
Abstract
Pancreatic cancer is one of the most lethal forms of cancer and has proven to be difficult to treat through conventional methods, including surgery and chemotherapy. Gene therapy serves as a potential novel treatment to interfere with genes that make this cancer so aggressive, but free nucleic acids have low cell uptake due to their negative charge and are unstable in circulation. Nanoparticles can serve as an effective carrier for a wide variety of gene therapies for pancreatic cancer as they can improve the circulation time, decrease the recognition by the immune system, and be functionalized to target specific surface proteins. In this review, we focus on therapeutic strategies using nanoparticles as carriers of small interfering RNA (siRNA), microRNA (miRNA), and gene augmentation (DNA) therapies in the context of pancreatic cancer. Lastly, we discuss the future outlook of nanoparticle-based therapies, including challenges in the clinical setting.
Introduction
In 2018, approximately 55,400 people will be diagnosed with and an estimated 44,300 will die from pancreatic adenocarcinoma in the United States alone. 1 With a 5-year survival rate of 8%, 1 pancreatic cancer is among the most lethal of all cancers and is one of the few cancers for which patient survival has not substantially improved over the past 40 years. 1 Although many advancements have been made in pancreatic cancer research, the incidence of this disease continues to increase without knowing an exact cause. 2
The poor prognosis of pancreatic cancer can primarily be attributed to its typically late-stage diagnosis. More than 80% of presented cases have already reached metastasis or a locally advanced state. 3 Furthermore, pancreatic cancer can be controlled only if it is found before it has spread and can be resected completely. 4 As a result, the only current effective way of combating the high death rates is to effectively diagnose and treat the cancer at an earlier stage. 3
Pancreatic cancer typically originates as a firm, highly sclerotic mass of infiltrating adenocarcinomas. 5 The edges of these cancers consist of long tongues of carcinoma that extend long beyond the main tumor mass, resulting in poorly defined edges of the cancers. 5 Pancreatic cancers are infiltrative neoplasms and often have vascular and perineural invasion, 5 leading to an early propensity for metastasis in numerous areas of the body. 6 The vast majority of the cancers develop from microscopic precursor lesions called pancreatic intraepithelial neoplasia (PanIN) and originate in small pancreatic ducts. 7 Whereas normal ducts are characterized by a low cuboidal epithelium, PanIN-1 lesions are characterized by the presence of mucinous hyperplasia. As the lesions become increasingly cancerous, they become characterized by cytological atypia (i.e., abnormal cell shape) and cells in the lesions develop enlarged nuclei and become increasingly crowded, causing papillary infolding ( Fig. 1 ). 7
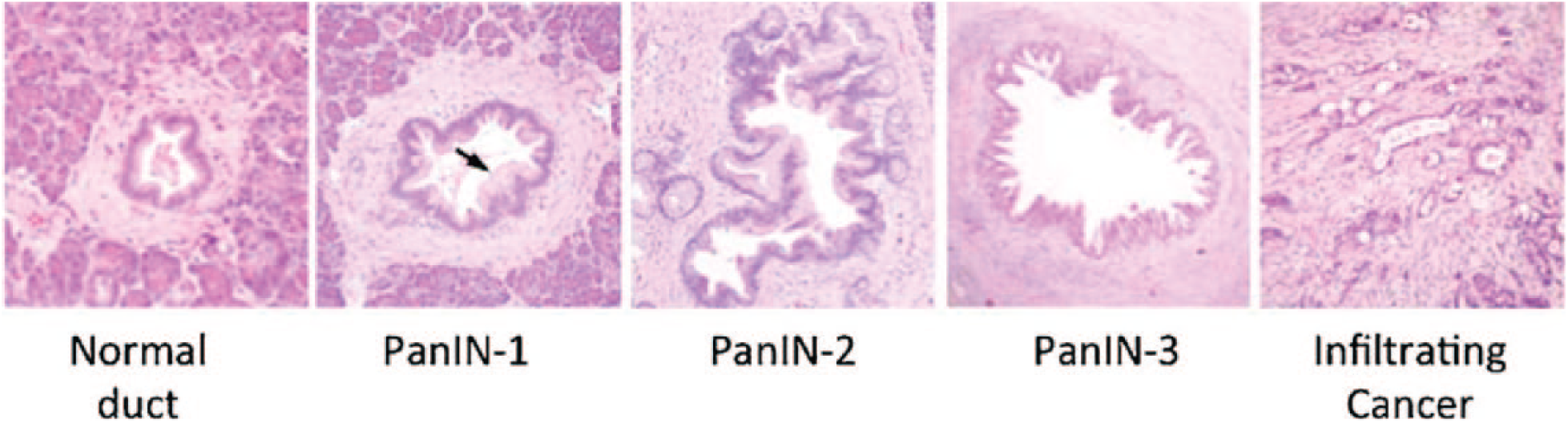
Histology shows the progression of PanINs in pancreatic cancer. Reprinted with permission from Macgregor-Das et al. 7 J. Surg. Oncol. 107(1): 8–14. © 2012 Wiley Periodicals, Inc.
