
Abstract
Biofoundries have enabled the ability to automate the construction of genetic constructs using computer-aided design. In this study, we have developed the methodology required to abstract and automate the construction of yeast-compatible designs. We demonstrate the use of our in-house software tool, AMOS, to coordinate with design software, JMP, and robotic liquid handling platforms to successfully manage the construction of a library of 88 yeast expression plasmids. In this proof-of-principle study, we used three fluorescent genes as proxy for three enzyme coding sequences. Our platform has been designed to quickly iterate around a design cycle of four protein coding sequences per plasmid, with larger numbers possible with multiplexed genome integrations in Saccharomyces cerevisiae. This work highlights how developing scalable new biotechnology applications requires a close integration between software development, liquid handling robotics, and protocol development.
Keywords
Introduction
Around the world, biofoundries (also termed genome foundries or DNA foundries) have hugely increased the scale and scope with which DNA constructs can be built for applications such as engineering microorganisms to manufacture high-value chemical, biological, and pharmaceutical products. At the heart of these foundries are automation robots and the protocols optimized for DNA assembly. 1 Fields such as synthetic biology and metabolic engineering increasingly use standardized building blocks of DNA parts. 2 DNA assembly techniques such as Gibson Assembly, 3 Golden Gate, 4 and BASIC 5 have enabled the ability to easily build designed DNA constructs from these standardized constituent parts.
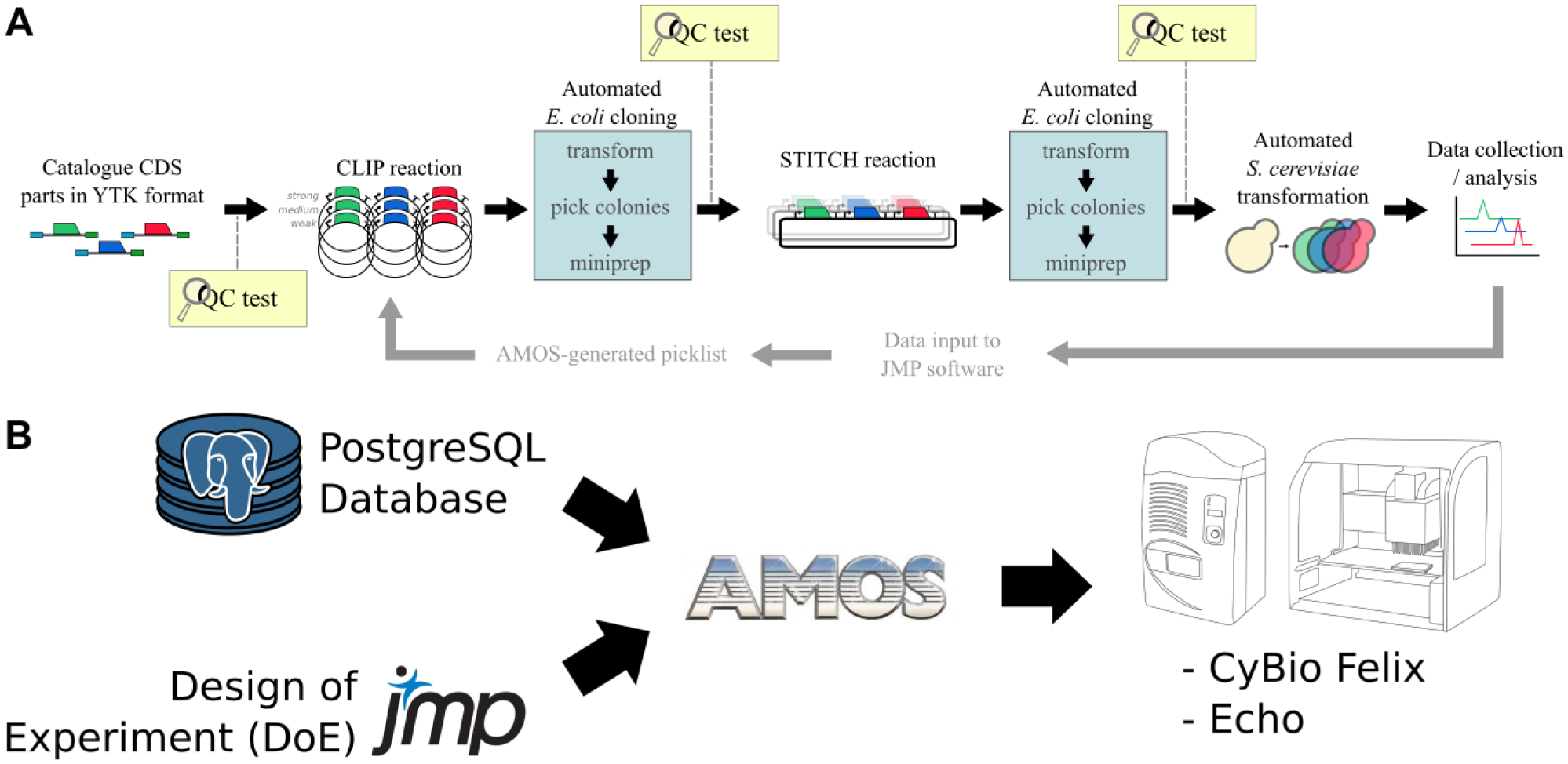
The MoClo (Golden Gate)—Yeast Toolkit (YTK) 4 uses two tiers of DNA assembly, involving different type II restriction enzymes and two rounds of amplification in Escherichia coli, prior to transformation of the DNA constructs into Saccharomyces cerevisiae. The YTK contains a set of standardized and well-characterized promoters, terminators, coding sequences, assembly connectors, E. coli markers and origins of replication (ORIs), and yeast markers and ORIs (including genome integration). The first-tier assembly is made up of single-transcription unit (TU) constructs (containing a promoter, coding sequence, and terminator), here defined as “clips.” These are then amplified in E. coli and subjected to a restriction digest-based quality control (QC) screen. These clips are combined into the desired combinations into a yeast expression plasmid to form a “stitch” that, after amplification in E. coli, is subjected to further QC. Finally, these stitches are transformed into yeast and characterized. However, this is difficult to scale by hand. Hence, this necessitate the development of a software LIMS (Laboratory Information Management System) platform, to enable a scalable DNA assembly process, together with an automated transformation protocol for S. cerevisiae.
