported by MIT under Air Force contract no. FA8721-05-C-0002 and/or FA8702-15-D-0001. Any opinions, findings, conclusions, or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of MIT.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.