
Abstract
An evolving union of high throughput screening (HTS) and absorption, distribution, metabolism, excretion, and toxicology (ADMET) technologies have transformed drug discovery. Human tissue-based, in vitro ADMET assays can efficiently generate reliable profiles for structure-activity or structure-property relationships of compounds from screening “hit sets” or libraries. The process of identifying discovery compounds with desirable “druglike” properties has consequently become increasingly data-driven. Chemists and biologists initiate the process by submitting requests to our laboratory through an intranet database. A Caco-2 cell model, automated on a Tecan Genesis workstation, evaluates the intestinal absorption of drug candidates. Distribution properties are determined with a high-throughput equilibrium dialysis technique for measuring plasma protein binding. Drug metabolism can be evaluated on a Genesis workstation via measurements of metabolic stability in liver microsomes. Drug-drug interactions can be predicted with HTS techniques using human recombinant hepatic CYP450 isoforms. A Genesis workstation, integrated with a fluorescence plate reader, executes CYP450 inhibition assays. Cell toxicity assays using human hepatocytes can serve as early, high-throughput indicators of potential systemic drug toxicity. The early availability of ADMET profiling data can now enable discovery scientists to quickly evaluate the factors that influence the pharmacodynamic and pharmacokinetic properties of compounds in lead optimization.
Introduction
High throughput screening (HTS) has been routinely employed for the past 15 years as a strategic method to discover compounds that elicit a “hit” activity in a particular molecular target or cellular screen. As research organizations have entered more and more compounds into development that were initially discovered and optimized from specific molecular targets, a trend towards higher rates of failure has been observed. These failure rates cannot be attributed to one specific cause, but rather appear to be spread out among several factors including lack of efficacy (30%) and poor pharmacokinetics or toxicity (50%), which are actually all interrelated. 1 With drug discovery and development costs now running between $800–900 million over 10–15 years, these failure rates have become unacceptable to the industry. 1 A shift in strategy for the drug discovery process has occurred, with the search for acceptable pharmacokinetics/pharmacodynamics (PK/PD) and toxicity now assuming near-equal importance to the goals of potency, efficacy, and selectivity. 2 The goal now is to identify and design away potential liabilities with absorption, distribution, metabolism, excretion, and toxicology (ADMET) early in the drug discovery process to reduce attrition later in drug development.
Effective acceleration of this new paradigm for lead optimization requires a rapid turnaround of hundreds of requests for ADMET data per week from multiple projects, all the while maintaining the high level of data quality previously provided for a relatively limited number of compounds. A collaborative union of HTS and ADMET technologies in recent years has transformed the hit-to-lead and lead optimization stages of drug discovery. 3,5 Technologies and industrialization techniques, originally developed from HTS, have successfully provided pharmaceutical companies with early ADMET data. Assay throughputs, higher than previously achieved, have been realized by adapting the assay protocols to microplate formats, including the use of scaled-down volumes and benchtop automation. High-throughput assay platforms and laboratory automation have been applied to established methods for CYP450 inhibition, 6,7 liver metabolic stability, 7,9 plasma protein binding, 10,11 drug absorption, 12,13 and cell toxicity. 14
The contemporary HTS paradigm now routinely encompasses the profiling of compounds in vitro for ADMET properties. Human or animal tissue-based, in vitro ADMET screens can efficiently generate reliable profiles for structure-activity or structure-property relationships. These assays had been traditionally performed manually on a very limited number of compounds in drug development for candidate selection. Now, data profiles can be assembled using automatable HTS assays that successfully balance quality (i.e., reproducibility and predictive value), quantity (i.e., throughput and turnaround time), and cost (i.e., sample size, supplies, capital equipment, and manpower). These early data have been used to rank compounds on a relative scale for acceptable druglike characteristics. Such profiles can be used prospectively for prediction of PK/PD properties, or retrospectively for ad hoc problem solving with compounds already in lead optimization. The process of identifying discovery compounds with desirable druglike properties has consequently become more data driven than ever before.
We have employed commercially available HTS assay technologies, laboratory automation, and user-friendly information processing systems to assemble and operate a comprehensive panel of in vitro ADMET assays for cost-effective use in the hit-to-lead and lead optimization stages of drug discovery.
Materials and Methods Caco-2 Cell Permeability Assay
A Tecan Genesis CPW 150 workstation (Tecan US, Durham, NC) was used as the automation platform. This robot has two arms: a liquid handling (LiHa) arm and a robotic manipulator (RoMa) arm. The LiHa is comprised of eight, independently controlled pipetting tips. The RoMa can grasp objects off the surface of the deck (e.g., microplates) and carry them to associated peripherals, such as the incubator or the microplate hotels. The workstation was programmed with Gemini software (Version 3.4), and all procedures (i.e., liquid handling, plate manipulation, and sample collection) were custom-written.
