
Abstract
Cystic fibrosis is a genetic disease affecting more than 70,000 people worldwide. Caused by a mutation in the CFTR gene, cystic fibrosis can result in difficulty breathing, widespread bacterial infections, edema, malnutrition, pancreatitis, and death. Current drug-based treatments struggle to reach the site of action due to the thick mucus, and only manage symptoms such as blocked airways, lung infections, and limited ability to digest food. Nanotechnology opens up possibilities for improved treatment strategies by focusing on drug penetration through the mucus lining, eliminating resulting bacterial infections, and targeting the underlying genetic cause of the disease. In this review, we present recent nanoparticle developments for cystic fibrosis, challenges in nanomedicine therapeutics, and future research directions in gene editing and nonviral vectors for gene delivery.
Introduction
Cystic fibrosis is a recessive genetic disease that is caused by the mutation of the CFTR (cystic fibrosis transmembrane conductance regulator) protein. 1 More than 70,000 people are affected globally, and most patients are diagnosed at 2 years old. 1 With more than 1900 mutations and a median survival age of 40 years, there is a need to develop more effective treatments and therapy methods.1,2
CFTR
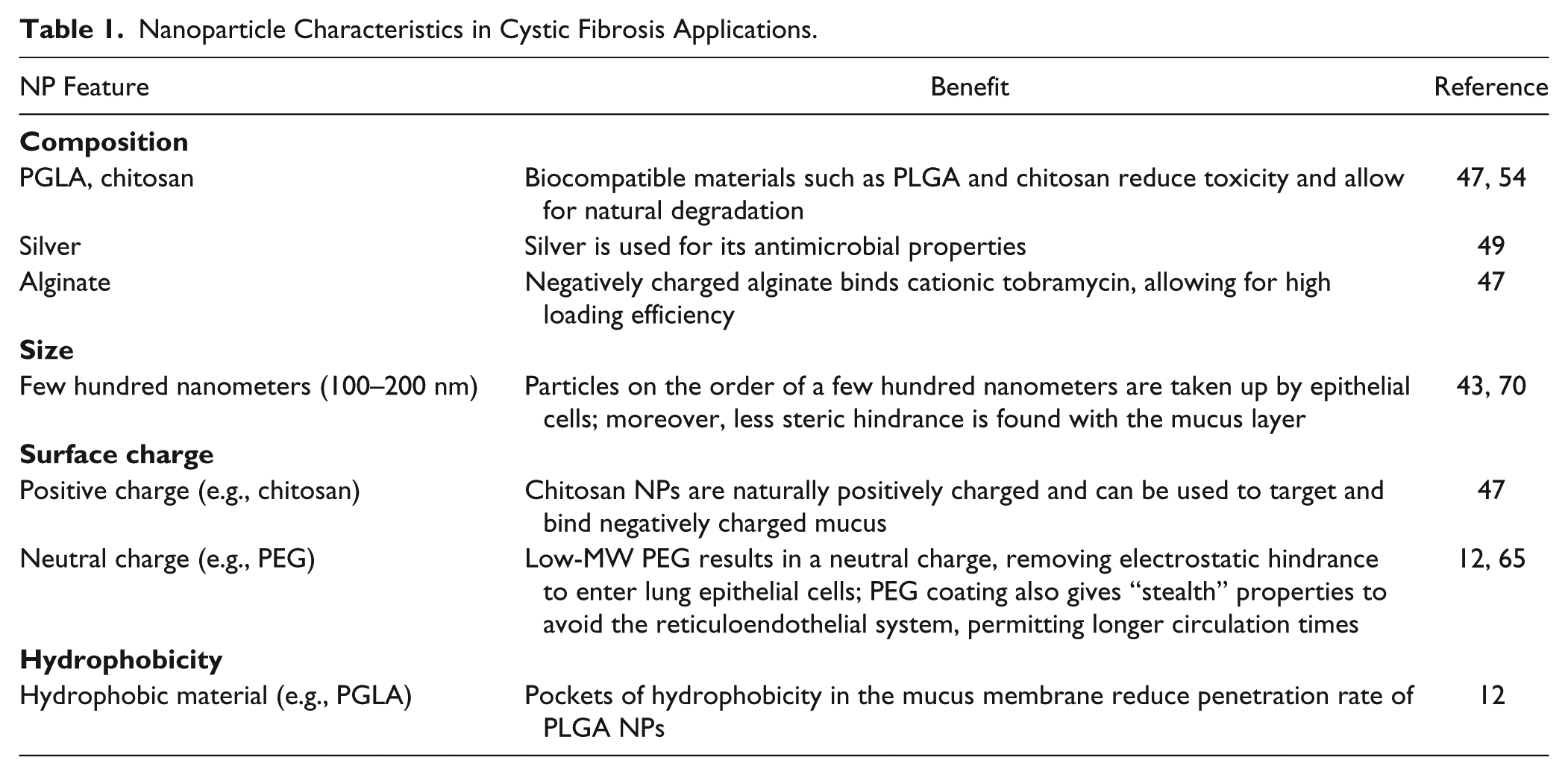
The CFTR protein is a chloride channel located in the surface membrane of epithelial cells; its function is to regulate the secretion of chloride ions out of cells that produce mucus, sweat, tears, saliva, and digestive enzymes, ultimately regulating fluid flow across the epithelial membrane. 3 Abnormal fluid flow across the epithelial membranes results in thick mucus production, which negatively affects organs by blocking ducts and harboring bacteria. 4 This mucus layer is a major barrier for drug delivery. 5 Due to their engineering flexibility, nanoparticles (NPs) as a vehicle for gene delivery are currently being investigated as a means to overcome these difficulties. Penetration can be enhanced by reducing NP size and neutralizing their surface charges, external conjugates can be used to target cells and reduce mucus viscosity, and a variety of therapeutic payloads can be delivered, including genetic therapies.6–9 Some of these are summarized in Table 1 .
Nanoparticle Characteristics in Cystic Fibrosis Applications.
Symptoms
The symptoms of cystic fibrosis are most apparent in the lungs and pancreas. In the airways of healthy individuals, the mucus layer, or periciliary layer, is a few tens of microns thick.10,11 However, with a dysfunctional CFTR protein channel, an abnormal amount of mucus builds up in various organs throughout the body. In the lungs, this accumulation of mucus can block airways, contribute to bacterial lung infections, and lead to respiratory failure. 1 While normal mucus functions to protect the exposed surfaces of internal organs, imbalanced ion transport in the lung airways in cystic fibrosis results in mucus with increased adhesion and viscoelasticity that becomes a substantial barrier for drug delivery, particularly through the inhalation route, the most straightforward method to target the lungs.12–14 Hence, designing mucus-penetrating particles or mucus-cleaving mucolytics, drugs that degrade mucus polymers, is crucial for more effective drug delivery.
