
Abstract
Synthetic biology depends on the ability to rapidly produce strains with improved phenotypes but is limited by the ability to rapidly produce strain collections with directed mutations. Here, we present a system capable of overcoming this limitation through automated P1-phage transductions of Escherichia coli. By combining the Keio collection of single-gene deletion E. coli mutants with P1-phage, it is possible to generate an engineered host-strain collection consisting of every possible gene deletion mutant. This strategy was tested by transducing 355 genetic markers from the Keio collection into five different host strains, and it achieved a 98% success rate. This method offers an improved mechanism for rapidly engineering collections of microbes and provides one method for rapidly deploying a broader synthetic biology effort.
Introduction
The efficient exploitation of Escherichia coli through synthetic biology calls for the rapid optimization of host metabolism to achieve optimal target performance. Through DNA manipulation techniques, new strains may be engineered that exceed the metabolic potential of the native organism as seen in engineering of E. coli secondary metabolite pathways for the production of various compounds including biofuels and pharmaceuticals. 1,2 In most cases, the resulting strains were not suited for commercial use because of low levels of product formation, and continued engineering of the strains' metabolism was required. Ideally, an engineered strain directs a maximal amount of feedstock nutrients to product formation while simultaneously minimizing the accumulation of secondary products and excess biomass. Strain optimization is often performed by eliminating metabolic pathways that lead to undesirable products (including biomass) and by upregulating the product formation pathways. One successful application of this engineering paradigm is the construction of an E. coli strain for the efficient production of 1,3-propanediol. 3 In this example, the final strain incorporated a combination of heterologously expressed genes and modified native metabolism.
The generation of optimized production strains, however, is severely limited by current methods for identifying and implementing genetic changes in a host. Although identification of the primary pathways responsible for product formation is often trivial, the redirection of flux through those pathways is often very challenging. Frequently, researchers use rational or model-driven design to identify likely candidate pathways for modification, 4,5 and there are several strategies for achieving the desired changes in a native host. Random mutation strategies (e.g., transposon mutagenesis) can be used to improve strains. 6 Such mutant libraries permit the exploration of large numbers of metabolic modifications, but the screening required to ensure sufficient genome coverage can consume valuable resources. Strain evolution strategies that rely on natural selection can also be used to produce ideal strains, 7 but its application is limited to cases where improved growth and improved product formation will emerge because of the same evolutionary pressure. Directed modification of strains can also be used. However, although significant progress can be made through directed means, gaps in our understanding of metabolism can limit the success of this engineering approach when used as a de novo strategy.
Regardless of which of the aforementioned engineering techniques is used to identify gene-modification targets, strain engineering often requires gene knockouts in the native host. These knockouts can be generated via recombineering techniques that permit nearly scar-free excision of chromosomal regions. 8 Alternately, the Keio collection of strains provides a ready source of E. coli strains with a single deleted gene, 9 and these deletions can be transferred to the recipient host via transduction by the P1-phage. Directed genome-modification efforts often use P1-phage transduction to mobilize genetic elements between E. coli strains. Transduction permits the rapid transfer of genetic markers without concern for trans mutations. Before the availability of the E. coli Keio Knockout collection, 10 gene deletions were first created using another technique such as that described by Datsenko and Wanner. 8 However, with the advent of the Keio collection, construction of gene-knockout strains now requires only a single transduction event. In addition, the collection is available in an arrayed and annotated form that amenable to automated applications.
Here, we demonstrate a system for rapidly producing a collection of directed mutants through automated P1-phage transduction. This system offers an alternative to both rational and random methods of strain engineering. Phage transduction is one of the oldest genetic techniques, 7,11 but it can be a laborious and difficult to scale manual method. The small particle size and high-specific activity of the phage make cross contamination a constant concern during manual transductions. Together, these factors limit the number of parallel transductions that one person can perform. Automating P1 transduction has several advantages when compared with automating other genetic techniques, such as chemical transformation. 12 Chemical transformation requires chilled sample holders and very precise heat-shock timing, and the quality of chemically competent cells seriously degrades once the cells are defrosted. Transduction is a robust process that tolerates some deviation in process variables and strain backgrounds. Every step in the phage preparation and transduction process can be performed under conditions that are easily achieved with an automated system. The phage stocks and recipient cells are stable throughout the day. The P1-phage's dependence on calcium ions for activity also facilitates both the containment of the phage and the development of a liquid-phase process.