Many genetic studies have identified several key genes and molecular pathways that are altered through this process and drive the formation of pancreatic cancer; the most common of these mutations is proto-oncogene KRAS2, which accounts for 90%–95% of total pancreatic cancer cases. 5 The activation of KRAS allows for rapid cell proliferation and is therefore considered an initiating event of pancreatic cancer. Similarly, the inactivation of p16/CDKN2a and p53, cell cycle regulators, has demonstrated the ability to inhibit tumor suppression capabilities. 5 In more than 90% of pancreatic ductal adenocarcinomas, CDKN2a has been found to be inactivated, with the vast majority occurring in the early PanIN stages. 7 Because of the large influence of genetics on pancreatic cancer development, it is estimated that the overall risk of pancreatic cancer is strongly correlated with familial risk, with a 6.79-fold increase in the likelihood of developing pancreatic cancer when a relative had pancreatic cancer. 3
Despite a general understanding of the pathology of pancreatic cancer, the current available treatments still face numerous shortcomings. Pancreatic cancer is difficult to detect and diagnose early due to its lack of noticeable symptoms during initial stages. Furthermore, only around 20% of tumors appear confined to the pancreas, and even fewer are resectable. 8 Surgery currently offers the only realistic chance to cure exocrine pancreatic cancer, 8 but this is only possible in a minority of cases. 2 Another method of treating pancreatic cancer is chemotherapy, with the two most commonly used chemotherapy drugs being fluorouracil and gemcitabine (GEM). 9 GEM, the current first-line chemotherapeutic drug for pancreatic cancer, has demonstrated a 23% 5-year survival in patients 10 and can be combined with albumin-bound paclitaxel as a potential treatment with low toxicity for metastatic pancreatic cancer. 11 FOLFIRINOX, a combination of 5-flurouracil, leucovorin, irinotecan, and oxaliplatin, is also commonly considered a standard treatment option for patients with metastatic pancreatic cancer. 9 However, despite an extension in median survival by 4.3 months, there are significant concerns regarding toxicity and severe side effects. 11 Thus, it is clear that there is a need for novel therapeutics for pancreatic cancer, as the current standard of care is not effective enough or without toxicity.
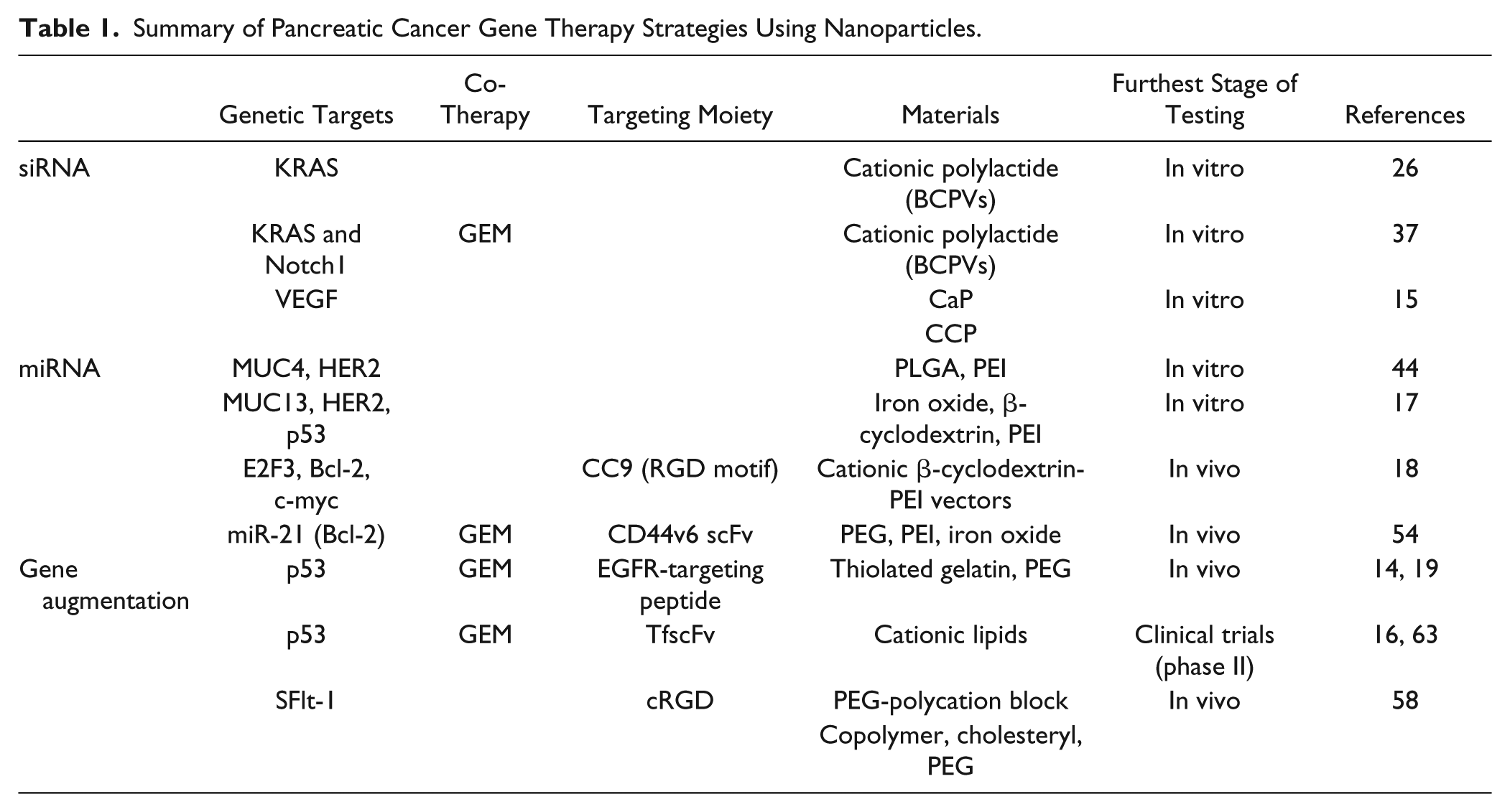
In this review, we highlight the use of nanoparticles as a carrier for gene therapies, which could potentially be a novel therapeutic that can advance patient care for pancreatic cancer. The goal of gene therapy in cancer is to provide a safe and efficient treatment to cancer patients by specifically targeting and deterring cancer cells within the body. 12 Gene therapy has large potential in pancreatic cancer treatment, whether it be turning on genes that induce apoptosis or turning off genes that increase proliferation. Nanoparticles offer an ideal platform for incorporating desirable characteristics into a single gene delivery system, 13 and have wide applications for the purposes of controlled drug release and cancer cell targeting. 12 Furthermore, the surfaces of nanoparticles can be modified to improve the effectiveness of cancer targeting. 12 Herein, we elaborate on nanoparticle carriers constructed from a variety of materials, including gelatin, 14 polymers such as polyethylene glycol (PEG), 15 and lipids. 16 The nanoparticles typically range in size from 100 to 200 nm,17–19 but can be as large as 400 nm, 16 and target various cell surface receptors, such as epidermal growth factor receptor (EGFR), while carrying different classes of gene therapy for distinct genes such as KRAS and p53 ( Table 1 ). Herein, we provide an overview of the latest developments in nanoparticle-based gene therapy for pancreatic cancers. First, we discuss small interfering RNA (siRNA) therapies, which are often used in the silencing of pathological genes and have recently been used in combination with nanoparticle carriers for more effective drug delivery. We then focus on microRNA (miRNA) therapies, which can propagate several gene regulation mechanisms and potentially provide new therapies in pancreatic cancer. Finally, we consider gene augmentation and conclude by discussing future perspectives and challenges.
Summary of Pancreatic Cancer Gene Therapy Strategies Using Nanoparticles.
siRNA