In the pancreas, cystic fibrosis-induced mucus inhibits the release of digestive enzymes by blocking the pancreatic duct. This makes it difficult for the body to break down food and absorb nutrients, ultimately leading to malnutrition and poor growth during early development. 1 Despite the blocked pancreatic duct, digestive enzymes continue to be produced and the increased concentration of enzymes can lead to edema in the surrounding acinar tissue and impair blood flow in the organ, resulting in damaged pancreatic tissue and pancreatitis.15,16 Long-term, chronic pancreatitis is associated with an increased risk of pancreatic cancer, and cystic fibrosis patients have been found to have an increased risk for pancreatic cancer. 17 In a correlation study, CFTR mutation carriers were found to have an increased risk of pancreatitis and patients with pancreatitis have been found to be 26.3 times more likely to develop pancreatic cancer.18,19 Hence, it is not surprising that patients with heterozygous CFTR mutations and pancreatitis have a moderately increased risk for pancreatic cancer. 17
Diagnosis
Testing for cystic fibrosis is commonly carried out in newborns by assessing the amount of the pancreatic enzyme precursor, immunoreactive trypsinogen (IRT), in the blood. 1 High levels of IRT can be indicative of cystic fibrosis and are used to determine whether the young patient should be formally diagnosed. 20 In the United States, newborn screening can have two or three stages, contingent on the results of the first IRT test. In some states, a second IRT test and/or a genetic test for one or more of the most common mutations are used. 21
Among other age groups, the gold standard for diagnosing cystic fibrosis is the sweat test. 22 Pilocarpine, a cholinergic agonist, is applied to the skin, and a mild electrical current pulses the drug into the skin to stimulate the secretion of sweat, which is collected and analyzed for salt content. 22 The CFTR protein, which modulates secretion of chloride ions, regulates ENaC, an epithelial sodium channel. In the skin, CFTR has a stimulatory effect on ENaC, causing sodium to be reabsorbed from the skin; however, when CFTR function is impaired, the stimulatory effect is removed and sodium reabsorption is decreased. 23 This causes the sweat of cystic fibrosis patients to be abnormally salty, which is one of the indicators of the disease. In the lungs and pancreas, CFTR and ENaC are inversely related: decreased CFTR function enhances sodium absorption from the airways and pancreatic duct, osmotically drawing water and inhibiting ENaC activity. 23 Consequently, the mucus outside of the epithelium becomes very dehydrated and sticky, a prime environment for bacterial growth leading to infection, inflammation, organ damage, and sepsis.
Current Treatments
The majority of current treatments address the symptoms of cystic fibrosis, but there are currently no cures to stop its progression. Once a patient is diagnosed, treatment plans revolve around maintaining lung function, with nutritional therapy as a secondary priority. 24 To remove the mucus and keep airways clear, bronchodilators, chest physical therapy, and mucolytics (e.g., Pulmozyme) are administered. 24 Nebulized hypertonic saline is used to decrease mucus viscosity in the lungs, increasing the clearance rate to maintain function. 25 Antibiotics are used to prevent infection. 1 With regard to nutritional therapy, enzyme supplements, multivitamins, and mineral supplements are frequently employed to encourage healthy development and maintenance in children and adults. 24
CFTR Modulators
Small-molecule drugs called CFTR modulators are currently on the market to rescue CFTR protein function. The most common CFTR modulators are ivacaftor, lumacaftor, and tezacaftor. 1 Ivacaftor is a potentiator-type modifier that addresses the specific G551D gating mutation. Potentiators serve to enhance chloride ion flow through the channel, assuming the channel is properly located in the plasma membrane. The CFTR protein is a member of the ATP-binding cassette family, relying on ATP hydrolysis to supply energy to drive the gating cycle of CFTR.12,14 However, in individuals with the G551D mutation, a glycine is replaced by an aspartate, afflicting ATP hydrolysis and preventing efficient ion transfer. Ivacaftor addresses this by extracellularly maintaining the chloride channel open state, allowing the CFTR gating cycle to operate independently of ATP hydrolysis, restoring chloride transport.12,14,26 Though ivacaftor is effective in improving weight gain and FEV1 (forced expiratory volume within 1 s, a measure of lung health), the drug is not widely applicable to cystic fibrosis patients because gating mutations only make up about 5% of all CFTR mutations.27–29
Lumacaftor/ivacaftor is a combination therapy specifically for patients with two copies of the ΔF508 mutation, which results in misfolded and misprocessed proteins that remain in the endoplasmic reticulum (ER) of the epithelial cell. 30 The lumacaftor portion of the therapy is a “corrector,” a modulator that improves intracellular trafficking of the CFTR protein. It allows the ΔF508-CFTR to form a tighter, more protease-resistant configuration that can be sent from the ER to the Golgi apparatus for eventual trafficking to the cell surface. 30 With the protein now in the cellular membrane, ivacaftor can act to hold the channel open as described above. 28 Tezacaftor/ivacaftor is another combination therapy for patients with two copies of the ΔF508 mutation. 28 This combination has been shown to rescue lung function with less adverse side effects than lumacaftor/ivacaftor, rendering tezacaftor/ivacaftor an improvement. 28 However, while effective at treating these common mutations, many additional, not-yet-treatable mutations remain. 29
Gene Therapies
Gene therapy involves delivering nucleic acids into a cell to replace, augment, or repress defective genes or biological functions. Gene therapy can be especially effective in treating diseases that are caused by mutations of a single gene, such as in the case with cystic fibrosis; however, there are many challenges, including evading the body’s immune system while delivering the correct gene. 31
The CFTR gene was successfully cloned in 1989, and clinical trials regarding CFTR gene transfer in cystic fibrosis patients were conducted in 1993. 31 Studies have used the amount of CFTR mRNA and electric potential difference across epithelial membranes to measure the efficacy of gene transfection; however, it has not been firmly established whether mRNA levels correlate with clinical outcomes due to low mRNA assay sensitivity.31,32 However, electric potential differences have been found to show reproducibility for diagnosing cystic fibrosis.33–35
Many advancements have been made for treating cystic fibrosis, but challenges still remain. Perhaps the most significant challenge in new therapies for cystic fibrosis is drug delivery.