Materials and Methods
Bacterial Strains and Collections
Transductions were tested using five ethanologenic recipient strains derived from E. coli strain MG1655: MT203 (Δack ΔldhA pfl::pdcZm adhBZm catR ), MJD372 (Δack ΔldhA ΔsucC pfl::pdcZm adhBZm catR ), MJD373 (Δack ΔldhA Δzwf pfl::pdcZm adhBZm catR ), MJD374 (Δack ΔldhA Δpgi pfl::pdcZm adhBZm catR ), and MJD375 (Δack ΔldhA Δmdh pfl::pdcZm adhBZm catR). All transduction donor strains were taken from the E. coli Keio Knockout collection (Open Biosytems, Huntsville, AL). 9 Supplemental Table 1 lists the Keio collection clones used in this study. The transduction donor strains were selected from a variety of metabolic functionalities with the intention of eventually assaying the effect of specific mutations on biofuel production. The recipient host-strain MT203 was a gift from David Keating and Mary Tremaine. The remaining recipient host strains were derived from MT203 using the manual P1 transduction method (described below) with selected Keio collection strains. 9
Unsuccessful transduction reactions by host and locus
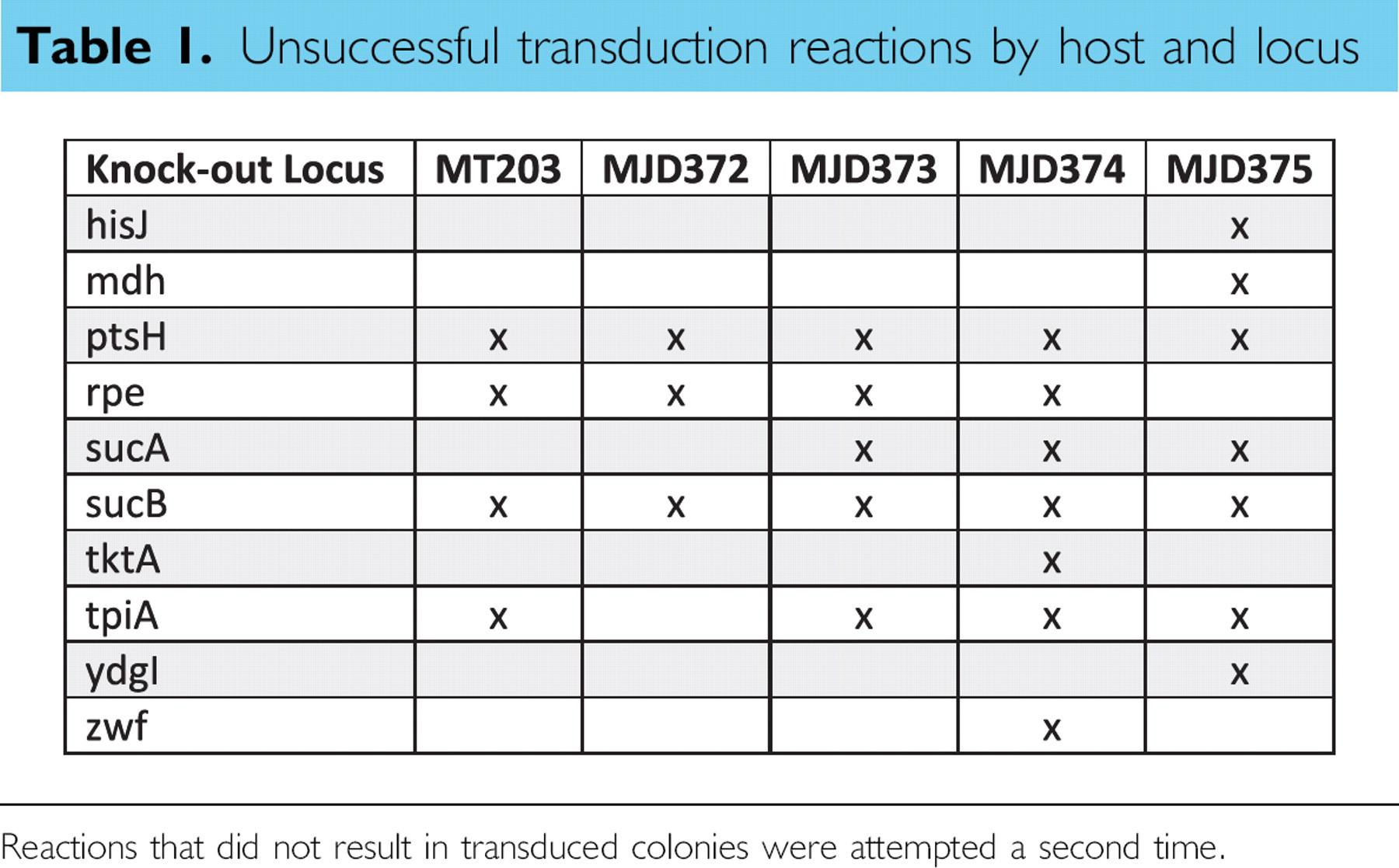
Reactions that did not result in transduced colonies were attempted a second time.