We chose JMP as our experimental design and analysis tool, which has been used extensively in engineering and quality by design (QbD) process development. While other design tools exist in synthetic biology, such as BioCoder, 6 AquaCore, 7 PaR-PaR, 8 Antha, 9 BioBlocks, 10 SBOL designer, 11 Cello, 12 Leaf LIMS, 13 and Aquarium, 14 the JMP custom design tool offers an intuitive way to create a high-level description of any possible set of experimental factors and response variables, while generating rigorous design of experiment (DoE) studies with integrated modeling and analysis capabilities. For workcell protocol development, our approach integrates at the highest possible level using picklists, with well locations and barcodes, after an optimized protocol has been created, rather than generating precise liquid handling instructions that may be prone to error or not transferable between different manufacturer’s liquid handling platforms. Therefore, the requirements for our custom LIMS, called AMOS, were to develop a platform that can translate a design into a set of physical parts and picklists, while scaling correctly for larger processes within the precise capabilities of our robotic systems. In this study, we report how we developed a scalable, robust rapid prototyping biotechnology platform for S. cerevisiae that brings together a number of automated and semiautomated processes driven by an integrated web application software environment.
Materials and Methods
Strains and Media
S288c BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) 15 S. cerevisiae was grown in 1× YPD (yeast extract peptone dextrose) medium. However, yeast cells used for transformation were grown in 2× YPAD medium (adenine hemisulfate 80 mg/L) and selected in yeast synthetic dropout medium without uracil. E. coli cells were grown in either lysogeny broth (LB), Super Optimal Broth with catabolite repression (SOC) media, or Terrific Broth (TB) medium with appropriate antibiotic selection.
DNA Part Preparation and Verification
The part plasmids containing promoters, terminators, and the low-copy uracil vector with defined assembly connectors used in this study were from the YTK.
4
We used Sanger sequencing to verify preassembled cassettes (backbone plasmid with unique overhangs that defines clip positions), fluorescence genes, spacer cassettes (defunct arbitrary cassettes acting as space holders for designs with fewer than four clips), and all YTK parts used in this study using primers as summarized in
We cloned clips and stitches into E. coli using a 96-well semiautomated E. coli transformation protocol.