Delivery Barriers
In addition to typical biological barriers to drug delivery, such as limited bioavailability and systematic side effects, cystic fibrosis presents an additional challenge in the form of a thick mucus layer blocking access to diseased cells. 5 Genetic therapies must also avoid the immune response and further penetrate the nuclear membrane.7–9 As summarized in Table 1 , NPs can be developed with a variety of materials, in various shapes and sizes, surface charges, therapeutics, and with specific targeting moieties. In the context of cystic fibrosis, NPs can be fine-tuned to overcome barriers that free drugs struggle to overcome.
As the majority of cystic fibrosis deaths are caused by lung disease, the rest of this review addresses how NPs have been used in the improvement of drug delivery and treatment, specifically to the lungs.29,31 The mucus layer is a primary focus, as it is characteristic of and a major challenge in cystic fibrosis.
Mucus-Penetrating NPs
Bacterial infections and pulmonary complications from cystic fibrosis, such as hemoptysis, lead to high mortality and morbidity rates worldwide. 30 Thus, the respiratory tract has become the primary target for delivering drug and gene therapies. 13 As mentioned, one of the greatest challenges in treating cystic fibrosis with NP drug delivery is penetrating the disease’s characteristic thick, sticky mucus, which prevents cilia from eliminating bacteria, leading to infections.10,12,13,25,36–39
The mucus architecture is a mesh structure filled with a low-viscosity fluid composed of mostly mucin fibers (~70%–80%) that are highly cross-linked by disulfide bonds and hydrophobic interactions.8,35 Additionally, actin and DNA and other macromolecules present in the mucus layer create hydrophobic and electrostatic barriers that greatly inhibit the penetration of inhaled therapies.8,35 The average mesh size is reduced to 100–400 nm compared with greater than 500 nm in healthy individuals, which increases steric hindrance and reduces drug delivery efficacy.13,40 Due to their size, NPs have emerged as promising vehicles to package and move drugs through the mucus, enhancing penetration and delivery rates. Smaller particles decrease the effect of steric hindrance by the mucus mesh.6,13 Electrostatic interactions can be eliminated by coating NPs with electrostatically neutral molecules such as polyethylene glycol (PEG). 6 The relationship between size, surface charge, and penetration is shown in Figure 1 . Penetration can be enhanced by equipping drug-bearing NPs with mucus-inert attachments or with mucolytics. 38 Moreover, NPs can be used to enhance the delivery of antibiotics such as tobramycin, as well as gene therapy to address symptoms or the genetic root of cystic fibrosis.
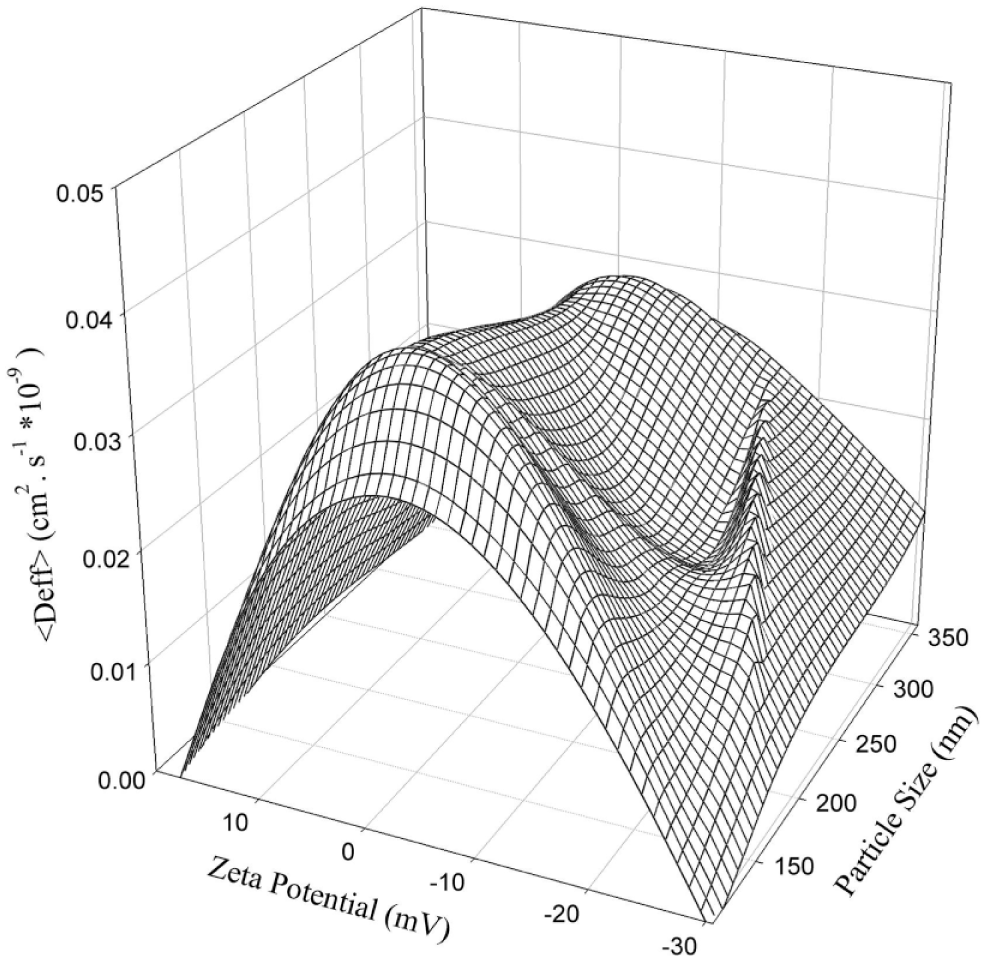
The relationship between NP size, surface charge, and diffusion through mucus. Smaller and more neutral NPs display higher diffusivity. Reprinted from European Journal of Pharmaceutics and Biopharmaceutics, 97, Abdulkarim, M.; Agulló, N.; Cattoz, B.; Griffiths, P.; Bernkop-Schnürch, A.; Borros, S.; Gumbleton, M., Nanoparticle Diffusion within Intestinal Mucus: Three-Dimensional Response Analysis Dissecting the Impact of Particle Surface Charge, Size and Heterogeneity across Polyelectrolyte, PEGylated and Viral Particles, 230–238. Copyright 2015, with permission from Elsevier.