Caco-2 cells (American Type Culture Collection, Rockville, MD) were maintained at 37 °C in complete Dulbecco's modified Eagle's medium, containing 10% fetal bovine serum, 1.1% nonessential amino acids, 100 units/mL penicillin, and 100 μg/mL streptomycin, in an atmosphere of 5% CO2 and 90% relative humidity. For permeability experiments, cells were seeded at a density of 80,000 cells/cm2 in Costar 12-or 24-well plates (Corning, Inc., Corning, NY) on Transwell polycarbonate filters (12 or 6.5 mm diameter, respectively, and with 0.4 μm pore size). The medium was manually changed every other day for the first seven days and every day thereafter. The cells were allowed to grow and differentiate for 21-25 days.
Culture medium was aspirated from the cell monolayers prior to permeability experiments. All cell monolayers were rinsed once with 1X Hank's balanced salt solution (HBSS), pH 7.4, and allowed to equilibrate at 37 °C for 30 min. Monolayers with transepithelial electrical resistance (TEER) values greater than 400 Ωcm2 were used in assays.
Dosing solutions for test compounds were prepared in 1X HBSS, pH 7.4, at a nominal concentration of 25 μM, and allowed to equilibrate overnight at room temperature while being mixed on a magnetic stirrer. Prior to the experiment, the dosing solutions were filtered through a 0.45 μm polyvinylidene fluoride filter. The actual concentration was determined by analyzing the dose solutions for parent compound.
For the experiment, the dosing solution was added to the donor side of the monolayers, the apical side for an A → B experiment, and the basolateral side for a B → A experiment. 1X HBSS, pH 7.4, was added to the receiver side of the monolayers. At each time point, samples were collected from the receiver side of the monolayers. Bidirectional (A → B and B → A) permeability studies were conducted in duplicate over the course of 150 min. Samples were collected at 15, 30, 60, 90, 120, and 150 min. Volumes in the apical and basolateral compartments were maintained constant by replenishing the 1X HBSS after each sampling.
Analysis was done by liquid chromatography-mass spectrometry-mass spectrometry (LC-MS-MS), and the apparent permeability coefficients (cm/s) were determined as
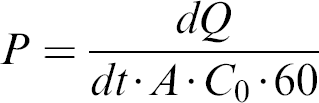
where dQ/dt · 60 is the slope of the permeation profile across the Caco-2 cell monolayers, A is the surface area of the insert (1.13 cm2), and C0 is the initial dosing concentration of the donor.
Plasma Protein Binding Assay
The approximate in vitro fractional binding of test compounds to human and animal plasma proteins was determined in 96-well equilibrium dialysis plates (Harvard/ Amika, Holliston, MA). Each well consists of a top (plasma) chamber and a bottom (buffer) chamber, separated by an ultrathin, semipermeable membrane with a 10 kDa molecular weight cutoff. Stock solutions for each test compound were prepared in acetonitrile at 200 μg/mL. The subsequent dialysis suspensions were prepared in pooled plasma (with K ethylenediamine tetraacetic acid) from male and female donors at 1000 ng/mL and dispensed into the plasma chambers in quadruplicate aliquots of 250 μL. The reciprocal buffer chambers were filled with of 250 μL of 100 mM potassium phosphate buffer, pH 7.4. Dialysis was performed at 37 °C for 5 h, with the plates mounted on an automated rotator (Harvard Bioscience, Holliston, MA) set at 20 rpm.
Samples of 150 μL were collected at the end of the dialysis period from all chambers. Each sample was diluted 1:1 with the reciprocal matrix to give a plasma-to-buffer ratio of 1:1. Sample extraction was performed by protein precipitation with 3 volumes of acetonitrile and centrifugation at 3000 rpm for 20 min. The supernatant was removed for analysis by LC-MS-MS. Percent bound is calculated with the formula:
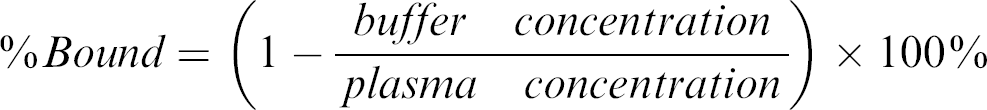
Liver Microsomal Metabolic Stability Assay
A Tecan Genesis 150 workstation (Tecan US, Durham, NC), programmed with Gemini software (Version 3.4) was used as the automation platform. This robot has one liquid handling (LiHa) arm and an orbital shaker that is equipped with a water-jacketed aluminum heating manifold. Procedures for liquid handling and incubation were custom-written.
Test compounds were incubated in duplicate Matrix MultiScreen minitubes (Matrix Technologies, Hudson, NH) with liver microsomes (Xenotech, Lenexa, KS) to measure the first order rate of consumption. Each assay was performed in 50 mM potassium phosphate buffer, pH 7.4, and 2.5 mM NADPH. Compounds were tested at a final assay concentration of 1.0 μM. The protein concentration in the reaction mix was 1 mg/mL. Compounds were preincubated for 5 min at 37 °C, and the metabolic reactions were initiated by the addition of NADPH. Aliquots of 80 μL were removed from the incubation mix at 0, 3, 6, 10, 15, 20, 40, and 60 min after the start of the reaction for profiling data, or at 0 and 30 min after the start of the reaction for screening data. Each aliquot was added to 160 μL acetonitrile for extraction by protein precipitation. These samples were mixed for 1 min by vortexing, and a volume of the mixture was filtered through wells in 0.25 mm glass fiber filter plates by centrifugation at 3000 rpm for 5 min. Sample extracts were analyzed by LC-MS-MS to determine parent compound levels. Percent loss of parent compound was calculated from the peak area at each time point to determine the half-life (t1/2, min) for test compounds.