siRNA is a type of double-stranded, interfering RNA (RNAi) that is a 21- to 23-nucleotide sequence, and has recently been used in combination with nanoparticle carriers for more effective drug delivery. 20 The nanoparticle carriers are crucial to the effective delivery of siRNA to cells, as RNA therapeutics are oftentimes rapidly degraded when injected alone into the bloodstream. 21 Once internalized, siRNA forms the activated RNA-induced silencing complex (RISC) in the cytoplasm by integrating with the RISC multiprotein complex, and can be considered a “guide strand.” 22 The RISC then binds completely to the mRNA of interest, effectively silencing the gene by preventing expression of mRNA by means of endonucleolytic cleavage.22,23 Due to its ability to target a variety of genes, siRNA is being used to target oncogenes, cell cycle regulatory genes, and growth factors. 20
Encapsulating the siRNA into a nanoparticle carrier can be beneficial in multiple ways: decreased recognition as a foreign particle by the immune system, enhanced tumor targeting, improved cell uptake, and longer circulation time—which in turn allows for increased accumulation in the tumor by the enhanced permeability and retention (EPR) effect. 24 Over time, nanoparticles will accumulate in the tumor site due to this increased permeability in tumor vasculature compared with normal tissue, which allows particle penetration into the tumor site, while poor lymphatic drainage allows nanoparticle retention. 24 When combined with the desirable delivery aspects associated with nanoparticle encapsulation, siRNAs have the potential to become a highly effective therapeutic for pancreatic cancer. Since siRNAs can target single genes, they can play a crucial role in manipulating the tumor environment with strong anticancer effects when targeting mutated genes that cause unregulated cell growth. 23
One of the potential targets for siRNA in the realm of pancreatic cancer therapy is oncogenes. An oncogene is the mutated form of a proto-oncogene—a gene that normally controls cell proliferation. Mutations in proto-oncogenes allow for uncontrolled cellular proliferation as the normal regulatory mechanism of the gene is altered. 25 As mentioned, KRAS is one such proto-oncogene and represents an excellent target for pancreatic cancer gene therapy as mutations in this gene are present in about 90% of pancreatic carcinomas. 26 The activation of the KRAS oncogene is thought to enhance the growth of tumors by altering cellular signal transduction. 26 Additionally, Notch signaling has been shown to play an important role in the development of pancreatic cancer, and overexpression of Notch1 has been reported to contribute to the epithelial–mesenchymal transition (EMT) process.27,28 The EMT process involves epithelial cells losing their junctions and basal polarity, which in turn converts cells to have a mesenchymal phenotype with enhanced migration, invasion, and resistance to apoptosis.27,28 This phenotypic transition is the key step change for tumor metastasis, and contributes to the development of chemotherapy drug resistance.29–32 Furthermore, Notch1 is a downstream receptor of KRAS signaling and functions collaboratively with KRAS to induce pancreatic cancer.33–37 However, without KRAS present, Notch1 alone is not sufficient to promote tumorigenesis. 35
Yang et al. synthesized biodegradable charged polyester-based vectors (BCPVs) and tested the transfection efficacy and the cytotoxicity to cells. 26 These BCPVs are cationic polylactides, which have a well-defined structure and are able to easily form complexes with siRNA due to electrostatic interaction. An MTT assay was used to show that BCPV caused no cytotoxicity to MiaPaCa-2 human pancreatic cancer cells, even at doses up to 160 µg/mL. Five different conjugates were created by varying the BCPV/siRNA weight ratio from 1:1 to 16:1 and were tested for transfection compared to Oligofectamine (Oligo), a known transfection reagent. A weight ratio of 8:1 BCPV/siRNA had similar transfection efficiency and KRAS knockdown to the Oligo-siRNA positive control. To test cell migration of the transfected cells, an in vitro wound healing assay was performed. Decreased proliferation (40% wound closure) was demonstrated for the BCPV-siRNA construct and the Oligo-siRNA after 72 h, compared with 80% wound closure for the free siRNA and the negative control blank. This decreased proliferation demonstrates how the knockdown of KRAS in the cancer cells decreases the rapid growth typically associated with cancer. 20 Additionally, the BCPV-siRNA also showed decreased invasiveness and a doubled rate of apoptosis compared with the controls. 26 Overall, their study demonstrated KRAS knockdown by biocompatible, biodegradable nanoparticle carriers. 26