Kanamycin resistance markers were excised from the host-strain chromosomes using the method described by Datsenko and Wanner. 8 Briefly, transduced strains were made electrocompetent and transformed with the plasmid, pCP20. Transformants were grown overnight at 30 °C in Luria Bertani (LB) medium supplemented with 100 μg/mL ampicillin. The resulting cultures were plated on LB agar medium and grown at 42 °C. Individual colonies were tested for ampicillin and kanamycin sensitivity. The gene deletions and subsequent kanamycin marker removal were verified by polymerase chain reaction (PCR) (described below).
Lysate Generation
P1-phage lysates were generated manually (Fig. 1A). Selected Keio collection clones were grown at 37 °C overnight in 1 mL LB supplemented with 50 μg/mL kanamycin in a 2 mL 96-deep-well plate (Nunc 278743). Fifty microliters of overnight culture was diluted in 950 μL LB supplemented with 0.2% glucose, 5 mM CaCl2, and 5% phage stock. Cell lysis proceeded at 37 °C for 2 h while shaking. One hundred microliters of chloroform was added to each well, and the plates were vortexed for 5 min. The plates were then centrifuged in a Beckman-Coulter Allegra 25R centrifuge at 4100 rpm for 7 min. The supernanant was then transferred into a fresh 96-well plate, covered with breathable sealing tape (Nunc 241205), and stored in the dark at 4 °C.
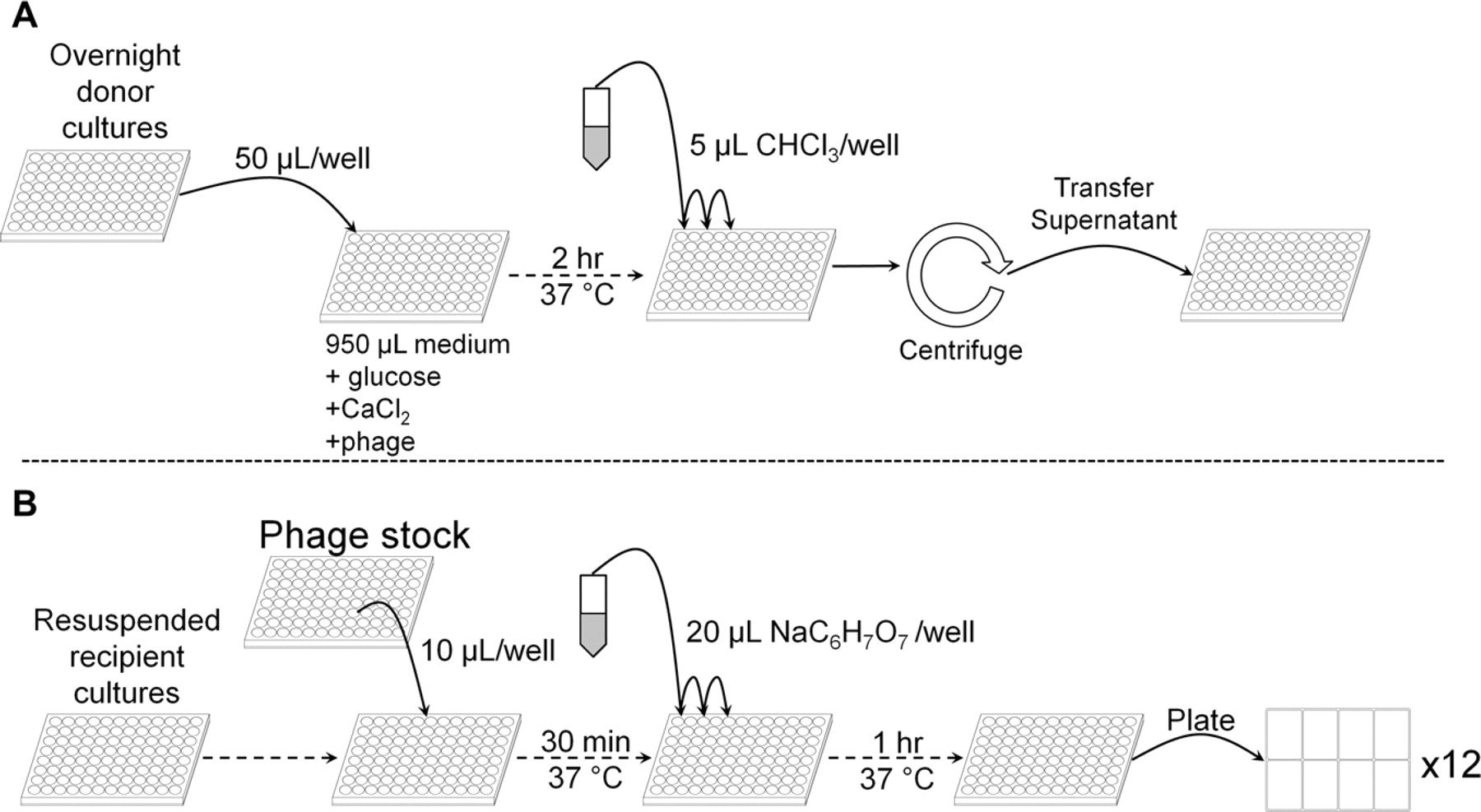
Flow diagram for 96-well (A) lysate generation and (B) transduction. Dotted lines indicate that a single plate is progressing through the process (e.g., through an incubation step). Solid lines indicate the transfer of material between reservoirs or plates. (A) Overnight