LC-MS-MS Instrumentation
Sample analyses were performed by LC-MS-MS. Incubation plates were transferred off-line for quantitative analysis. High-performance liquid chromatography was accomplished on an Agilent 1100 Series HPLC (Agilent Technologies, Waldbronn, Germany). The system was interfaced with a Gilson 215 liquid handler and a Gilson 819 injection valve actuator (Gilson, Inc., Middleton, WI) to inject samples onto a reverse-phase column. Mobile phase A, with 0.1% (v/v) formic acid in water, and mobile phase B, with 0.1% (v/v) formic acid in acetonitrile were used in the HPLC gradient conditions.
Chromatography for samples collected from the permeability and protein binding assays was done on Polaris C18 columns (30 × 2 mm i.d., 5 μm; Varian, Walnut Creek, CA) at a flow rate of 0.4 mL/min, with 10 μL injection volumes. Gradient conditions were varied to accommodate a variety of compounds. Mass spectrometry for these samples was performed by a Waters Micromass Quattro micro with electrospray ionization in either multireaction monitoring (MRM), or selected reaction monitoring (SRM), for optimum sensitivity.
The chromatography for liver microsome metabolic stability samples was carried out with injection volumes of 10 μL, typically added to Zorbax SB C8-A (30 × 4.6 mm i.d., 3.5 μm; Phenomenex, Torrance, CA) or Polaris C18 (20 × 2 mm i.d., 5 μm; Varian, Walnut Creek, CA) columns at flow rates of 1.5 mL/min, or an Aquasil C18 (30 × 2 mm i.d., 3 mm; ThermoHypersil, Keystone, PA) column at a flow rate of 0.5 mL/min. Mass spectrometry was usually performed in positive mode with a Waters Micromass Quattro Ultima (Waters Corp., Milford, MA). MS methods for test compounds were automatically developed using MassLynx software (Version 4.0) with QuanOptimize, which selected the most intense precursor and product ion (ESI+, ESI−), and optimized the cone voltage and collision energy for multiple reaction monitoring (MRM) conditions. Compounds that failed this method development were analyzed with atmospheric pressure chemical ionization using selected reaction monitoring (SRM).
Cytochrome P450 lnhibition Assay
A Tecan Genesis 150 workstation (Tecan US, Durham, NC), programmed with Gemini software (Version 4.0) was used as the automation platform. This robot has one liquid handling (LiHa) arm and one robotic manipulator (RoMa) arm, and was integrated with a Tecan Genios Pro multifunctional plate reader using Magellan (Version 4.00) software. The procedures for liquid handling, plate manipulation, and incubation were custom-written.
This assay measured the IC50 of test compounds to inhibit isoforms of the human hepatic xenobiotic metabolizing enzymes CYP1A2, 2C9, 2C19, 2D6, and 3A4. Eight concentrations (0.01-30 μM in half-log intervals) of each test compound were tested in 200 mM KPO4, 1.3 mM NADP+, 3.3 mM glucose-6-phosphate, 3.3 mM MgCl2, and 0.4 units/mL glucose-6-phosphate dehydrogenase. Test compounds that were previously dissolved in acetonitrile were separately diluted in assay buffer. Duplicate 100 μL aliquots of diluted test compound were dispensed into Falcon Microtest 96-well assay plates (BD Biosciences, Franklin Lakes, NJ), and the plates were preincubated for at least 10 min at 37 °C. A 100 μL volume of human recombinant CYP isoforms (Supersomes, BD Gentest, Woburn, MA; 2.5-15.0 pmol/mL) and substrate (1.5-75 μM) were then added to each test well. The reaction was incubated for 15-45 min at 37 °C. Standard inhibition curves using assay-specific reference inhibitors (BD Gentest, Woburn, MA) were run with each assay plate. The reaction was terminated with the addition of 75 μL 80% acetonitrile/20% 0.5 M Tris base. The assay plate was read with the fluorescent plate reader at product-specific excitation and emission wavelengths.
Multiparameter Toxicity Panel
All cell toxicity assays were performed with the ACTIV-Tox human hepatocyte cell line (Amphioxus Cell Technologies, Houston, TX). Following receipt of cell plates from Amphioxus Cell Technologies, the cells were acclimated to 37 °C and the medium in the wells was replaced. After a 24 h incubation at 37 °C in an atmosphere of 5% CO2 and 90% relative humidity, test compounds were incubated with the cells for 24-48 h, depending on the particular assay conditions.
