
Abstract
The reproducibility of high-throughput cell-based assays is dependent on having a consistent source of cells for each experiment. Developing an understanding of the nature of cells growing in vitro and factors that influence their responsiveness to test compounds will contribute to the development of reproducible cell-based assays. Using good cell culture practices and establishing standard operating procedures (SOPs) for handling cultures can eliminate several potential contributors to variability in the responsiveness and performance of cells. The SOPs for handling each cell type must have clear and detailed instructions that can be understood and followed among different laboratories. The SOPs should include documenting the source of cells and authenticating their identity, both of which have become required to achieve peer acceptance of experimental data. Variability caused by biological issues such as phenotypic drift can be reduced by using standardized subculture procedures or using cryopreserved cells to set up experiments. Variability caused by inconsistent dispensing of cells per well and edge effects can be identified by measuring how many cells are present and whether they are alive or dead. Multiplex methods for real-time measurement of viable or dead cell number in each sample can be used for normalizing data and determining if proliferation or cytotoxicity has occurred during the experiment. Following good cell culture practices will go a long way toward executing reproducible cell-based assays. Resources will be included describing good cell culture practices, cell line authentication, and multiplex determination of cell number as an internal control.
Introduction
This article focuses on the use of established cell lines that are most often used for screening assays. The use of primary cells prepared from animals has many additional considerations, some of which are mentioned in the reference materials but will not be covered in this article.
Cells in culture are often used as an in vitro model system to predict in vivo responsiveness of humans. For many researchers, these in vitro model systems represent only a small component of an overall research program. Researchers may receive extensive training to become experts in measuring molecular events regulating signal transduction pathways at the single-cell level; however, training often overlooks basic requirements for maintaining cultured cells in a condition necessary to provide reproducible results. Many researchers assume cultures of continuous cell lines to be stable and consistent; however, subtle changes in the conditions used to culture cells can lead to variability in response to test compounds.
Many research scientists do not fully appreciate the importance of considering the true biological nature of cells growing in vitro as a model system. It is common to see anthropomorphic statements used to describe cultured cells (e.g., “Cells are happy in the presence of extracellular matrix components” or “Cells have a strategy to develop drug resistance”). Cells in culture are often described as being “happy” when they exhibit a high percent viability or when they exhibit a rapid rate of proliferation. When cells in stock culture are observed to grow rapidly, the proliferation rate may result in the population doubling every 16–24 h. Rapid growth is not normal for most cell types in vivo. One should consider if a culture showing a rapid proliferation rate is a good predictive model of the in vivo situation being studied, or is it just convenient for obtaining an abundance of cells to use for experiments.
Cells do not strategize about how to develop drug resistance. A population of cells exhibiting drug resistance is more likely the result of selective survival of a subpopulation rather than the result of a series of strategic decisions requiring cells in culture to demonstrate higher-level conscious thought. Use of such anthropomorphic descriptions contributes to the misunderstanding of the nature of cells cultured in vitro. A basic misunderstanding of the nature of cells grown in vitro can lead to poor assay design and a lack of reproducibility.
A major challenge with using cell-based assays is that cultured cells are inherently variable. A better understanding of parameters that contribute to variability in cell-based assays can lead to designing assays with improved reproducibility. Even within a clonal cell line, there is variation among individual members of the population. The selective survival of a subpopulation of cells under certain culture conditions can lead to a change in the phenotype of the population. Selection pressure during long-term culture results in the survival of the fittest and evolution of the population to an altered phenotype. New characteristics and the responsiveness of cells exposed to a particular modulator can change over time in culture as the passage number increases. That change is called phenotypic drift. The phenotypic changes may be the result of genetic mutations, chromosome instability, epigenetic changes, or an altered metabolism resulting from depletion of an energy source (e.g., glucose or glutamine) or lack of oxygen. Realizing the potential effects of slight variation in the culture conditions is an early step toward achieving improved reproducibility of cell-based assays.
To achieve reproducibility and to retain the parent cell line performance, it is critical that cells are passaged as few times as possible. A change in responsiveness can occur within a single passage if the stock cultures used to set up an assay are treated differently. It is important to treat stock cultures of cells the same way each time an experimental assay is set up. Cells have been documented to respond differently due to changes in culture conditions as simple as cell density 1 or contact inhibition. Figure 1 illustrates results of a caspase assay using HL-60 cells that were grown to three different densities in parent stock cultures and then used to prepare 96-well assay plates each containing the same number of cells per well. The results show that the cells from the lowest-density stock culture were more responsive to vinblastine treatment. Cells from the higher-density stock cultures showed less induction of caspase activity. These data demonstrate the importance of considering the effect of stock culture density on the responsiveness of cells to toxin.