Two common ways to overcome the cystic fibrosis delivery barrier are decreasing particle adhesion to the mucus by surface-coating NPs with mucus-inert polymers such as PEG and reducing mucosal viscoelasticity with mucolytics. 38
PEGylation
Suk et al. illustrated the effect of PEG coating on the NP ability to penetrate the microstructure of cystic fibrosis mucus. 13 They compared the diffusivities, transport rates, and displacements of 100, 200, and 500 nm diameter PEG-coated NPs with those of uncoated NPs. 13 The PEG coating increased penetration of the porous mucosal barrier for the 200 nm particle, compared with the uncoated particle, confirming PEG’s mucus resistance in facilitating NP transport, and the 200 nm size was found to be optimal for mucus penetration. 13 Moreover, PEGylation was also found to increase NP mobility in Burkholderia multivorans and Pseudomonas aeruginosa biofilms, which are a result of bacterial infections and common in the airways of cystic fibrosis patients. 41
The effect of PEGylation degree and PEG chain conformation was further investigated by Craparo et al., who synthesized mucus-penetrating fluorescent NPs (FNPs) to deliver nonsteroidal anti-inflammatory ibuprofen.
37
α,β-Poly(N-2-hydroxyethyl)-
Porsio et al. further developed PEGylated PHEA-RhB-PLA-PEG FNPs with cell-penetrating peptides (CPPs) to deliver ivacaftor through the pulmonary route. 5 They promoted ivacaftor cell internalization within the lung by introducing transactivating transcriptional activator peptide (Tat), a type of CPP. 5 Specifically, PEGylated and Tat-decorated FNPs (Iva_Tat-PEG FNP) of ~70 nm size were synthesized by nanoprecipitation and loaded with ivacaftor to test the drug release profile and uptake capacity in the presence of cystic fibrosis artificial mucus (CF-AM) on human bronchial epithelial (16-HBE) cells. 5 Faster penetration and higher ivacaftor release (12%) were achieved by Iva_Tat-PEG FNPs compared with 6% for the free ivacaftor control. 5 A significant enhancement (18.5% vs 5.3%) in ivacaftor cellular uptake of Tat-PEG FNPs was also observed, with 16-HBE cells approximately 90% viable after 24 h compared with 80% viable for free ivacaftor ( Fig. 2 ). 5 In light of these results, the PEGylated and Tat-decorated FNPs show great potential for delivering CFTR modulators through pulmonary administration to treat cystic fibrosis patients.
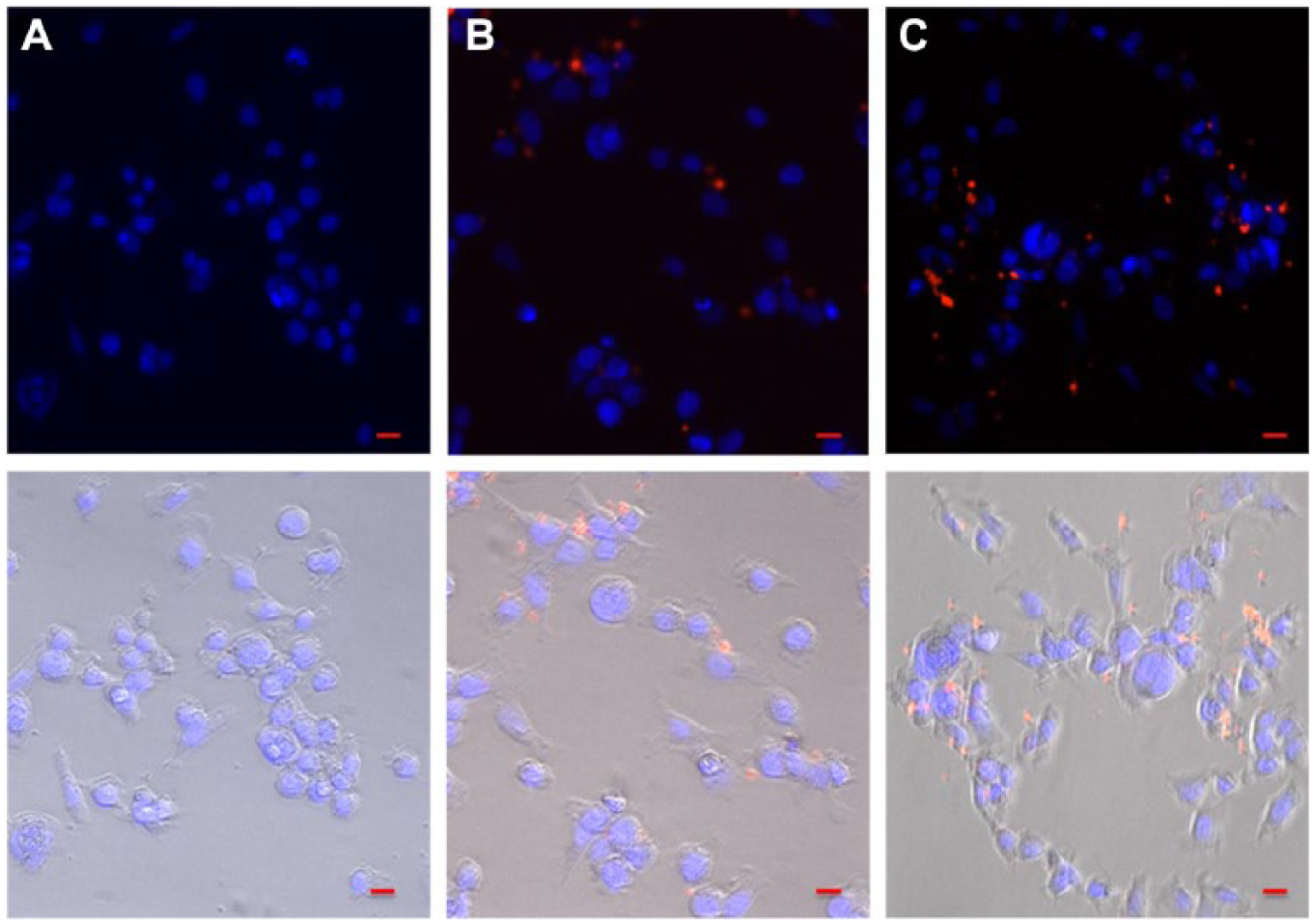
Fluorescence microscopy images describing uptake of (
Mucolytics
Mucolytic agents enhance cystic fibrosis mucus penetration by cleaving disulfide bonds of mucin fibers to reduce mucus viscoelasticity and improve NP mobility. 12 Gene therapy for cystic fibrosis relies on endocytosis to cross cell membranes and initiate transcription in the cell nucleus. 2 To achieve this, mucolytics such as N-acetyl cysteine (NAC) and recombinant human DNase (rhDNase) have been used to improve the delivery of nonviral gene carriers. 2
In another study by Suk and colleagues, NAC in combination with a PEG coating on NP carriers was found to enhance mucus penetration across cystic fibrosis mucus.