Given these proof-of-concept studies regarding BCPV nanocarriers as a potential therapy for pancreatic cancer, Yang et al. developed the BCPV nanocarrier system with both KRAS and Notch1 siRNA. They tested these gene therapy nanocarriers in conjunction with GEM on the MiaPaCa-2 human pancreatic cancer cell line, to examine the silencing efficacy and reduced drug resistance of GEM. 37 BCPVs were made to have tunable zeta potential and size by changing the weight ratio of BCPVs to siRNAs. The ratio of 8:1 BCPV/siRNA was found to have maximum loading of siRNA with an average diameter of 104 ± 15 nm and a zeta potential of 40 ± 5 mV. Through fluorescence imaging, the authors confirmed the internalization and endosomal escape of BCPV-siRNA through the proton sponge effect, and silencing efficacy of BCPV-siRNA by RT-PCR. It was found that both KRAS and Notch1 expression had both significantly decreased by approximately 85% and 70%, respectively, coupled with decreased cell proliferation of up to 60% and a cell morphology change to epithelial-like cells. GEM sensitivity was then increased upon co-delivery with siRNA as at a 2000 nM dose of GEM, growth was inhibited by 92% ± 4.1%, as opposed to 28% ± 1.7% with 2000 nM GEM treatment only. Also, increased apoptosis of 50% was observed with the combination therapy compared with GEM monotherapy, in which only 20% of the cells were apoptotic at the highest 2000 nM dose. 37 The authors successfully showed that KRAS and Notch1 knockdown in pancreatic cancer cells in vitro constrained cell proliferation and increased GEM sensitivity, and thus apoptosis. 37 The authors concluded that the BCPV nanocarrier could serve as a promising potential carrier for siRNA therapy and chemotherapy co-delivery due to its high stability and proven gene silencing ability with both Notch1 and KRAS.
Another target for siRNA gene therapy is methyl-CpG binding domain protein 1 (MBD1), which is a transcriptional regulator that binds to the methylated CpG islands, which results in repression of tumor suppressor genes. Abnormal methylation of tumor suppressor genes via improper chromatin state, achieved in part by MBD1, contributes significantly to the development of cancer.38,39 While it is common to have overexpression of MBD1 in pancreatic carcinomas in humans, its role in the development of pancreatic cancer is not well defined. 40 Thus, repressing MBD1 using siRNA delivered by nanoparticles could potentially have therapeutic benefits. Though MBD1 and KRAS are involved in uncontrolled cell proliferation, angiogenesis is a key process that leads to rapid growth of cancer and its rapid metastasis, and genes related to angiogenesis are also potential targets for RNAi therapy.41,42 Vascular endothelial growth factor (VEGF) is one of the most significant contributors to angiogenesis, and is highly expressed in pancreatic cancer cells.42,43 Pancreatic cancer patients positive for VEGF-RII were reported to live 8.5 ± 1.2 months postresection, while those who were VEGF-RII-negative had a median survival of 16.7 ± 1.8 months postresection, and VEGF-RII treatment led to significant growth stimulation in VEGF-negative cells. 41
To silence VEGF, Pittella et al. developed a platform using a novel PEG-block charge-conversional polymer (CCP) for siRNA interference of VEGF in human pancreatic cancer cells (PANC-1). 15 Calcium phosphate (CaP) was used to form a stable core that can incorporate polyanions such as nucleic acids, while the PEG-block polymer prevented the agglomeration of the CaP and enhances internalization and transfection efficiency. The CCP leads to strong membrane destabilization under the acidic environment inside the endosome and improves endosomal escape by switching from the mono-protonated form to the di-protonated form. The authors successfully synthesized and characterized the hybrid CaP-siRNA with PEG-block polymer nanoparticles, which had a uniform size of 42 ± 5 nm. A high siRNA encapsulation percentage of 80% was achieved and no cytotoxicity in PANC-1 cells was observed with the nanoparticle itself. Eighty-two percent gene knockdown of VEGF in PANC-1 cells was observed and found to be statistically significant compared with naked siRNA or nanoparticles without the PEG-block polymer ( Fig. 2 ). 15 Although in vivo experiments were not provided, these studies lay a concrete foundation of siRNA delivery in pancreatic cancer cells for future studies in vivo.