Lactate dehydrogenase (LDH) release, inhibition of cell proliferation, cellular adenosine triphosphate depletion, and caspase activation were determined with commercial assay kits (CytoTox96, CellTiter 96, CellTiter-Glo and Apo-ONE, respectively, Promega Corp., Madison, WI) that were supplied with the cells.
Data Analysis
Compound tracking and the analysis of data were accomplished with ActivityBase Version 5.2.2.49 (ID Business Solutions, Guilford, United Kingdom), which incorporated either the XLFit software (Version 4.1.1) or custom-written Microsoft Excel 2002 macros to process compound dispensary worklists and data from instrument output files, and automatically upload analyzed data to a central corporate R&D database.
Results and Discussion
HTS assay technologies, integrated with laboratory automation and information processing, can be effective in addressing ADMET issues early in drug discovery. In particular, the attrition rate due to failure of compounds with in vitro potency to exhibit in vivo efficacy, or potential liability to elicit drug-drug interactions, or systemic toxicity may be reduced with early in vitro profiling.
Chemists and biologists initiate the process on behalf of project teams by submitting requests to our laboratory through a corporate intranet database (eRoom Software (Version 7), Documentum Inc., Pleasanton, CA). A standardized request form enables a time-stamped request for assays to be entered compound-by-compound, with supporting physicochemical data (e.g., salt form, molecular weight, structure, solubility). The form is updated as assay requests are fulfilled, with a final time stamp when the entire request is completed. This permits designated team members to monitor the progress of the request in real time, and allows the data to be previewed prior to upload to the central corporate database.
It is possible to request data from all available assays in order to obtain a broad profile of activity or property data on a particular compound or series of compounds. Alternatively, assays may be intentionally staggered in an iterative design to provide additional or higher resolution data as a compound successfully progresses along the work plan. This strategy allows for certain assays to be performed in a truncated, high throughput screening format (e.g., reduced number of time points or test concentrations; refer to the discussion of the metabolic stability assays below) for rapid filtering out of compounds with obvious undesirable properties. These findings would then be followed up on request with more definitive, high-content profiling assay formats. These options may be specifically requested; the experimental design and data collection are then performed following distinct templates.
Drug absorption considers the path of a molecule from its site of administration to the systemic circulation. The mechanism of absorption will govern the onset and duration of drug action, as well as the level of systemic exposure. For orally administered drugs, this path passes through gastrointestinal epithelial cells and into the enterohepatic circulation. The predominant route is via passive diffusion through cells, although paracellular diffusion along the tight junctions between cells and carrier-mediated active transport are also significant. Drug absorption may also be limited by efflux transporters, such as p-glycoprotein. Consequently, drug absorption is a complex process dependent upon solubility and permeability, formulation variables, and physiological factors including regional differences in gastrointestinal pH, permeability, and motility.
Monolayers of the human colon carcinoma cell line, Caco-2, are regarded as the industry standard in vitro model system for studying mechanisms of drug transport across the intestinal epithelium. 2,12,13,15,16 These human intestinal enterocytes express transporter and efflux pumps and possess tight junctions and brush border enzymes (Fig. 1). For molecules that are largely absorbed through passive diffusion, a good correlation exists between apparent permeability coefficient and lipophilicity (measured by the distribution coefficient between aqueous and organic solvents). An assay that measures bidirectional flux from one side of the Caco-2 cell monolayer to the other, readily automated on a Tecan Genesis CPW 150 workstation (Fig. 2), can effectively profile hundreds of compounds per month.
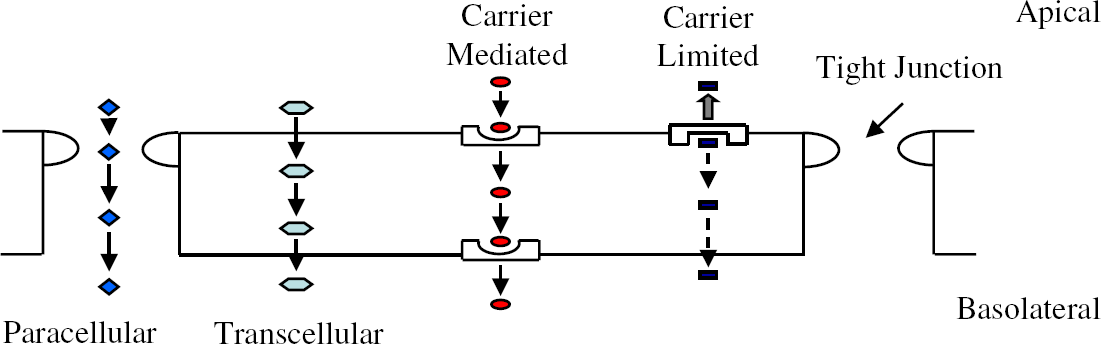
Schematic overview of the various parallel mechanisms of drug transport across a prototypical Caco-2 cell monolayer.