38
Moreover, Suk et al. further evaluated the penetrating ability of rod-shaped NPs that were ~350 nm in length and ~10 nm in diameter across cystic fibrosis mucus.
43
Specifically, the NP consisted of poly-
Broughton-Head et al. developed a diffusion assay to analyze the diffusion-limiting and diffusion-enhancing properties of mucolytics, such as DNase and NAC. 36 They measured the penetration of 200 nm fluorescent nanospheres through synthetic biogels consisting of DNA, mucin, and actin, which mimics cystic fibrosis mucus composition. 36 Results showed that biogels with 5 mg/mL actin significantly inhibited the ability of NAC and DNase to enhance NP diffusion, compared with biogels with lesser actin concentrations. 36 The authors concluded that actin tightly binds to DNase, reducing DNase activity or attenuating DNA potentiating effects of NAC mucolysis. 36
Symptomatic Treatments—Bacterial Infection
In addition to preventing conventional drug treatments from penetrating the lung epithelium, the mucus provides a breeding ground for bacteria. 44 Infections can further exacerbate the patient’s condition and cause inflammation. 1 P. aeruginosa is the most common lung infection found in cystic fibrosis patients, and certain strains adapted for mucosal environments have been shown to easily proliferate and form a biofilm over the extracellular matrix of epithelial cells lining the lungs. 44 The most common method of treating these infections is through inhalation of antibiotics, typically tobramycin. However, in its free form, tobramycin cannot reach a therapeutic level because it is rapidly cleared and is unable to penetrate the mucosal membrane. 44
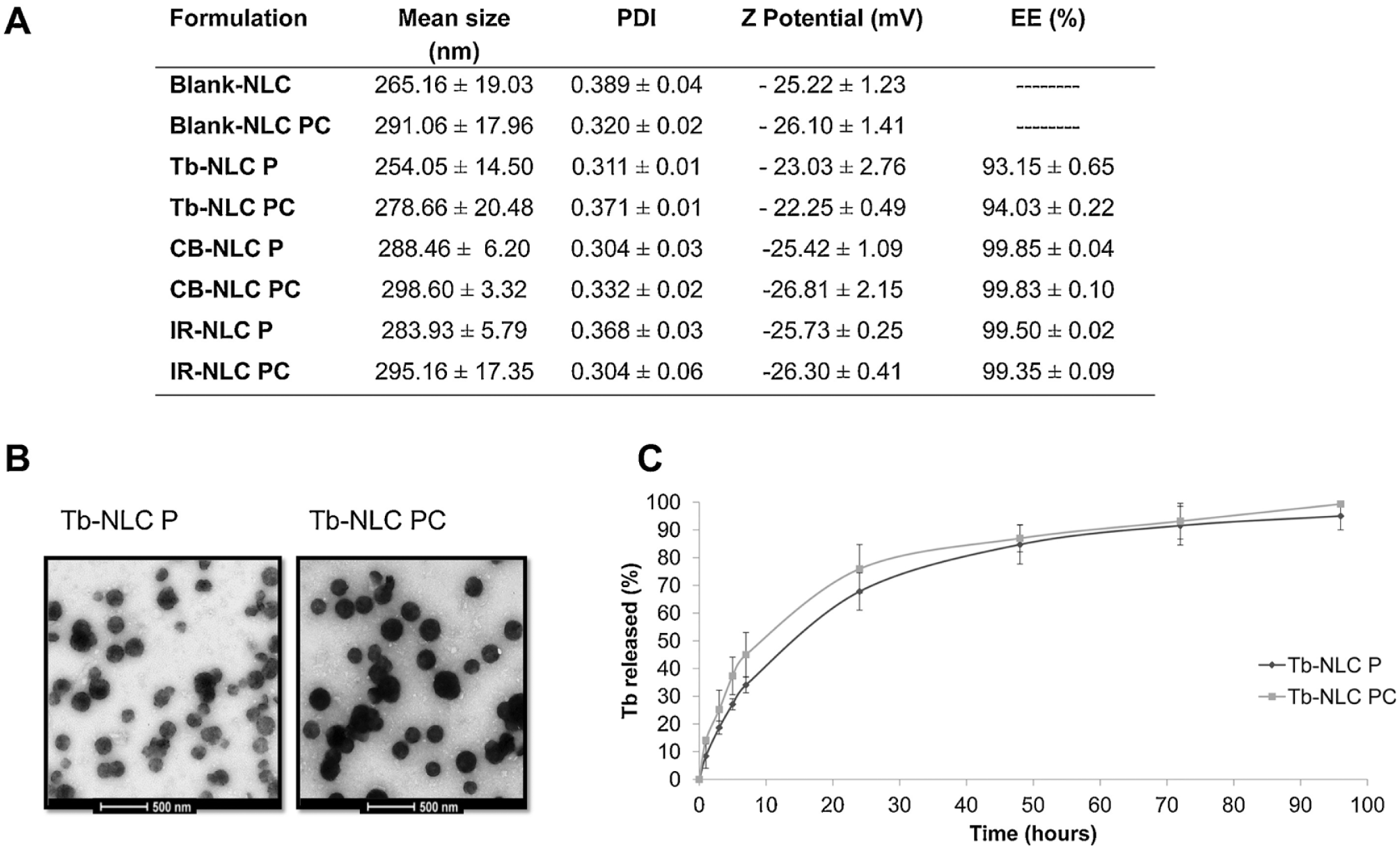
To enhance drug delivery, Moreno-Sastre et al. developed tobramycin-loaded nanostructured lipid carriers (Tb-NLCs) in order to bypass the mucosal membrane and increase tobramycin retention. 44 The NP cores were made with either the solid lipid Precirol ATO 5 (NLC P) or a 50:50 mixture of Precirol ATO 5 and Compritol ATO 888 (NLC PC), another solid lipid. 44 NLCs that were combined with tobramycin were found to be 250 nm in diameter through transmission electron microscopy and had a negative zeta potential, both of which are mucus-penetrating properties.44–46 Both NLCs released 80% of the drug in the first 24 h, followed by sustained release for up to 92 h. 44 Almost 100% of the loaded tobramycin was released in both NLC P and NLC PC ( Fig. 3 ). 