16
Preassembled cassettes and the low-copy uracil vector consist of a green fluorescent protein (GFP) dropout cassette that is cut out during clip and stitch assembly to allow green/white selection. Putative clones are all white colonies that are able to grow on appropriate antibiotic selection. The PureLink Pro Quick96 Plasmid Purification Kit (Thermo Fisher, Waltham, MA) was used to isolate plasmids from putative clones picked manually.
17
Isolated DNA parts were assayed using the PicoGreen dsDNA reagent (Thermo Fisher) to ensure that each DNA part could be transferred successfully using acoustic liquid handling (Echo 550/525, Labcyte, San Jose, CA). Parts that passed QC were entered into library storage in 96-deep-well plates (DeepWell 1 mL, Fisherbrand, Thermo Fisher), registered in the database, and given ID numbers (
(
Design: DoE Using JMP
Experimental variables (continuous, discrete numeric, and categorical) and the appropriate responses were defined using the JMP custom design tool (SAS Institute, Cary, NC). We developed the controlled vocabulary based on a base set size of four (or fewer) transcriptional units with variable promoter strengths (constitutive and inducible promoters), auxotroph types, copy number, and genomic integration of transcriptional units at multiple sites (
As an example of the possible design space for a stitch in yeast, we selected three promoter classes (high, medium, and low) per coding sequence in this proof-of-principle study (
Build: AMOS
We used the Flask framework 18 to develop a Python-based web application and PostgreSQL open-source database system to create the AMOS software platform. 19 The application can be accessed using a web browser and multiple users can run jobs concurrently. The system has a storage database showing the well position, plate barcode, and characterization metadata required to identify the specific parts to produce a design. The system is split into two build sections. First, a constructor package takes the design from JMP as an input and parses this into a format that groups the parts together so it can be built in stages. Second, the job dashboard section calculates the quantities of each part that are required to complete the job and generates the instructions for liquid handling ( Fig. 1B ).
The AMOS software workflow is summarized in
The job dashboard parses the design and generates all the necessary liquid handling instructions across automation workcells and rounds of assembly. The system looks ahead to the final number of stitches required to complete the study and, on that basis, calculates the set of unique clips required based on the working volume of the low-dead-volume (LDV) plates and the volume required for each Golden Gate reaction. Similarly, the volume and number of unique parts required are calculated based on the set of clips required. The algorithm allows the system to expand in build size, while using the lowest amount of DNA possible across multiple wells and plates as required. The job dashboard then generates a picklist that takes parts from long-term storage into the first available well of 384-well Echo LDV plates. The destination well chosen by the CybioFelix is then uploaded into the job dashboard, where the subsequent picklists for acoustic dispensing (Echo 550/525) for the clip, stitch, and enzyme master mix reactions can occur. Golden Gate reactions were performed according to existing protocols (
High-Throughput S. cerevisiae Transformation and Selection
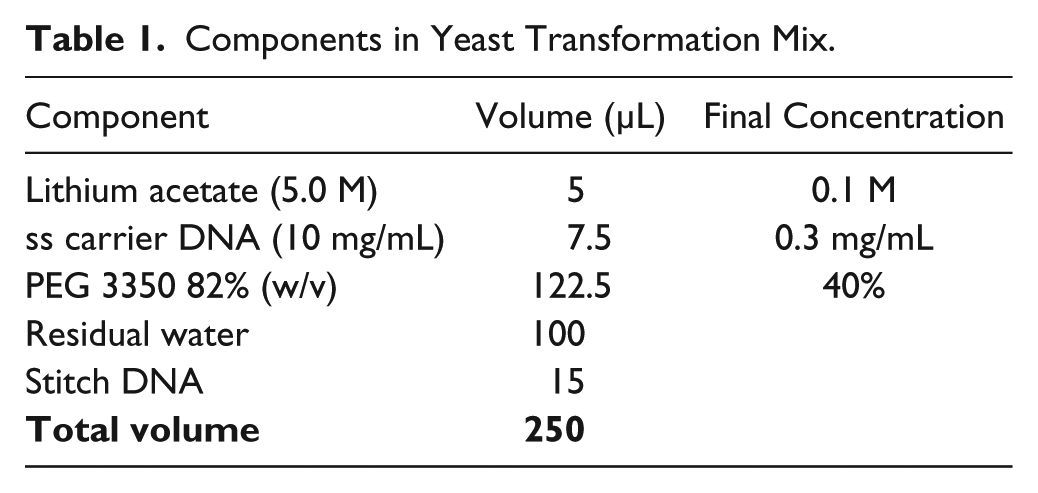
Frozen yeast cells with high transformation efficiency were prepared according to an existing protocol. 20 Overnight cultures were grown for 16 h and then inoculated into prewarmed 500 mL of 2× YPAD medium to make the final titer of 5 × 108 cells/mL. A multidrop combi (Thermo Fisher) was used to aliquot 100 µL of cells into a 96-deep-well plate (Masterblock 2 mL V bottom, Greiner Bio-One, Monroe, NC) and stored at −80 °C for later use. A standard lithium acetate-based transformation method was used to transform cells 21 with the components listed in Table 1 . Tip-based mixing for complete resuspension of yeast cells proved inefficient; therefore, shaking was used for resuspension. Six 3 µL droplets (18 µL total) from each well were arrayed onto selective agar in an Omnitray (Thermo Fisher) with a 4 min drying time after each dispense.