cultures are diluted 1:20 in 1 mL of phage-containing medium. After 2 h at 37 °C, chloroform is dispensed into each well, the plates are centrifuged, and the supernatants are held at 4 °C. (B) Phage stock is dispensed into wells containing resuspended transduction host cells. After 30 min at 37 °C, sodium citrate is dispensed into each well followed by another hour at 37 °C. The completed transduction reactions are then plated on eight-well agar dishes.
Liquid Handling
All liquid-handling operations were performed on an automated system consisting of a Tecan Freedom EVO 200 fitted with 8-channel pipetting, 96-channel pipetting, and robotic manipulator arms (Fig. 2). On-deck temperature-controlled plate carriers provided nonshaking incubation, and shaking incubation was supplied by an integrated Liconic StoreX 40HR humidified incubator. An integrated Liconic LPX220 carousel delivered consumables and samples to the robotic manipulator arm for placement on the deck. All automated components were controlled by the Tecan EVOware Plus v2.3 software package. Two reagent reservoirs on the robotic deck supplied 1 M sodium citrate buffer (pH 5.5) and LB medium supplemented with 10 mM MgSO4 and 5 mM CaCl2. Additional integrated components visible in Figure 1 were not relevant to this study.

Robotic system based on a Tecan Freedom EVO 200 liquid-handling platform: (a) 8-channel pipetting arm, (b) transport to carousel, (c) 96-channel pipetting arm, (d) heated incubation position at 37 °C, (e) transport to STX44 shaking incubator at 37 °C, and (f) robotic manipulator arm. Additional integrated components shown in the image were not relevant to this study.