44 In addition, the NLCs were shown to have similar minimum inhibitory concentrations (MICs) to free tobramycin, meaning the encapsulation did not affect its antimicrobial properties. 44 Notably, in vivo biodistribution imaging studies qualitatively showed that NLCs loaded with the infrared imaging agent, IR-783 (IR-NLCs), were able to remain in the lungs to a greater extent than free IR-783 at both 24 and 48 h. 44 IR signals from the free IR-783 disappeared from the lungs within 48 h via liver and renal elimination, while the signals from the IR-NLCs were concentrated and maintained in the lungs. 44 Moreover, compared with free tobramycin, both types of Tb-NLCs displayed increased in vitro efficacy against P. aeruginosa as well as an increase in in vivo mucus penetration and pulmonary distribution in mice ( Fig. 4 ). 44
(
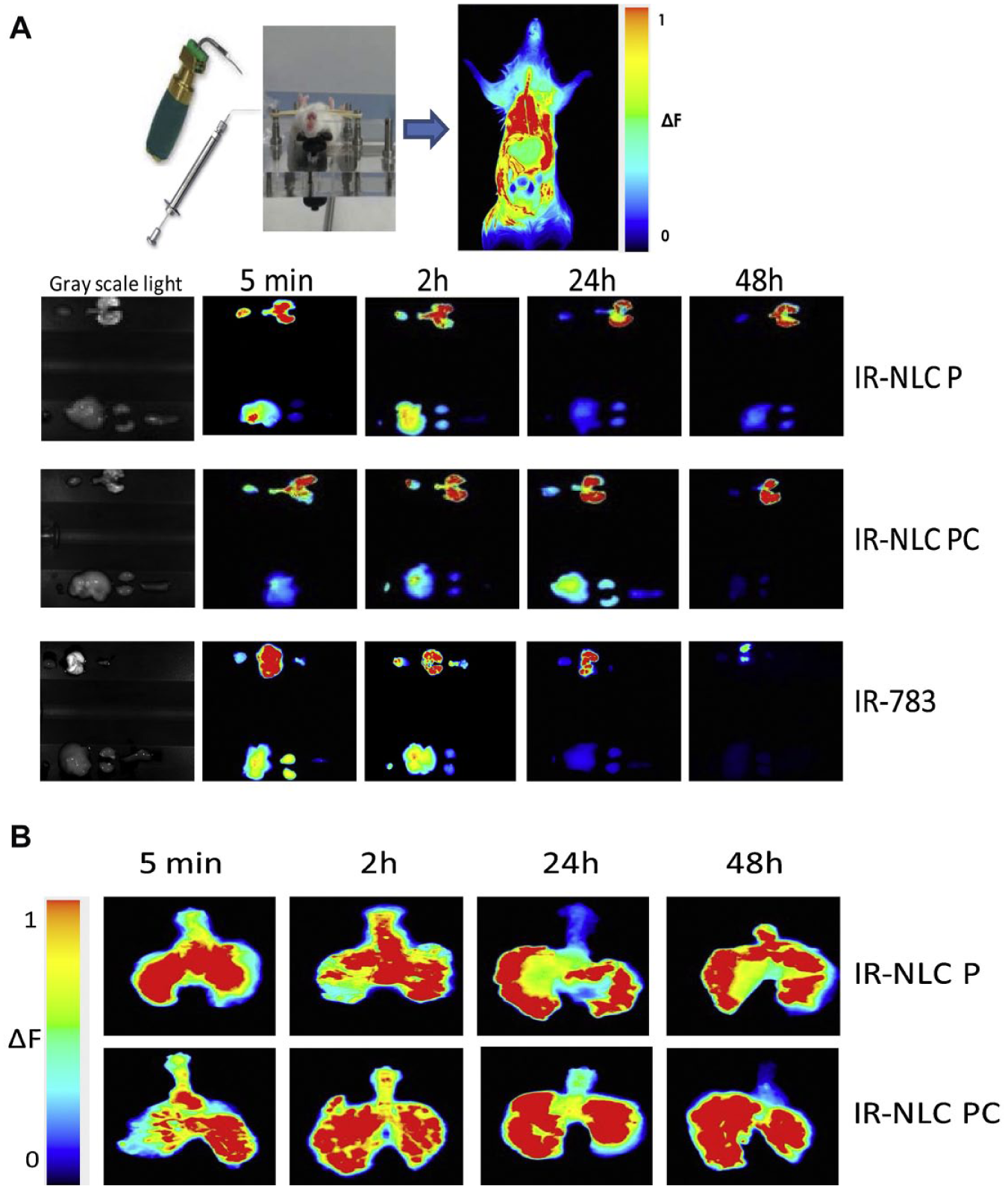
(
Deacon et al. developed a different method of encapsulating tobramycin in NPs with the same goal of increasing mucus membrane penetration. 47 They discovered a strong interaction via isothermal titration calorimetry between alginate and tobramycin. In addition to the drug and alginate, chitosan was added as a stabilizing polymer and NPs were functionalized with DNase to help break down the DNA polymers that contribute to the mucus. 47 The tobramycin NPs (T-NPs), both DNase functionalized and nonfunctionalized, were around 500 nm in diameter and the zeta potentials of both NPs were about –25 mV ( Fig. 5 ). In addition, the MIC of T-NPs was not significantly different from the MIC of free tobramycin. The NPs were also found to have low cytotoxicity, with a 90% survival rate after 96 h postincubation using Galleria mellonella larvae, a model system that mimics the progression of human infections. 48 After G. mellonella were infected with P. aeruginosa, their survival rate doubled upon T-NP treatment compared with the untreated group after 96 h (80% vs 40%). In addition, the effects of DNase functionalization were tested on four patient mucus samples. While not statistically significant, the functionalized T-NPs reduced the proportion of P. aeruginosa that survived. 47 Overall, this study provides evidence that the incorporation of tobramycin into alginate and chitosan-based NPs shows efficacy against infections of P. aeruginosa toward cystic fibrosis treatment.