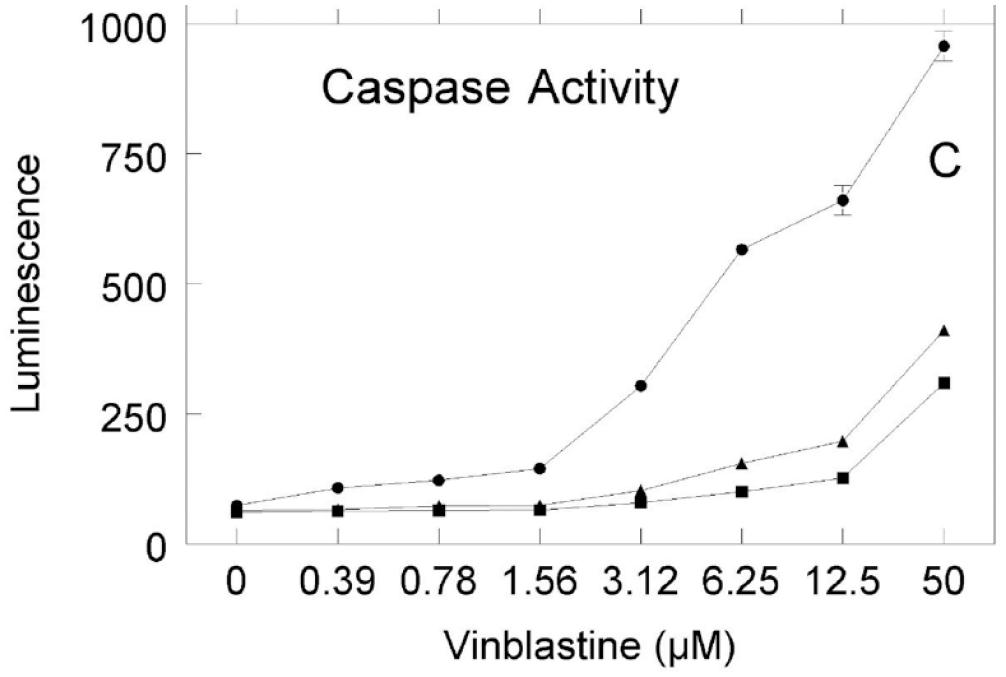
Individual stock cultures of HL-60 cells were grown to three different densities, approximately 4 × 105/mL (circles), 2 × 106/mL (triangles), and 3 × 106/mL (squares), and used to set up three separate 96-well assay plates, each containing 25,000 cells/well in 90 µL of RPMI 1640 medium supplemented with 10% fetal bovine serum. Cells were equilibrated to culture conditions for 15 min. Various concentrations of a vinblastine stock solution in DMSO were diluted into RPMI medium, and 10 µL was added to each well to achieve final concentrations of 0–50 µM. The plates were cultured for 4 h in the presence of vinblastine or vehicle control. Caspase-3 activity was detected using a luminogenic Caspase-Glo 3/7 Assay System (Promega Corporation, Madison, WI). Data points shown represent the mean ± SD (n = 3). Reprinted with permission from Assay and Drug Development Technologies, Mary Ann Liebert, Inc., publisher.
When results cannot be repeated during assay development, scientists frequently reach out to commercial vendors to ask, “What is wrong with the assay kit I bought?” Most variability in cell-based assays is a result of the cells and culture conditions rather than detection reagents. Variability among batches of assay reagents can be minimized by using rigorous quality control (QC) testing to ensure that relevant performance specifications are met. Setting QC specifications that predict assay performance can be accomplished for individual assay components by relying on quantifiable physical parameters such as concentration, chemical purity, or chemical composition.