Transductions
Figure 1B outlines the automated transduction process. Overnight cultures of the recipient strain were dispensed, 100 μL/well, into V-bottom polypropylene 96-well plates (Nunc 422587). The cells were then pelleted at 4100 × g for 5 min in a Beckman-Coulter Allegra 25R centrifuge. The supernatants were discarded. Phage aliquots were manually dispensed from stocks into V-bottom polypropylene 96-well plates. Six wells on each phage-aliquot plate were left empty and served as no-phage controls. Recipient-pellet plates and phage-aliquot plates were loaded onto the carousel.
The 96-well pipetting head resuspended the pelleted recipient cells with 100 μL supplemented LB from the on-deck reservoir. Phage-stock aliquots were mixed into each well containing resuspended recipient strains. For transducing strains MT203, MJD372, and MJD373, 10 μL of phage stock was used per reaction. For the remaining recipient strains, 20 μL of stock was used per reaction. The robotic manipulator arm moved the cell/phage-suspension plate to the temperature-controlled plate carrier, and the plates were incubated for 30 min at 37 °C. After incubation, the 96-channel pipetting arm added 20 μL of 1 M sodium citrate (pH 5.5) to each well. This was followed by 1 h of shaking incubation at 37 °C in the StoreX incubator. Each completed transduction reaction was plated in 1 well of an 8-well dish (Nunc 267062) filled with 5 mL LB agar medium supplemented with 50 μg/mL kanamycin. The 8-well dishes were prepared by manually pipetting 5 mL agar medium into each well. The reactions were plated by spotting 100 μL of reaction product as an array of 12 equal spots across the agar surface (i.e., 8.3 μL/spot) (Fig. 3). In this way, cells from one 96-well reaction plate were plated onto twelve 8-well agar dishes. To account for any differences in agar height, we used the liquid sensing functionality of the eight-channel pipetting arm to detect the level of the agar medium. Each spot was dispensed 0.5 mm above the detected medium level. The entire process was tracked using Tecan Sample Tracking software. All of the transduction and eight-well dishes were bar-code labeled. The Sample Tracking software built in reporting functionality provided reports mapping each plated sample to a transduction event. Manual labeling of plates was also used in the event of software failure. The agar dishes were incubated overnight at 37 °C. The resulting colonies were manually picked into deep-well plates containing LB supplemented with 50 μg/mL kanamycin and grown overnight at 37 °C. Overnight stocks were supplemented to 20% glycerol and stored at −80 °C.

Plating layout. Each complete transduction reaction was dispensed as 12 equal spots onto 5 mL of solid medium in 8-well dishes. In this way, the transductions from a single 96-well plate were plated onto twelve 8-well dishes. Tecan Sample Tracking software was used to map the plated cells to specific transductions.
Manual transductions were carried out using a method similar to the aforementioned protocol. One hundred microliters of an overnight culture of the recipient strain was pelleted at 6000 × g in a microcentrifuge (Eppendorf, Hamburg, Germany). The supernatant was discarded, and the pellet was resuspended in 100 μL of LB supplemented with 10 mM MgSO4 and 5 mM CaCl2. Ten microliters of the donor phage stock was added to the resuspended recipient strains. After 30 min of incubation at 37 °C, the reaction was stopped via addition of 20 μL of 1 M sodium citrate buffer (pH 5.5). After a 1-h outgrowth, the entire contents of the reaction tube were plated on LB agar supplemented with 50 μg/mL kanamycin.