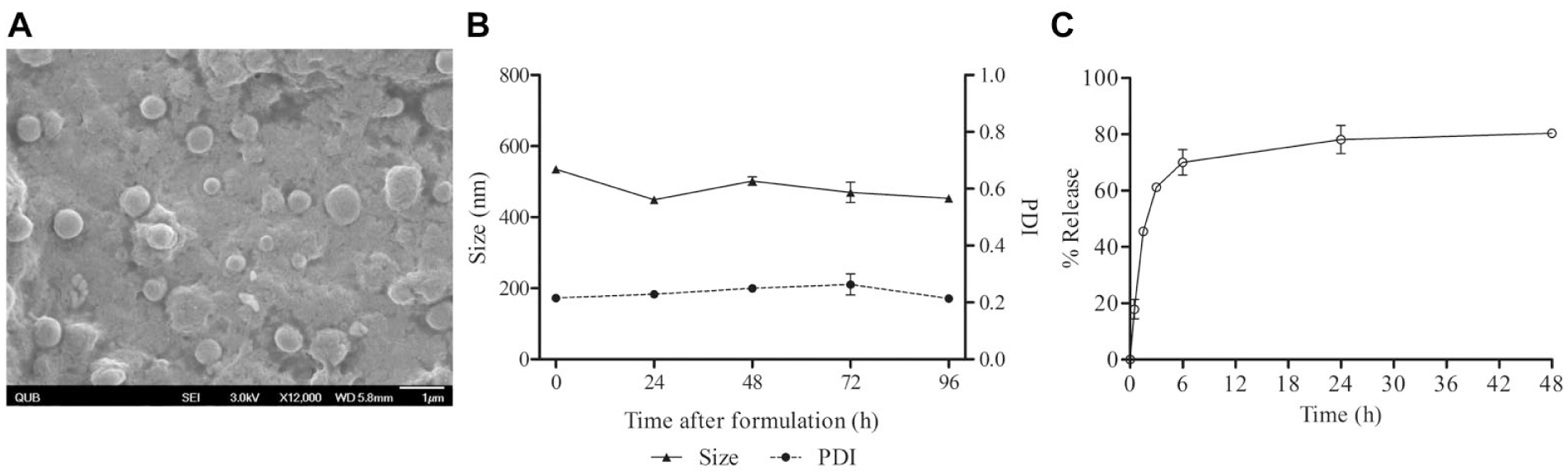
T-NP characteristics. (
Tripathi et al. synthesized silver nanoballs for their antimicrobial properties against gram-negative bacteria, including P. aeruginosa, by mixing silver nitrate and sodium dodecyl sulfate. 49 The mixture was reduced with hydrazine hydrate and sodium citrate to form nanoballs of 12 nm. 49 Silver nanoballs present positive charges on their surfaces in solution, which attracts them to gram-negative bacteria. 49 However, the positive charge also causes stronger interactions with the mucus, decreasing the interaction they have with the bacteria. 49 The antimicrobial properties of the silver nanoballs were measured on Escherichia coli, Salmonella typhimurium, Bacillus subtilis, and P. aeruginosa after 24 h. 49 For each type of bacteria, the optical density decreased from an absorbance of 0.5 without nanoballs to 0.1 with 40 µg/mL of nanoballs, indicating successful antimicrobial effects. 49
Gene Therapeutics
There are more than 1900 identified CFTR mutations, broadly categorized into six classes: defects in protein production (class 1), protein processing (class 2), protein regulation (class 3), protein conduction (class 4), reduced amounts of functional CFTR (class 5), and normal amounts of functional CFTR with high turnover (class 6). 2 Most drug therapies, such as ivacaftor, can temporarily alleviate cystic fibrosis symptoms through a single mechanism of action and are suitable for treating only a particular type of CFTR mutation. Hence, current research in gene therapies aims to treat the disease by restoring the mutated CFTR gene. 2 Much research is focused on the ΔF508 deletion, the most common cystic fibrosis mutation. 15 Gene therapy is promising, and clinical studies have shown some efficacy; however, progress has been slow due to difficultly in delivering the therapy to the lung.2,50 In addition to previously discussed delivery barriers, gene therapies must also penetrate the nuclear membrane. 51 Viral vectors that stimulate a very low immune response, such as adeno-associated viral vectors and lentiviruses, are being investigated, although each poses challenges in repeated-administration immune responses and lung epithelial cell penetration, including the relative absence of particular receptors that viruses exploit on the more easily accessible apical side of the epithelium, compared with the basolateral side.2,51,52 Viral vectors are also difficult to mass-produce and have constraints regarding gene size, limiting scalability and flexibility for clinical translation. 53 As a delivery method, NPs can be engineered to mitigate these issues while maintaining gene transfection efficiency.8,53
Much research has focused on using polylactic-co-glycolic acid (PLGA) NPs, as the particles are biodegradable, can be aerosolized and loaded with therapeutic proteins, have shown evidence of cellular uptake and transport to the cytoplasm and Golgi, and are easily functionalized with targeting moieties and PEG.54,55 Aerosolization is important to consider during NP synthesis as it has potential to be used for the inhalation method of delivery, a noninvasive and convenient route for self-administered medication that is particularly convenient for patients with chronic diseases such as cystic fibrosis. 55
Addressing the CFTR Gene
Although previous reports show that directly addressing the CFTR gene may be an effective method of treating cystic fibrosis, the use of immune-stimulating vectors has made the long-term delivery of therapeutic doses difficult.23,56 Oglesby et al. found that the overexpression of microRNA-145, -223, and -494 was correlated with cystic fibrosis incidence. 57 Fernández Fernández et al. developed a process to create PLGA and chitosan NPs to be loaded with locked nucleic acids (LNAs) to target, bind, and block the activity of these microRNAs.7,58 Oglesby et al. used double-emulsion and self-assembly methods to synthesize PLGA and chitosan particles loaded with two different 16-base LNAs. 7 The NPs were approximately 200 nm in diameter, making them more likely to be endocytosed rather than targeted by monocytes.7,59 These NPs were loaded with LNAs with up to 70% efficiency, and nebulization for lung delivery did not affect their physiochemical properties.7,54,55
Alton et al. conducted a phase IIb clinical trial to determine the efficacy of a nonviral gene therapy for cystic fibrosis.50,60 The GL67A cationic lipid comprises 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE); 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine-n-(methoxy [PEG5000]) (ammonium salt) (DMPE-PEG5000); and the GL67 lipid.50,61 The GL67A solution was mixed with equal amounts of the pGM169 drug (a plasmid containing wild-type CFTR gene) to form complexes and was administered to cystic fibrosis-affected patients once a month for a year with a nebulizer. 50 FEV1, forced vital capacity (FVC), midexpiratory flow 25%–75%, lung clearance index (LCI), computed tomography (CT) lung scans (inspecting gas trapping, a symptom of the mutated CFTR gene), quality of life scores, exercise capacity, gas transfer, and inflammatory markers as secondary outcomes were assessed. 50 Results showed statistically significant improvements in FEV1 (3.7%), FVC, and gas trapping on CT scans compared with the saline control, and although not significant, positive results were seen in other secondary outcomes irrespective of age, sex, or CFTR mutation class. 50 However, the small improvements in FEV1, FVC, and CT did not greatly affect the quality of life for treated individuals. 50 The greatest improvements were seen in subjects with the lowest baseline FEV1 and may be because increased obstruction allows for increased drug deposition and retention in the proximal airways.