Components in Yeast Transformation Mix.
Test: Characterization
Putative yeast colonies were manually picked with no prior screening and grown to exponential phase at 30 °C in 500 µL of selective media in 96-deep-well plates (Masterblock 2 mL V bottom, Greiner Bio-One), sealed using a gas-permeable seal (BeatheEasy Merck KGaA, Darmstadt, Germany) before flow cytometry analysis (LSR Fortessa, Becton Dickinson, NJ). FlowJo (Treestar, San Carlos, CA) and JMP (SAS Institute) were used for analysis of cytometry data.
Results and Discussion
Testing Build Process: Golden Gate Assembly Using YTK DNA Parts
In order to test the efficiency of this process, we picked two replicates of each clip reaction for plasmid isolation and used the BsmBI restriction enzyme for verification, which showed a 100% success rate (
High-Throughput Yeast Transformation
We decided that creating large batches of frozen yeast cells stored in 96-well plates would ensure robust transformation across multiple runs. We used warm sterile water, 1× YPAD, or phosphate-buffered saline (PBS) to thaw the cells. We observed that only water or PBS, but not 1× YPAD media, allowed cell resuspension by shaking ( Fig. 2A ). Cells must be resuspended in 33% PEG-3350 for lithium acetate transformation; however, it was not possible to do so directly from a frozen cell pellet. Therefore, we found that flash thawing the cells in water, pelleting, and removing only 90% of the supernatant meant that the cells could be resuspended in the remaining volume by shaking. We then added PEG-3350 to give a final concentration of 33%. However, we found that the 33% final concentration of PEG did not give consistent transformation efficiency across a 96-well plate. By increasing the final concentration of PEG-3350 to 40%, we found that cells did not aggregate and the colonies per microgram DNA increased ( Fig. 2B ). We used a shaking incubator at 42 °C to heat shock yeast cells with the transformation mix. This step was increased from the standard methods to 3 h, which improved the transformation efficiency across the plate.