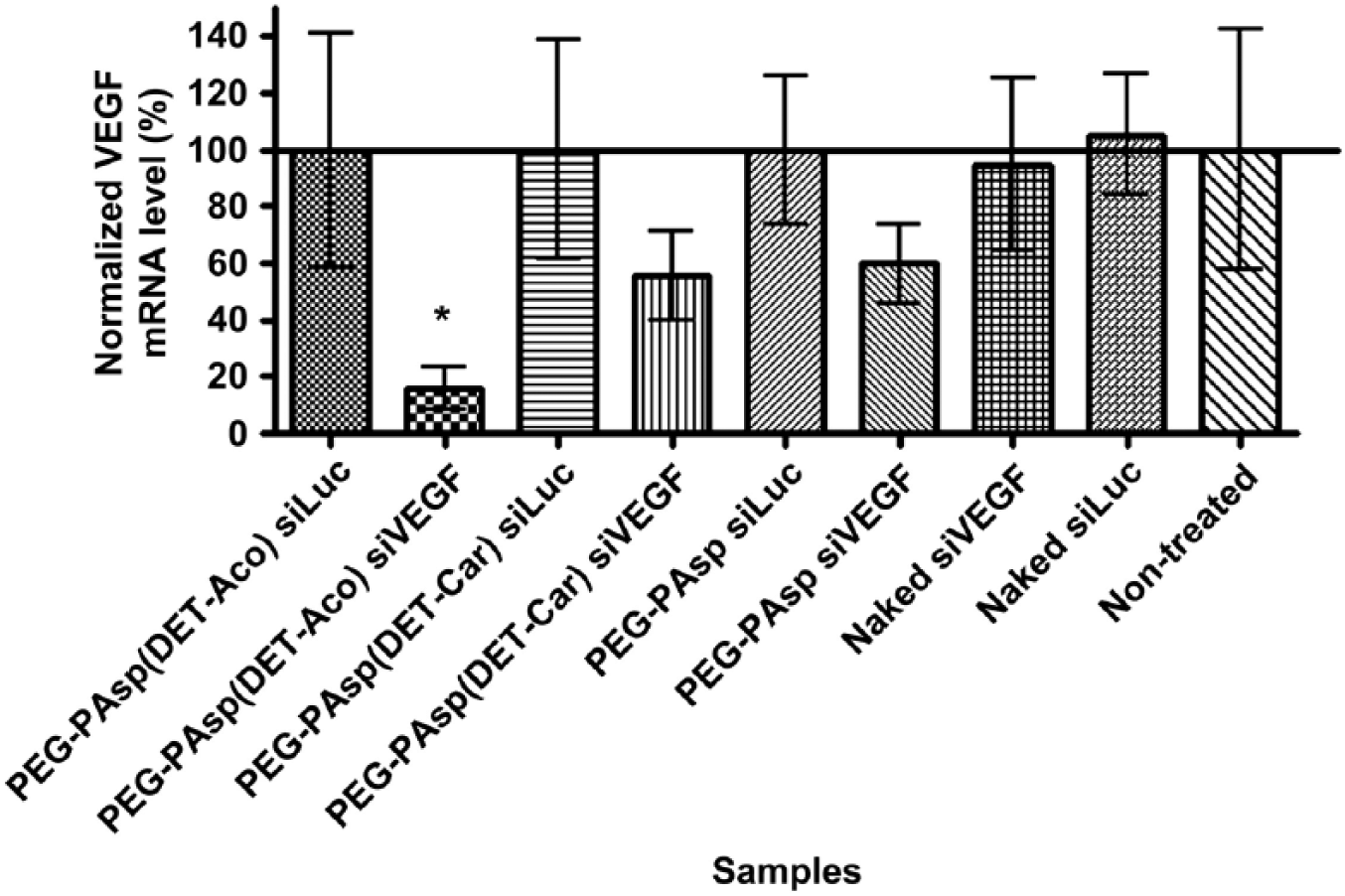
Gene knockdown of VEGF at 24 h after PANC-1 cells were exposed to the treatment groups for 3 h. The silencing efficiency of VEGF was greatest for the CCP group. The nontargeted control sequence had no effect on the VEGF expression. PEGPAsp(DET-Aco) = CCP group; PEGPAsp(DET-Car) = non-CCP group; siLuc = nontargeted control sequence. Reprinted with permission from Pittella et al. 15 Biomaterials 32: 3106–3114. © 2011 Elsevier.
miRNA
miRNAs are 19–25 nucleotides long and are endogenous noncoding RNA molecules found in plants and animals. 23 miRNAs influence the output of many protein-coding genes by partial complementary base pairing to mRNA, subsequently silencing the mRNA. miRNA propagates several gene regulation mechanisms, including translational repression, mRNA degradation, and mRNA endonucleolytic cleavage. 23 As a result, miRNAs have significant roles in cancer initiation, progression, metastasis, and therapeutic efficacy through their interactions on pathologically relevant gene targets. 44 Since miRNA binds only partially complementary to mRNA, multiple mRNAs can be targeted rather than just one, as in the case with siRNA.
The advent of miRNAs as agents in pancreatic cancer propagation provides new therapies through the inhibition of overexpressed oncogenic miRNAs or restoration of downregulated tumor suppressor miRNAs in cancer cells. miR-150 is a tumor suppressor miRNA that was found by Arora et al. to be downregulated in the majority of pancreatic tumor cases based on 20 patient pancreatic cancer tissue specimens.
44
Restoration of miR-150 could make for an effective approach for therapy; however, exogenous delivery of miRNA is limited and, similar to siRNA, and complicated by its physiochemical properties, particularly its negative charge, hydrophilicity, vulnerability to nuclease degradation, and inefficient uptake.