Tecan Genesis CPW 150 workstation supporting Caco-2 cell in vitro drug permeability assays.
Additional data on the effect of transporter mechanisms may be subsequently collected by testing particular compounds for A → B and B → A flux in the absence and presence of known transporter inhibitors. 17 We have observed, for example, that the apparent permeability coefficients of the anticancer agent vinblastine were modulated by a pglycoprotein inhibitor, suggesting that this drug and its therapeutic effect can be attenuated by efflux transport (Table 1). The antihistamine chlorpheniramine, on the other hand, was unaffected by the p-glycoprotein inhibitor.
Effect of a p-glycoprotein inhibitor on the apparent permeability coefficients for test compounds across Caco-2 cell monolayers. Data are from duplicate measurements collected in the absence and presence of 2 μM inhibitor.

Drug distribution considers the movement of drug from the systemic circulation to the extravascular space, which is often the location of the therapeutic target. The process of drug distribution is largely a function of passive diffusion and protein binding in blood and tissues. Drug molecules that are protein bound have their distribution restricted and are therefore pharmacologically inactive. Equilibrium dialysis is the most common method in the industry for measuring drug-protein binding and thus estimating effective drug concentration at the pharmacological target. 2,10,11 This technique measures binding at equilibrium, and unlike ultrafiltration or column chromatography, is less susceptible to artifacts of nonspecific binding to filter membranes and other experimental surfaces, or changes in plasma protein concentration during ultracentrifugation.
The throughput of the equilibrium dialysis was effectively scaled up with 96-well microplate technology and benchtop automation to profile hundreds of compounds per month. Our in vitro assay produced binding data (Fig. 3) that correlated well with human clinical data for a test set of thirteen drugs. The assay was particularly effective for binning test compounds as poorly (< 80%), moderately, and highly (> 95%) protein bound. The assay was also robust enough to accommodate plasma samples from common experimental animal species (Table 2). This capability can thus provide for species scaling and comparison of data among experimental pharmacological or toxicological models.
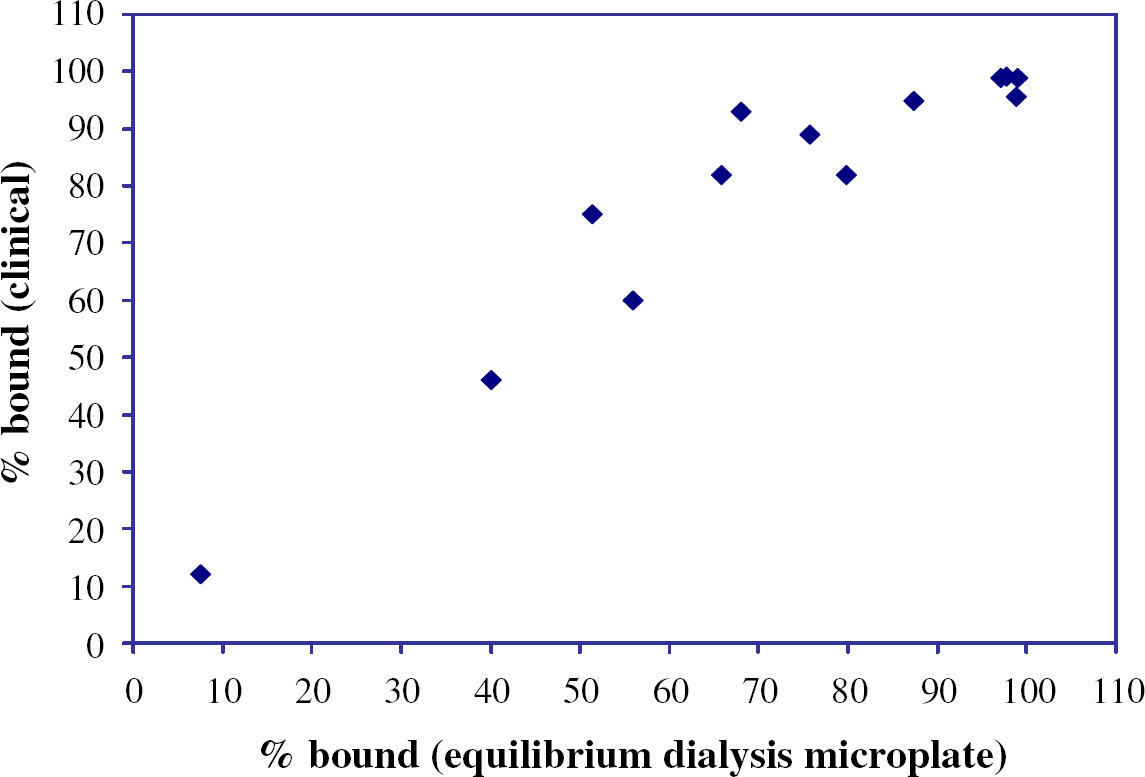
Comparison of human plasma protein binding measurements from the in vitro equilibrium dialysis microplate method and clinical data for quadruplicate determinations with 13 test compounds (amitriptyline, desipramine, methotrexate, nevirapine, ontazolast, pirenzepine, prednisone, propranolol, quinine, rifampicin, warfarin, compound 7, and compound 8).