(
After heat shock, cells were washed and arrayed onto agar. We used a tip-based liquid handling robot to dispense 3 µL of cells on prewarmed synthetic dropout media (without uracil) agar plates. The pipetting height and liquid class were determined to produce a droplet at the tip, which was then transferred to the surface of the agar. Each droplet or “spot” was allowed to dry for approximately 4 min before spotting again, with the number of spotting events determined by the transformation efficiency. To maintain cells in suspension between spotting events, a number of timed mixing loops were used ( Fig. 2C ). Figure 3 summarizes the whole flow of work of the high-throughput yeast transformation and selection steps. Using our established yeast transformation protocol, we are able to transform and select putative yeast transformants in a 96-well format within 3 days. Conceivably, high-throughput yeast transformation could facilitate the rapid building of very large DNA constructs for higher eukaryotic hosts. 22
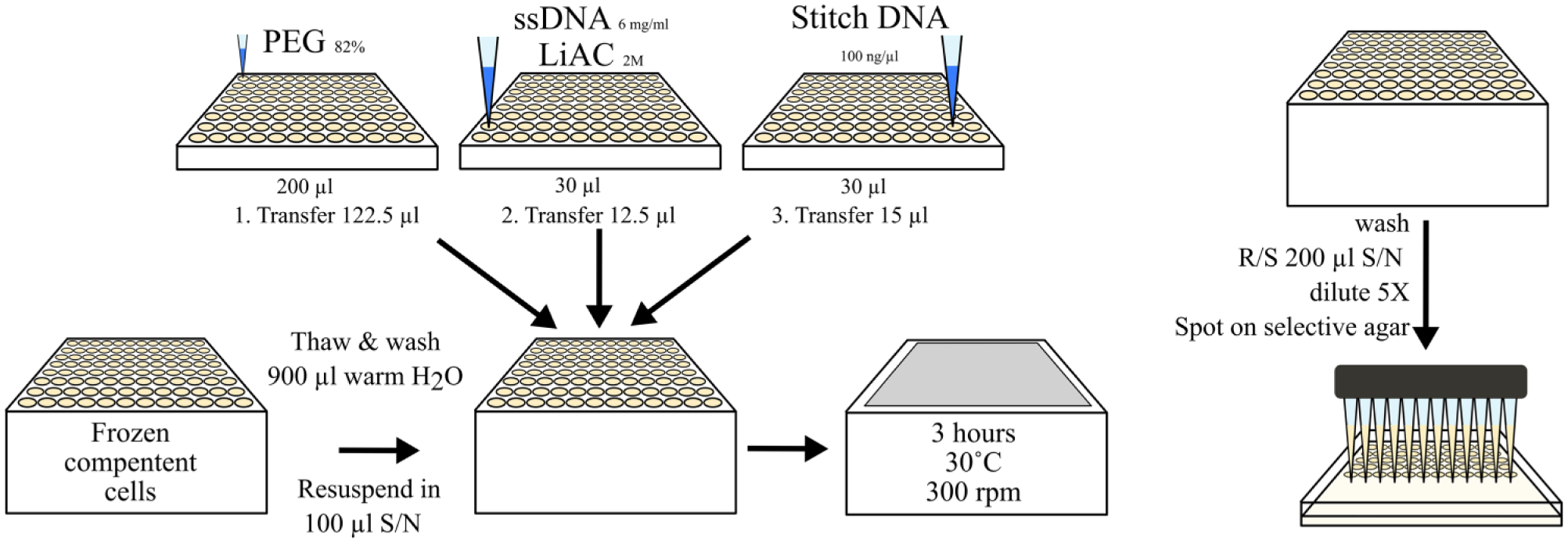
Schematic diagram illustrating the robotic steps involved in creating a high-throughput yeast transformation protocol compatible with the London DNA Foundry workcell setup. The full method is extensively described in the Results and Discussion section. R/S = resuspend; S/N = supernatant.
Analyzing Fluorescence Data in JMP
We used flow cytometry to analyze the fluorescence of the three different fluorescent proteins produced by yeast transformed with the assembled stitches. sfGFP, mTagBFP, and mRuby2 were at fixed positions 1, 2, and 3 in all 88 stitches. Figure 4 shows that as promoter strength increases at all three positions in the stitch assembly, the expression of sfGFP, mTagBFP, and mRuby2 also increases, as expected from previous characterization of these promoters, confirming that the design–build–test behaves as expected. 4