However, designing predictive QC assays for a variable component such as cultured cells is much more challenging. To achieve reproducible results within and between laboratories, much care must be taken to ensure that consistent procedures are used for handling of the cell line used. Coecke et al. reported on good cell culture practices based on six operational principles. 2 The six operational principles of good cell culture practices can be summarized as (1) establishing a sufficient understanding of the in vitro system, (2) ensuring the quality of all materials to achieve reproducible results, (3) documenting the information necessary to enable the repeating of work, (4) ensuring that individuals and the environment are protected from any hazards, (5) complying with laws, regulations, and ethical principles, and (6) providing adequate training of individuals to achieve good cell culture practices. 2 The obvious way to improve reproducibility is to treat the cultures the same way each time they are handled. To accomplished that, a detailed standard operating procedure (SOP) for handling cells must be developed and rigorously followed.
Establishing an SOP
Often a starting point for establishing an SOP is to attempt to follow information in an existing in-house protocol or a Materials and Methods section of a scientific publication. The descriptions of protocols for culturing cells are often absent or poorly described in the Materials and Methods section. Repeating someone else’s work based on incomplete information can be difficult. There are several useful published resources providing guidance for items to include in the SOPs for handling cells in culture.3–9 Some of the key items to address in cell culture SOPs include culture medium, supplements and other additives, culture-ware, incubation conditions, and subculturing cells. 2
Culture Medium
The culture medium should be identified by a vendor part number and lot traced. For example, not all culture media reported as RPMI are the same. Formulations can vary in the amount of ingredients, such as phenol red, glucose, glutamine, and bicarbonate, or synthetic buffers. The bicarbonate level in the culture medium should be appropriately balanced with the percent CO2 used in the culture chamber. Minor medium formulation differences can be cytotoxic or have more subtle effects on the responsiveness or growth of cells and can lead to a selection pressure resulting in a different phenotype.
The culture medium should provide an adequate source of energy to support cell growth without severe stress between subculture passaging. The availability of glucose and glutamine that are commonly used by cells as an energy source can influence the metabolic pathways that cells rely on to generate energy to survive. Most culture media for mammalian cells contain approximately 5 mM glucose, mimicking the plasma concentration. Sometimes the energy source molecules can become depleted, resulting in stress and starvation of the culture. Monitoring the levels of key ingredients can provide insight into the metabolic status of the culture. Mass spectrometry or biochemical assays are available to measure the levels of energy source molecules and metabolites such as glucose, lactate, glutamine, and glutamate. 10 Care should be taken to not overcompensate for depletion of glucose by using what is commonly called “high-glucose medium,” which contains nonphysiological levels of glucose (~25 mM) that mimic a diabetic environment, which could interfere with the cell-based assay model system.
Eukaryotic culture medium is often supplemented with animal serum, most commonly 10% fetal bovine serum to support cell health and growth. Because animal-derived products are poorly defined and inconsistent, careful documentation is recommended to trace different lots as a potential cause for variability among assay results. Prevalidation of each lot of serum may be required to determine if it is fit for purpose; however, it is unlikely another laboratory halfway around the world will be able to obtain that same lot of serum a decade into the future. A chemically defined medium formulation is preferred.
Supplements
Biological supplements such as growth factors or cytokines should be clearly described and from a recombinant source if possible. Animal-derived materials are becoming far less common, but some examples still exist. Lot-to-lot variation in batches of animal-derived materials has been extensively documented and can lead to variability in results. The quantity of units of biological activity of such additives should be optimized, and the stability confirmed to ensure a uniform culture condition. It is recommended to report the specific activity of growth factors used based on international units of activity as defined by the National Institute of Biological Standards and Controls (NIBSC) when that information is available. 11 For example, from the NIBSC, “90/712 was established as the 1st International Standard (IS) for basic fibroblast growth factor (bFGF, FGF-2) in 1993. The bFGF used in this preparation is the human form of the molecule, synthesized in E. coli by recombinant DNA technology.” 12
Batch-to-batch variability of animal-derived scaffold ingredients such as collagens or Matrigel may lead to changes in differentiation patterns.13,14 Matrigel has also been shown to exhibit batch-to-batch variability in elastic modulus (stiffness), which can change assay results. Because Matrigel is a tumor extract, it is likely contaminated with DNA. Vital dyes to detect dead cells by binding of a fluorophore to DNA will result in high fluorescent background staining of cells cultured in Matrigel.