High-throughput equilibrium dialysis plasma protein binding measurements for a poorly bound drug (pirenzepine), a moderately bound drug (nevirapine), and a highly bound drug (ontazolast) 24 from quadruplicate measurements with plasma from various species

Drug metabolism considers the biotransformation of drugs, usually in the liver, to hydrophilic derivatives that may be more rapidly eliminated by the kidneys. Metabolic reactions that add or uncover polar groups, such as oxidation and hydrolysis, generally receive the most attention in drug discovery. Drug metabolism is a significant component of potential ADMET issues in drug development, including metabolic stability, drug-drug interactions, and drug toxicity.
Metabolic stability is a key determinant of the bioavailability and clearance of a drug. Liver microsomes are widely used as an in vitro model for drug metabolism, owing to their ease of preparation and use, high content of drug metabolizing enzymes, high rate of substrate turnover, and robustness. 18 Typically, a single concentration of test substrate (at levels well below the apparent KM of drug metabolizing enzymes) is incubated with microsomes and the appropriate cofactors so that the first order disappearance of the parent compound may be measured over multiple time points to calculate a half-life. 19 Microsomal metabolic stability can be a reliable predictor of in vivo drug metabolism 20 if one assumes that oxidative metabolism in the liver is the major route of drug clearance.
Our metabolic stability assay was performed on a Tecan Genesis 150 workstation (Fig. 4) and can test thousands of compounds per month with microsomes from human, monkey, dog, rat, and mouse. This versatility can thus enable species scaling and comparison of metabolism data among experimental pharmacological or toxicological models. The test method was designed to measure percent remaining parent compound at a single time point for rapid screening 21 or at multiple time points for follow-up profiling. The half-life data were comparable between the two methods (Table 3), which allows for quick discrimination of compounds with either brief or prolonged half-lives. Generally, low metabolic stability or a short half-life suggests highhepatic clearance and consequently poor in vivo exposure.
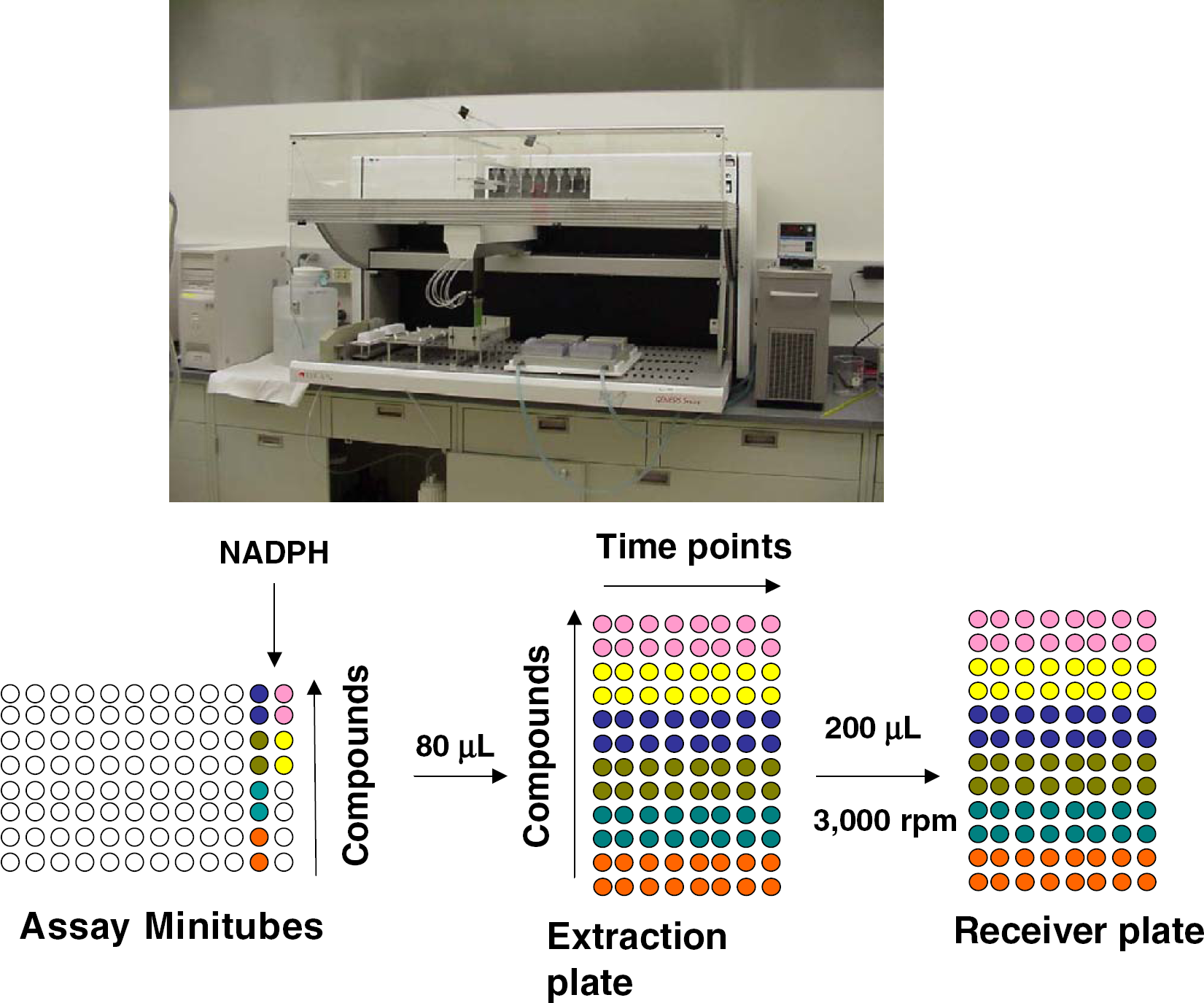
Tecan Genesis 150 workstation supporting in vitro liver microsome metabolic stability assays, and representation of the deck layout of the incubation, extraction, and filtration plates when the assay is conducted in duplicate with eight time points (0, 3, 6, 10, 15, 20, 40, 60 min).