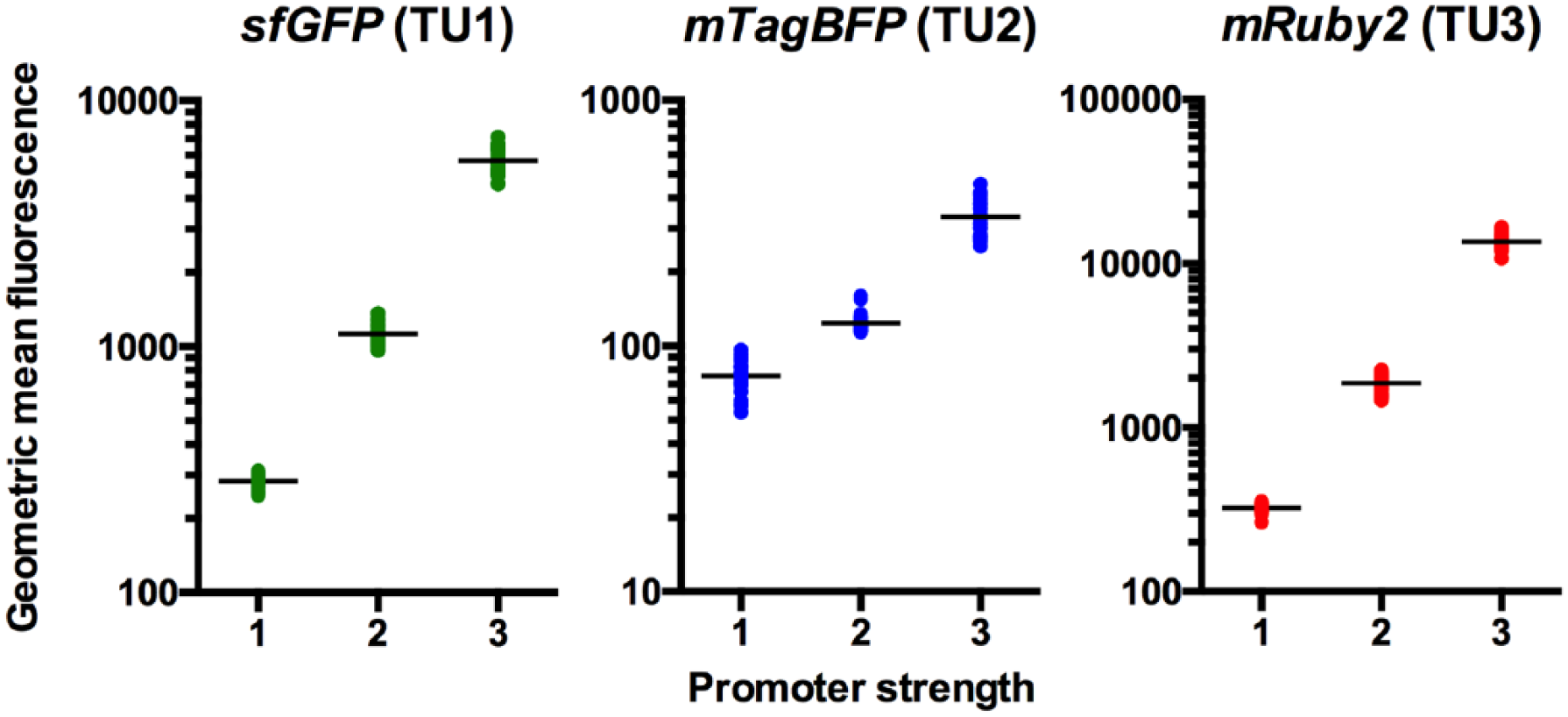
The expression of sfGFP, mTagBFP, and mRuby2 of all stitches analyzed using flow cytometry and JMP. As promoter strength increases, the expression of sfGFP, mTagBFP, and mRuby2 also increases, confirming that the design–build–test behaves as expected. sfGFP, mtagBFP, and mRuby2 were at fixed positions (TU1, TU2, and TU3) in all stitches.
In this study, we have demonstrated the automation of a parts-based yeast genetic engineering platform, driven by design and process management software. This has been established by integrating protocol development, software (AMOS), and automation platforms, and this framework can enable more rapid design–build–test cycles or larger more high-throughput applications using YTK-formatted DNA parts.
Supplemental Material
Rajakumar_Gowers_etal_supplementary – Supplemental material for Rapid Prototyping Platform for Saccharomyces cerevisiae Using Computer-Aided Genetic Design Enabled by Parallel Software and Workcell Platform Development
Supplemental material, Rajakumar_Gowers_etal_supplementary for Rapid Prototyping Platform for Saccharomyces cerevisiae Using Computer-Aided Genetic Design Enabled by Parallel Software and Workcell Platform Development by P. D. Rajakumar, G-O. F. Gowers, L. Suckling, A. Foster, T. Ellis, R. I. Kitney, D. W. McClymont and P. S. Freemont in SLAS Technology
Footnotes
Acknowledgements
We acknowledge the Dueber Lab (Berkeley) for distribution of the YTK parts and W. Shaw (Imperial) for his distribution of the YTK-compatible preassembled cassette vectors and expression vectors.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded through a BBSRC iCASE studentship grant (BB/P504579/1), a BBSRC SUGER grant (BB/K019791/1), and a BBSRC Foundry grant (BB/L027852/1).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.