Transduction Verification
Transduction was verified by colony PCR on 25 selected transductants. The transductants were chosen randomly from the list of all transduction reactions. The freezer-stocked cells were stamped onto LB Agar supplemented with 50 μg/mL kanamycin and grown at 37 °C overnight. The resulting colonies were picked for subsequent analysis. A small amount of the cells were resuspended in two tubes containing 50 μL of a PCR master mix (1X GoTaq reaction buffer, 400 μM dATP, 400 μM dGTP, 400 μM dCTP, 400 μM dTTP, 3 mM MgCl2, 0.2 μM forward and reverse primers, and 2.5 U of GoTaq polymerase). The thermal-cycling program was the following: 95 °C for 3 min; 10 cycles of 95 °C for 30 s, 60 °C-1.0 °C per cycle for 45 s, and 72 °C for 90 s; 20 cycles of 95 °C for 30 s, 50 °C for 45 s, and 72 °C for 90 s; and 72 °C for 3 min. The primers used for verification are listed in Supplemental Table 2. The two PCR reactions verified both the presence of the transduced locus and the location of that locus on the recipient host's chromosome. The first reaction product spanned the knockout locus. The second PCR reaction used one primer from outside of the knockout locus and one primer within the kanamycin resistance gene. Primers were designed from the E. coli strain BW25113 genome. Every reaction contained two sets of forward and reverse primers. The first set was an internal control for the ldhA deletion found in all of the recipient strains. The second set was specific to the transduction being verified. A no-cell control PCR reaction was performed for every verification reaction to control for cell carryover between reactions. PCR product sizes were verified by agarose gel electrophoresis.
PCR data for 25 random loci in five host strains
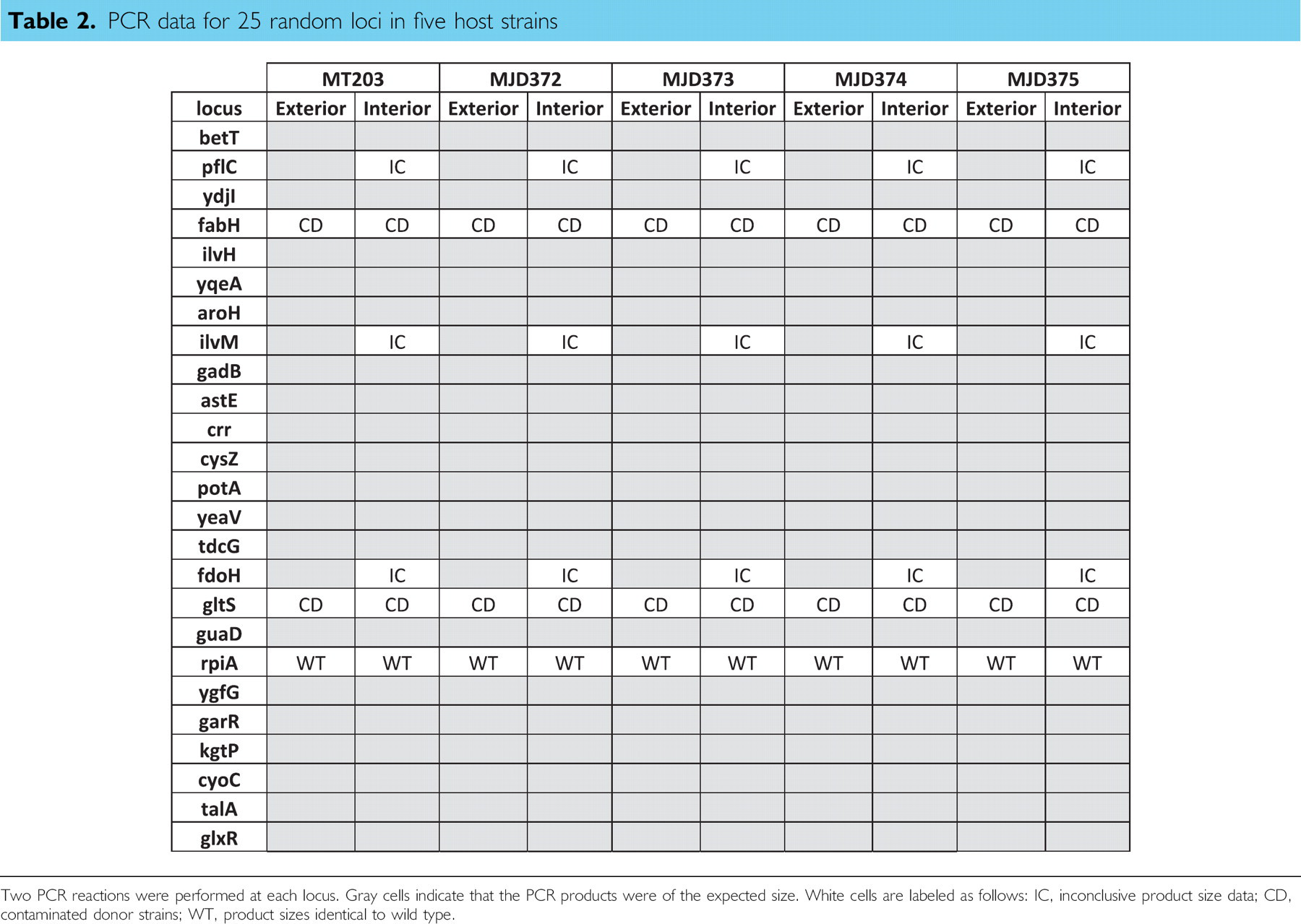
Two PCR reactions were performed at each locus. Gray cells indicate that the PCR products were of the expected size. White cells are labeled as follows: IC, inconclusive product size data; CD, contaminated donor strains; WT, product sizes identical to wild type.