Comparison of multiple time point and single time point methods for determining mouse liver microsome metabolic stability, expressed as first order half-lives for disappearance of parent compound from duplicate measurements

The most important drug metabolizing enzymes are a superfamily of heme-containing, membrane-bound mono-oxygenases, cytochrome P450 (CYP450). 2,18 The major human isoforms are designated 1A2, 2C9, 2C19, 2D6, and 3A4; over half of the drugs in current use are metabolized by CYP3A4. These enzymes have broad substrate specificities, catalyze multiple sites of oxidation, invoke very reactive oxygenating species, and are differentially expressed among various human populations. CYP450s may be inhibited (or induced) by drugs, which create potential liabilities for drugdrug interactions. On occasions when drugs are coadministered with CYP450 inhibitors, their plasma concentration may become elevated as a result of decreased clearance, which could result in overdose and toxicity.
Reliable, low-cost microplate-based assays for CYP450 inhibition have been developed using baculovirus-expressed recombinant human enzyme systems and fluorogenic probe substrates. 6,22 These probes undergo O-dealkylations, and their corresponding product fluorescence is not usually subject to interference from any background fluorescence or quenching by cofactors (e.g., NADPH) or test compounds. Multiple probe substrates are available for use with each CYP450, which can be helpful in accommodating the complex, non-Michaelis-Menten inhibition kinetics often observed with these enzymes. 23 Our CYP450 inhibition assays were fully automated on a Tecan Genesis 150 workstation (Fig. 5) and were capable of profiling thousands of compounds per month.

Tecan Genesis 150 workstation supporting in vitro human CYP450 inhibition assays, and representation of the deck layout of the incubation plates when the assay is conducted in duplicate with eight test compound concentrations (0.01, 0.03, 0.1, 0.3, 1,3, 10, 30 μM).
Drug-induced organ toxicity is a function of the intrinsic toxicity of the drug substance, the concentration of the drug at the target organ, and the relative influence of systemic detoxification (or bioactivation) mechanisms. Since intrinsic toxicity can be most directly related to chemical structure, it receives considerable attention in drug discovery. The liver can be a particularly sensitive end organ for toxicity as a consequence of the first-pass effect and the liver's predominant role in metabolic detoxification and activation. A suite of cell toxicity assays are frequently used to generate an “early warning” sentinel profile. 2,4 Toxicity profiling with hepatocytes can provide particular insights to metabolism-based toxicity.
Prototypical in vitro toxicity assays include measurement of the release of cytoplasmic enzymes, such as lactate dehydrogenase (LDH), as an indicator of cytoplasmic membrane integrity, measurement of mitochondrial activity or ATP levels as indicators of cellular viability and energy consumption, and measurement of caspase activity as a marker for the induction of apoptosis. 14 These assays can be miniaturized and automated in 96-well microplate formats, with fluorescent, luminescent, or colorimetric readouts. Figure 6 illustrates the effects of a test set of eight compounds on toxicity biomarkers in an immortalized line of HepG2/C3A hepatocytes. Hepatotoxicants such as amiodarone, diclofenac, tacrine, and tamoxifen were notably effective in most assays, while the negative control compound, acyclovir, was comparatively inactive. The patterns of response in these assays can provide a broad picture of the potential toxic interactions with test compounds.