45
The authors investigated the efficacy of a poly(
In addition to MUC4, MUC13 is another transmembrane glycoprotein that is aberrantly expressed in pancreatic cancer. MUC13 influences the expression of HER2, pAKTSer473, and p53 and is suppressed by miRNA-145 (miR-145). 47 miR-145 thus makes for a potentially effective therapy. Setua et al. explores the viability of using magnetic nanoparticles (MNPs) as a nanocarrier for the delivery of miR-145. 17 The MNPs effectively target pancreatic tumors through the EPR effect, and PEIs present on the molecule complex with negatively charged nucleic acids, making them effective delivery vehicles. The authors found that treatment of human red blood cells at varying concentrations of MNP formulation (MNPF) indicated little to no toxicity compared with the lipofectamine control. Additionally, unloaded MNPFs were added to pancreatic cancer cells and showed no toxicity compared with lipofectamine after 48 h, suggesting that the carrier itself is safe for clinical applications. 17 miR-145-MNPF-treated cells had significantly reduced cell viability in human pancreatic cancer cell lines, HPAF-II (72%) and AsPC-1 (26%) compared with the control (NC-MNPF) with near 100% viability. Moreover, immunoblotting data indicated that miR-145-MNPF inhibited MUC13, pAKTSer473, and HER2 expression while restoring p53 functionality. 17 Thus, miR-145-MNPF is a promising formulation for MUC13 targeting and modulation of its downstream oncogenic signaling cascade.
Downregulation of antiapoptosis factors presents another therapeutic opportunity to treat pancreatic cancer. For example, miR-34a targets several genes involved in cell proliferation and migration, including E2F3, Bcl-2, c-myc, and cyclin D1. 18 miR-34a can also trigger apoptosis via p53 activation and c-Met activation.48,49 Therefore, research on therapies that involve the restoration of miR-34a is promising because of its wide array of important functions in antitumor pathways. Hu et al. developed a novel miR-34a complex consisting of CC9 (CRGDKGPDC), a specific tumor-homing and penetrating bifunctional peptide, conjugated with cationic β-cyclodextrin-polyethylenimine (β-PEI-CD [PC]) vectors. 18 β-cyclodextrin crosslinked by 600 Da PEI is capable of mediating gene transfection in different tumor cell lines and mouse models, 50 and CC9 was used to target tumors through an RGD motif. Subsequently, in the tumor site, CC9 is proteolytically cleaved to CRGDK, which binds to neuropilin-1 (NRP-1) and allows the particle to be internalized. 18 Upon treatment with PC/miR-34a + CC9, cells in the G0/G1 phase increased from 26.08 ± 1.8% to 69.66 ± 1.7%. Comparatively, cells in the G2/M and S phases were 4.71 ± 2.5% and 26.63 ± 2.0%, respectively, which were lower than untreated cells, with 15.95 ± 1.3% and 57.97 ± 2.3% for the G2/M and S phases, respectively, indicating effective cell growth inhibition. To assess therapeutic efficiency of miR-34a delivery in vivo, 100 µL of the 15 µM miR-34a nanoparticle formulation was intravenously administered twice a week for 2 weeks to tumor-bearing mice with xenografts consisting of PANC-1 human pancreatic cancer cells. PET imaging confirmed that the tumor size decreased dramatically 3 weeks after the first injection, demonstrating the promise of the PC/miR-34a + CC9 delivery system for cancer inhibition. 18
In addition to miR therapies that target mRNA with antitumor effects, miRNAs, such as miR-21, that have oncogenic activity can be repressed as a strategy for pancreatic cancer therapy. miR-21 plays a key role in cell proliferation, differentiation, and survival as well as cancer initiation and propagation.51,52 It has been reported that miR-21 inhibition by antisense oligonucleotides (ASOs) increases sensitivity to GEM and downregulates other cancer-associated phenotypes. 53 Li et al. developed CD44v6-targeted PEG-PEI-IONPs carrying a combination drug of miR-21 antisense oligonucleotides (ASO-miR-21) and GEM for co-delivery into CD44v6-expressing human pancreatic cancer cells, a molecular target that is overexpressed in pancreatic cancer. Significant downregulation of Bcl-2 protein was observed in PANC-1 cells treated with the combination drug (34.38 ± 3.77%) and Mia PaCa-2 cells (26.30 ± 2.33%). As a result, the combined NP therapy showed enhanced cytotoxicity. Combination treatment of the ASO-miR-21 and GEM in mice resulted in tumor volumes of 145 ± 7.25 mm3, compared with ~600 mm3 for the GEM monotherapy and 1000 mm3 for the PBS-treated mice 21 days after treatment in a xenograft mice model using MiaPaCa-2 cells in BALB/c nude mice. 54
Gene Augmentation
Gene augmentation is a type of gene therapy that involves replacing a defective gene with the wild-type form. Pancreatic cancer is characterized by a small number of mutations that are in a very high percentage of cancers and that play key roles in the aggressiveness of the cancer. 55 As such, these specific mutated genes can be excellent targets for gene augmentation.
One of these common genes is p53, which is mutated in 75% of pancreatic cancers and is the third most common mutation in pancreatic cancer, behind KRAS2 and p16. 56 Functional p53 is a critical cell cycle regulator and recognizes damaged DNA. Missense mutations can cause cell proliferation and metastasis, and pancreatic cancers with p53 mutations have proven to be difficult to treat using chemotherapy. 57 Taken together, these characteristics make p53 an excellent target for gene augmentation therapy for pancreatic cancer.