Results and Discussion
Automated transductions were performed on five different recipient E. coli strains using lysates from 355 donor strains taken from the Keio collection. For host-strain MJD274 and MJD375, transduction efficiencies were improved by using 20 μL of donor lysate. The lysates used in this study were produced manually, but there is no reason why this step could not have been automated. In this study, it was not time efficient to automate the production of four plates of lysate stock. With storage at 4°C, phagestocksare stable over several months. This permitted us to reuse the same four plates of lysate stock throughout method development and the transductions described here. All other steps of the transduction, including plating for single colonies, were carried performed by the robotic system. Using the system described previously, it was possible to schedule up to 15 transduction plates per 24 h. With only a single shift of personnel, eight transduction plates could be processed per day. This throughput would permit complete coverage of the nonredundant Keio collection in approximately 1 week of run time.
Over the course of 355 transductions, plus the control reactions, performed on five different host strains, 96% of the transduction reactions produced kanamycin-resistant colonies on the first attempt. The failed reactions were performed again, and the total success rate increased to 98%. Table 1 summarizes the unsuccessful transduction events after two attempts. Some of the failures may have been because of the genotype of the resulting strains. One example is the transductions of the rpe locus. Lyngstadaas et al. 13 reported that rpe-pgi and rpe-zwf double mutants are extremely slow growing in a complex medium such as the LB used in this study. The transduction failures at the ptsH and sucB loci were likely caused by low-activity P1 lysates. This was an unavoidable consequence of performing a uniform lysate generation across all of the donor strains. The individual growth characteristics that affect lysate efficiency were not taken into account. The other failed reactions have not been predicted or reported as producing synthetic-lethal phenotypes. 14 Many of these reactions could likely be made successful with individual troubleshooting.
Several methods were attempted for the final step of plating the resulting transductants onto selective agar medium. Standard genetics practice dictates that one must produce single colonies derived from a single cell, thus producing colonies that arise from a single transduction event. Plating beads were tested because they are commonly used in manual methods to evenly distribute cell suspensions. Unfortunately, difficulties in dispensing the beads aseptically to each of the eight wells made this method untractable. The most successful method of plating involved dispensing the completed transduction reactions across the agar surface using the eight-channel pipetting arm. Each rectangular segment of the 8-well plate was defined in the controlling software as a 12 virtual wells. Then, the completed transduction reactions could be dispensed among all of the virtual wells. This distributed the resulting transductants sufficiently to obtain individual colonies without the use of custom equipment (Fig. 4A).

Typical plate results for (A) a transduction reaction well and (B) a control reaction well. Transduction reaction wells easily achieved single colonies in all cases. Typical transduction reactions produced 100 colonies per well.