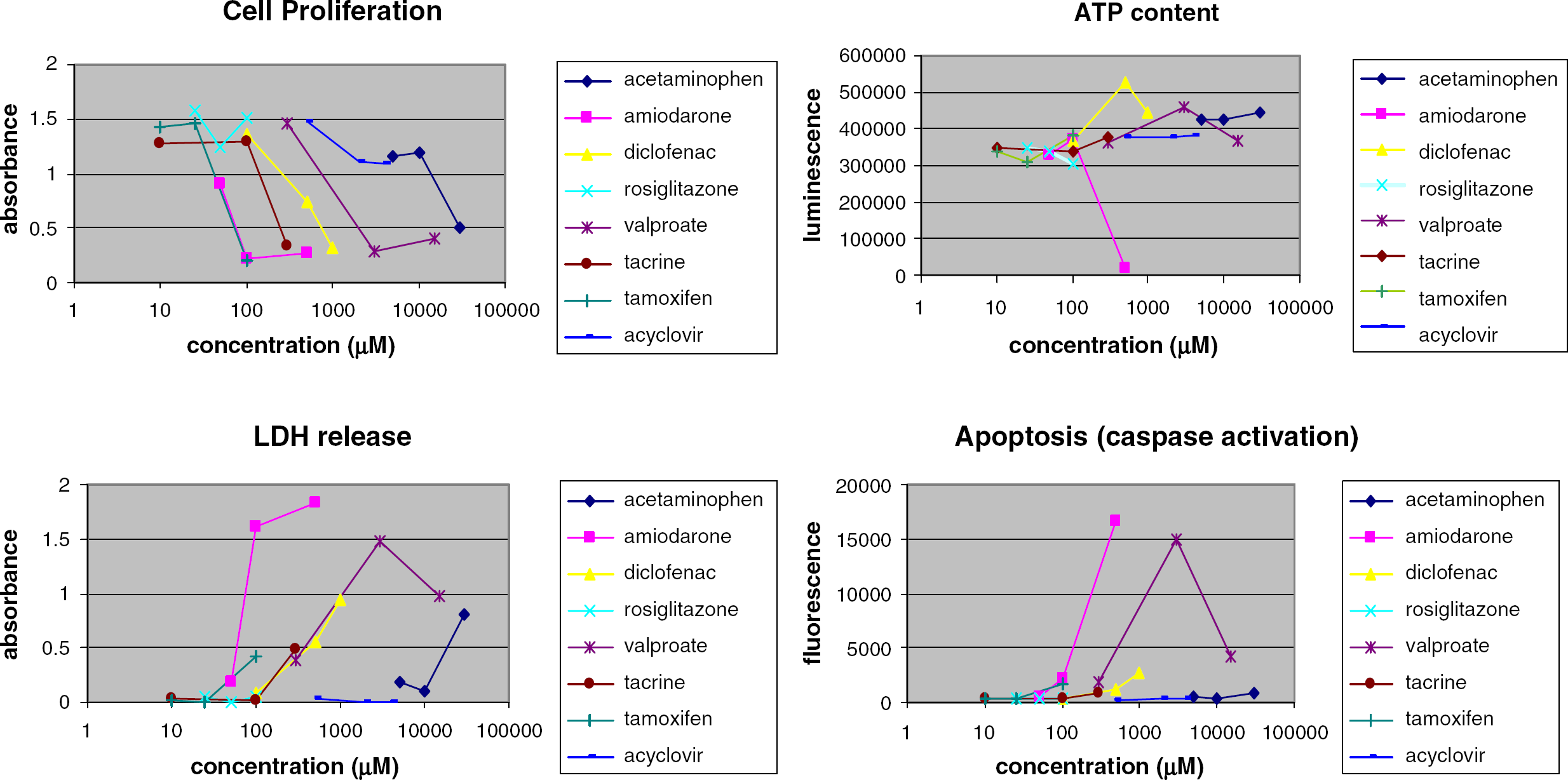
Concentration-dependent effect of test compounds on toxicity biomarkers after 24-48 h exposure to HepG2/C3A hepatocytes. Data are from duplicate measurements.
Conclusion
Pharmacokinetic, drug metabolism, pharmaceutics, and toxicology information on pharmacologically active compounds can now be acquired very shortly after synthesis, or after selection from screening hit sets or libraries. This work describes the use of commercially available individual workstations, hardware, and software to support a panel of in vitro ADMET assays. The automation and information processing technology take full advantage of the sensitivity, selectivity, and throughput available from microplatecompatible instrument readouts and contemporary liquid chromatography coupled with tandem mass spectrometry. The robust, miniaturized techniques improve assay reproducibility, reduce potential hazards from handling human biological media, and save time and money. The resulting early ADMET data serve as valuable predictors of pharmacokinetics and pharmacodynamics. Universal access to these profiling data via a central R&D database can accelerate the selection of druglike predevelopment candidates from current drug discovery programs and accrue added value for subsequent programs by enriching the data content of the corporate compound collection.
Acknowledgments
The authors would like to thank Carol Ann Homon, Mehran Yazdanian, Stephen Norris, Guanfa Gan, Donald Tweedie, Odette Fahmi, Scott Leonard, David Pilloise, Qihong Huang, and Supriya Jayadev from Boehringer Ingelheim Pharmaceuticals, and Norman Sussman and James Kelly from Amphioxus Cell Technologies for their collaboration and helpful discussions.