Xu and Amiji used EGFR-targeting gelatin nanoparticles as a delivery vector. 14 The p53 plasmid DNA was encapsulated in the thiolated gelatin by adding 200 mg of thiolated gelatin per milligram of p53 plasmid to form nanoparticles. The surface was PEGylated and functionalized with an EGFR-targeting peptide (YHWYGYTPQNVIGGGGC). Nanoparticles were found to be biocompatible with 80% viability at a high concentration of 6 mg/mL compared with 40% viability for PANC-1 cells treated with PEI. These particles were able to achieve a plasmid encapsulation efficiency of 94.8 ± 5.1%. 14 Using laser scanning confocal fluorescence microscopy, nanoparticles were found to be endocytosed and the plasmid released within 30 min of internalization. Upon nanoparticle incubation with human PANC-1 cells, which highly express EGFR, a fivefold increase in the number of apoptotic cells was found after 96 h compared with untreated cells, and was nearly twice as effective as the nontargeted nanoparticles. 14 These results show the potential of p53 gene augmentation at inducing apoptosis of pancreatic cancer cells and furthermore highlight the increased efficacy associated with EGFR targeting.
In a subsequent paper by Xu et al., this same nanoparticle was tested in vivo. First, this same carrier system was used to encapsulate GEM to be used as a dual therapy in conjunction with the gene therapy. 19 GEM-loaded gelatin nanoparticles were used due to their improved antitumoral activity in SCID mice bearing PANC-1 xenografts. Using the same mice model, at 33 days post-tumor injection, targeted PEGylated gelatin nanoparticles containing only the wild-type p53 (wt-p53) inhibited tumor growth by 50.1%, while the nontargeted PEGylated nanoparticles inhibited tumor growth by 38.3%. Critically, the naked wt-p53 plasmid DNA showed minimal growth inhibition, indicating that both targeting and long circulation time provided by a PEGylated nanoparticle were essential for intratumoral localization and therapeutic effect. Lastly, the combination therapy was tested using both GEM and p53 in their PEGylated EGFR-targeting nanoparticle formulations, which resulted in maximum tumor growth inhibition of 77.3%, compared with monotreatments using the identical delivery platform, which led to growth inhibition of 61.7% and 50.1%, respectively, for GEM and wt-p53. 19 This indicates synergistic effects of the chemotherapeutic and the gene therapy.
In addition to EGFR, the transferrin (Tf) receptor can be used to target pancreatic cancer. The Tf receptor is overexpressed in 93% of pancreatic cancers and is recycled during internalization during rapid cell growth, which can improve uptake of the targeted vectors. 16 Camp et al. used a single-chain antibody fragment (TFscFv) to target the Tf receptor and deliver wt-p53 via cationic liposomal nanoparticles (SGT-53). They further tested SGT-53 with GEM. In vitro, SGT-53 had no effect on the murine pancreatic cancer cell line, Panc02, while 0.5 µM GEM inhibited growth by 19% compared with the untreated group. The two therapies in combination inhibited growth by 49%. Using Panc02 murine pancreatic cancer cells to generate metastatic pancreatic cancer in C57BL/6 mice, the authors demonstrated a high degree of specificity of the nanoparticles due to a greater than threefold increase in the intensity of fluorescence of tumor-bearing tissue compared with the saline control. Moreover, the survival study showed a median survival of 21 days for the untreated group versus 37 days for the combination therapy group. Additionally, GEM alone improved median survival to 30 days, while SGT-53 only improved survival to 29 days ( Fig. 3 ). These results highlight the potential of targeted gene therapy in combination with GEM as a superior pancreatic cancer therapy. 16
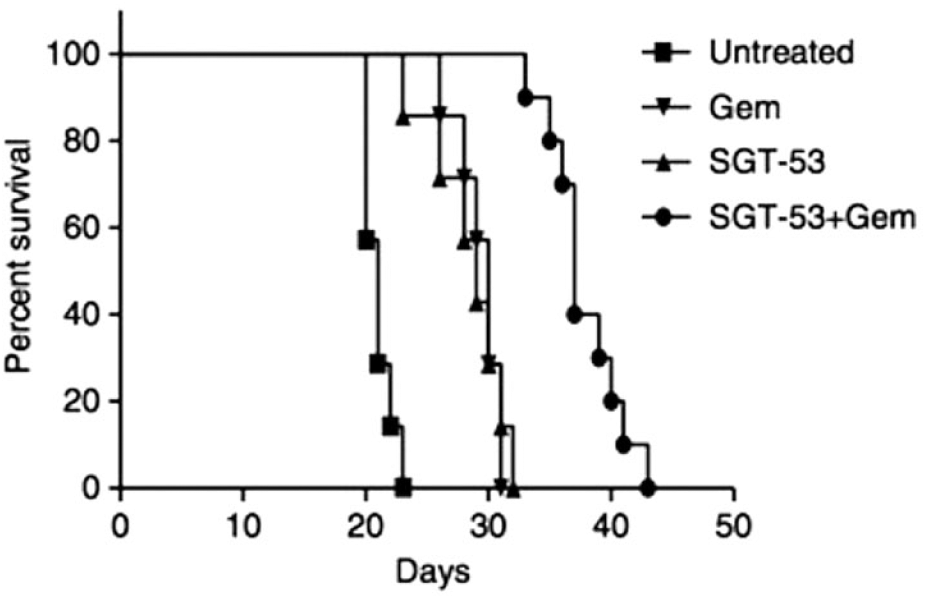
Survival curve of SGT-53 therapy and GEM demonstrating that the combination therapy had a median survival of 37 days compared with 21 for the untreated group, and 29–30 for the monotherapies. Reprinted with permission from Camp et al. 16 Cancer Gene Ther. 20: 222–228. © 2013 Springer Nature Limited.