Progress has been made with CRISPR/Cas9 gene editing technology delivered via NPs to directly correct the mutated CFTR gene. For instance, zwitterionic amino lipid NPs (ZNPs) have been shown to be able to co-transport single-guide RNA (sgRNA) and Cas9 RNA to the lungs with 95% reduction in protein expression. 9 McNeer et al. used site-specific gene editing to repair the ΔF508 mutation. 62 They used triplex-forming peptide nucleic acids (PNAs) to trigger DNA repair and were able to restore the CFTR gene to 25% of cells in vitro and saw a significant decrease in nasal potential differences (NPDs), a standard diagnostic tool for cystic fibrosis in vivo, with very low levels of off-target effects. 62 Researchers are also developing carbon NPs as alternative nonviral delivery vectors for gene editing. 63
Increasing Effectiveness of CFTR Protein
As mentioned, the ΔF508 mutation, present in approximately 70% of cystic fibrosis cases in the United States, reduces the ability for the CFTR protein to conduct sodium ions and triggers degradation of the protein in the ER before it can successfully translocate to the plasma membrane.15,64 Therefore, recent research has aimed to prevent CFTR protein degradation and allows its limited functionality as a way to partially alleviate the disease. 65
The FDA-approved drug PS-341 prevents protein degradation by inhibiting proteasome activity. 66 Attempting to rescue CFTR function, Vij et al. encapsulated PS-341 within PLGA-PEG NPs and intranasally administered NPs to mice with CF. 65 Fluorescence results revealed that the particles successfully localized to the lungs, greatly reducing immunosuppressive side effects resulting from PS-341 systemic administration, and released the drug over an 11-day period. 65 Proteasomal assay results showed an approximately twofold decrease in proteasome activity that correlated with a reduction in inflammation in cystic fibrosis lungs. 65
Egan et al. found that orally delivered curcumin, a naturally derived compound, allows the misfolded CFTR protein to escape the ER, although its mechanism is still unclear. 67 Cartiera et al. encapsulated curcumin in PLGA NPs to enhance its bioavailability. They orally administered NPs loaded with 3.75 mg of curcumin to cystic fibrosis murine models three times a day for 2 days at low (<3.75 mg released) and high (all 3.75 mg released) doses. 16 When NPD was used to measure the isoproterenol response (IR), a common indicator of sodium conductance for cystic fibrosis diagnosis, a 3.7 mV increase was observed for those treated with the curcumin-PLGA NP compared with free curcumin (IR changes greater than 2 mV are considered a “full response”).4,16,68 Figure 6 shows a healthy and cystic fibrosis NPD, with clear differences in IR. 4
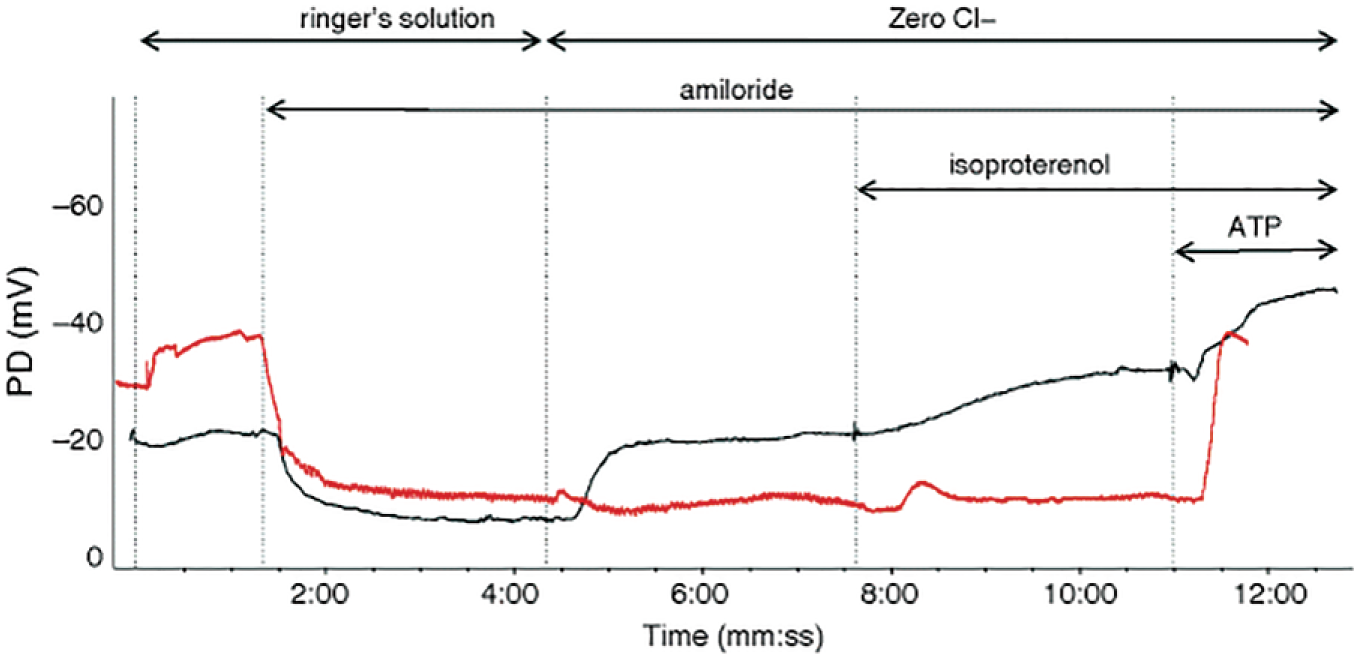
NPD measurements of cystic fibrosis-affected (red) and nonaffected (gray) patients. The lack of hyperpolarization in the IR is a common result in CF-affected patients. Reprinted with permission from Springer: SAGE Publications, Springer Nature, Nasal Potential Difference Measurements to Assess CFTR Ion Channel Activity, Rowe, Steve M., Clancy, John Paul, Wilschanski, Michael, 2018.
Future Directions
Current research in NP development aims to improve the delivery of conventional drugs to the lung and deliver new types of therapeutics.5,65 The development of mucolytics and PEGylated NPs is used to penetrate the thickened mucus layer and represents a significant step in more efficiently delivering drugs to cystic fibrosis patients. 38 Tobramycin-loaded NPs designed to penetrate the mucus layer have shown effectiveness in combating bacteria that accumulate in the mucus.44,47 Mucolytics for degrading actin have also been investigated. Gelsolin, a protein that degrades actin, was found to reduce mucous viscosity by 62% on average. 69 Early developments regarding therapies targeting the genetic and protein basis of cystic fibrosis are promising, with at least one undergoing clinical trials. 50 Together with genetic editing tools such as CRISPR/Cas9, burgeoning nonviral NP delivery mechanisms may have a wide variety of applications, including potentially safer CFTR gene correction.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.