As mentioned previously, cross contamination of manual-phage transductions is a well-known problem. On each transduction plate, several wells served as no-phage controls to identify any cross contamination of the phage particles. During the phage-stock preparation, those wells contained no cells to produce a phage lysate. Initial results indicated a persistent phage contamination throughout all control wells. This contamination resulted from carryover of phage through several steps of the phage-stock generation process. The phage stock dispensed across the plates during the phage-generation phase was itself the product of a kanamycin-resistant strain. The diluted phage in the control wells was then sufficiently active to produce a few transductants per well. The contamination was eliminated by repeating the phage-stock generation using a phage stock propagated on a kanamycin-sensitive strain (Fig. 4B). In cases where the transduction plates incubated for more than 24 h, small colonies would eventually arise in all wells. All of the late-arising colonies tested were kanamycin sensitive. Unfortunately, excluding late-arising colonies artificially reduces the success rate in cases such as the rpe mutant locus described earlier. Also, in a parallel system such as the one described herein, cross contamination can occur through repeated use of robotic multichannel pipets. Twenty-five transduction reactions were randomly chosen for PCR verification in each host background. This random sampling was used to identify the frequency of cross contamination during the transduction reactions. Table 2 summarizes the results of those PCR verification reactions. There were no cases where cross contamination could be attributed to the automated steps of the process. It is important to note that the negative or aberrant PCR results occurred identically across all five host strains. This implies that any cross contamination occurred before the automated steps in the process. For the pflC, ilvM, and fdoH loci, the first PCR using flanking primers returned the expected result. However, PCR reactions using both transduced strains and the original Keio donor strains resulted in products of different but incorrect sizes (data not shown). The error is likely a combination of primer misannealing and differences between MT203 and the Keio collection host, BW25113. Verifications of the fabH and gltS loci indicated problems with contaminated donor strains. PCR results from multiple colonies of the donor strains indicated a mixture of two different product sizes. The resulting fabH and gltS transductants possessed one of the two PCR product sizes indicating the faithful transduction from one of the two strains present in the original donor stock (data not shown). The rpiA locus was the only loci for which direct evidence of the method's failure to faithfully transduce the donor's genetic marker.
We have presented a new system for performing automated P1-phage transductions in E. coli. Using this method, we were able to dramatically increase the efficiency of directed genome engineering. By making use of available collections, such as the E. coli Keio Knockout collection, this method can be used to optimize synthetic biology efforts to for industrial production hosts. Using this collection, Typas et al. developed a similar system for generating double knockouts via use of High Frequency of Recombination (Hfr) E. coli. 10 This system has an impressive throughput, generating several thousand double knockouts over a few days. The resulting strains can be easily assayed for growth defects on solid medium by using an optical quantitation system. This process requires an Hfr donor strain not readily available in all collections, and it produces an Hfr strain that is not ideal for production strains. The method presented here complements this Hfr-based system by expanding the scope of high-throughput genome modifications beyond Hfr strains.
Through the application of high-throughput transductions, one can harness the advantages of both random and rational design for carbon-flux optimization. A collection of several hundred directed mutations can be produced in a single day, each with a redirected carbon flux around most of the nonessential metabolic pathways. An alternative approach would be to group mutations for related functions (e.g., amino acid synthesis) together for a single study. Mutants would be produced on a scale that screening, even by manual means, would not significantly slow the process. Any hits identified during screening could be immediately identified through careful sample tracking during transduction and library handling. This process can quickly provide several candidate knockouts to be recombined and tested further. If the donor strains originated from the Keio collection, as in this study, the antibiotic resistant cassettes can be easily excised to produce the next iteration of recipient hosts. This method permits a substantial parallel effort for creating and testing strains in an iterative engineering process. Iterative cycles of the high-throughput transduction method described here, in combination with screening, can be used to more rapidly produce optimized host strains.
The current focus of many synthetic biology efforts is directed toward alternative biofuels. 11,15 In E. coli, alternative metabolic products include acetate, lactate, and glycerol. With the extremely thin profit margins of the biofuels industry, little carbon can be spared for nonfuel products. The metabolic pathways to the alternative fermentation products are well known, but optimizing their production involves engineering additional metabolic pathways including ATP synthesis, redox balancing, and protein synthesis. Efforts are currently underway to apply this methodology to biofuels strain development.
ACKNOWLEDGMENTS
The authors would like to thank David Keating and Mary Tremaine for providing strain MT203. The list of Keio collection clones used in this study was suggested by Jennifer Reed and Joonhoon Kim. This work was funded by the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science DE-FC02–07ER64494).
Competing Interests Statement: The authors certify that they have no relevant financial interests in this article.
SUPPLEMENTARY DATA
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jala.2010.08.005.