Alternatively, Ge et al. sought to deliver SFlt-1, an antiangiogenic gene, using cRGD as a targeting moiety. 58 The authors used PEG-polycation block copolymer polyplex micelles, which self-assemble into rod-shaped nanoparticles, and added cRGD moieties to improve targeting and internalization. Using 20 kDa PEG (PEG20), the HeLa cells treated with the micelles that included the targeting moiety expressed SFlt-1 an order of magnitude higher than those cells treated by micelles without the targeting moiety. Cholesteryl was conjugated to the nanoparticles with the purpose of increasing the hydrophobicity of the nanoparticle core to allow for increased compaction of DNA and was able to improve the binding efficiency of the polymer to plasmid DNA by more than 85% as determined by the binding number of block copolymer per individual DNA. The PEG20 cholesteryl-conjugated cRDG nanoparticles slowed tumor growth by ~50% compared with the untreated group, while the nontargeted version slowed tumor growth by approximately 25% in a xenograft mice model using human BxPC3 pancreas cells. 58 What is noteworthy is that the SFlt-1 protein was found to be sequestered predominantly in the tumor stroma adjacent to the vascular endothelial cells and not in the tumor nests. Wt-SFlt-1 was able to restrict neovascular growth due to the antiangiogenic properties of the wild-type gene, which subsequently led to growth inhibition of the tumor itself. 58 The results convey that this gene therapy platform using the cholesteryl-conjugated polyplex micelles with 20 kDa PEGylation can be used as a gene delivery platform and can be modified with different targeting moieties to specifically target subgroups of pancreatic cancer.
Conclusions
Gene therapies encapsulated by nanoparticles serve as promising therapeutics for pancreatic cancer. When designing a gene delivery therapy carried by nanoparticles, the specific, common mutations of the cancer of interest are the critical consideration to the successful design of the gene therapy system. Common mutations will identify potential gene targets for the gene therapy and can inform the type of nucleic acid class chosen based on what is known about how to interfere with the mutant gene’s functioning or how to restore a functioning version of the given gene. Additionally, mutations that lead to the overexpression of cell surface proteins would indicate potential extracellular targets for targeting moieties, which can be used to improve the efficacy of the therapy. 14 Ideally, both the gene and extracellular target are expressed in a very high percentage of cases of the cancer of interest so that the designed therapy can be effective in a high percentage of cases.
These therapeutics have demonstrated tumor growth suppression and increases in survival compared with conventional chemotherapies, such as GEM. 19 Furthermore, synergistic effects have been demonstrated when using these gene therapy systems in combination with chemotherapy.16,19 However, for the most part, the gene therapies described herein are in their infancy and must be further developed for clinical application.
A vast majority of the testing for the therapies has only been completed in vitro. While in vitro systems are useful for understanding the mechanisms of gene therapies, they have less physiological relevance as humans and other living organisms have much more complex responses that involve interactions with organ systems and other cell types. In vivo models, particularly mice models (both xenograft and allograft), can give a better indication of how the therapy may operate in humans compared with in vitro models. Still, these models have flaws, such as differences in drug metabolism and the limited ability to recapitulate human de novo tumor development in mice, which make it difficult to identify the therapies that would be effective in humans. 59 Nonetheless, it is still important that these therapies be tested and optimized in vivo before they move to clinical trials because these models can be useful tools and offer important insight.
One reason why in vivo studies are so critical is to evaluate the toxicity of nanoparticles. There is evidence that nanoparticles can cause adverse events in organs, including the brain and skin, and further contribute to diseases such as cardiovascular disease. 60 In vitro and in vivo toxicity can vary significantly, and hence in vitro biocompatibility does not equate to in vivo safety. 60 One benefit of nanoparticles, however, is that the surfaces can be modified. Thus, if in vivo toxicity concerns become apparent, modifications to the nanoparticle could potentially minimize toxicity without sacrificing significant efficacy. 61
Of the therapies described, SGT-53—the Tf targeting p53 encapsulated by a cationic liposome—is currently being investigated in clinical trials. In phase I, doses were well tolerated up to 3.6 mg of DNA per infusion, and 5 of 12 patients had verifiable tumor shrinkage. 62 This therapy is currently in phase II clinical trials, being tested in combination with GEM and nab-paclitaxel. 63
While promising, several challenges must be addressed for widespread clinical application of gene therapies for pancreatic cancer. First, off-target effects can be problematic; however, targeting moieties can minimize these effects, increase uptake efficacy, and improve therapeutic efficacy. 64 Second, mass production of nanoparticles is challenging. The protocols to synthesize these nanoparticles often require extensive time, materials, and cost. 65 The ability to effectively scale production is essential to make it widely available and cost-effective to produce. Lastly, though these therapies are effective, they still have not led to remission ( Fig. 3 ). 16 As such, it may be useful in the future to consider combinations of gene therapies as a treatment regimen, similar to chemotherapy regimens including multiple, separate drugs.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.