The routine use of antibiotics should be avoided. Their presence can mask low-level contamination and hide poor culture technique. 2 In addition, antibiotics can alter the responsiveness of cells to test compounds. In some cases, stock cultures of cells are maintained in antibiotic-free medium, but high-throughput screening (HTS) assay protocols include the addition of antibiotics to reduce contamination on the deck of robotic liquid handlers not able to fit in a clean environment. If antibiotics are used, their influence on cells treated with control compounds should be documented, and any hits identified during HTS should be rescreened in antibiotic-free follow-up assays to rule out specific molecular interactions.
Culture-ware
The containers used to culture cells should not be taken for granted. The surface treatments of culture-ware can vary among different suppliers, resulting in changes in the physiology of stock cultures or responsiveness of cells in multiwell assay plates. Culture-ware should be specified in the SOPs and not changed without prior validation. There may be a general supply of culture-ware for your laboratory that is made available by a laboratory manager, or in some cases, a department-wide stockroom will carry an inventory of standard culture-ware or multiwell assay plates that were purchased in bulk to save costs. Purchasing agents with good intentions to save money by switching vendors may not be aware of the cost of validating a change. This is just one example where a subtle change can cause variability among cell-based assay results. This type of variability can be reduced by paying attention to detail and specifying the source of relevant materials in the SOPs.
The descriptions of specialized culture-ware for implementing individual cell-based assays do not need to be included in the general SOPs for handling cells; however, because similar designs may be available from multiple vendors, a detailed accounting including the vendor part number must be described in the individual assay protocols. Some examples of specialized plates include multiwell plates with an ultra-low binding surface that aides in the formation of three-dimensional spheroids, assay plates with thin glass bottoms designed for microscopic imaging, and assay plates with a thin gas permeable layer made from silicone that enables gas exchange between the culture medium and the atmosphere.
Incubation Conditions
Conditions for culturing most human cell lines are adjusted to 37 °C, 5% CO2 in air atmosphere, with a pan of water in the bottom of the incubator to attempt to maintain a humid environment and avoid problems associated with evaporation of culture medium. Routine maintenance should include cleaning the incubator and measuring the temperature and CO2 levels independent of the electronic indicators on the instrument. Good general resources describing routine maintenance include “Best Practices for CO2 Incubator Maintenance” 15 and “Proper Care and Maintenance for a Cell Culture Incubator.” 16
Cell culture incubators are routinely shared among multiple scientists in a laboratory. Sharing resources often dictates compromise. Multiple users result in more frequent opening and closing of the incubator door, which can lead to temporary changes in environmental conditions that often go unnoticed. Those subtle changes in incubation conditions can lead to outer-well variability or edge effects during extended incubations and can result in variation in cell-based assays. Evaporative gradients also can result from plate lid design. In some cases, edge effects can be reduced by using gas-permeable plate sealers. An especially critical time is the initial incubation period following subculturing or dispensing of cells into assay plates. Temperature gradients from stacking plates or vibrations caused by fan motors within the incubator also can change the environment and result in edge effects. Some have found success by leaving the cells undisturbed on the lab bench at room temperature for 30 min to achieve uniform cell attachment. 17
Subculturing Cells
Subculturing of cell lines is required to maintain a continuous state of growth and avoid overcrowding, depletion of nutrients, and accumulation of harmful waste products. Subculturing cells is a critical step in the handling process that must be documented in detail in the SOPs so it can be reproduced. Examples of some of the items that must be described include the trypsinization protocol (including the type of trypsin or other dissociation solution used to remove anchorage-dependent cells), the time and temperature of trypsin exposure, whether there is a calcium-free phosphate-buffered saline rinse prior to trypsinization, the process to neutralize trypsin activity to minimize cell damage, the number of cells harvested from the stock culture flask to document the rate of ongoing growth, the cell number per square centimeter used to reseed new flasks, the volume of culture medium used (depth above the layer of cells) that will alter the diffusion of gases available to the cell layer, and the type of culture vessel used and whether or not the cap is vented. Minor variations in any of these steps can potentially lead to phenotypic drift. Those items are often not documented in sufficient detail to ensure replicating assay results. It is advisable for each lab to create and adopt the use of a template for routine recording of data at each passage. The example in this link can serve as a starting point. 18 Even if the protocols are well documented, individual interpretation of detailed written instructions can result in variation of the process used.
Contamination
Variability in eukaryotic cell culture can be caused by contamination with bacteria, fungi, or other cell types. Growth of contaminating unwanted prokaryote organisms or other eukaryotic cells can go undetected and lead to erroneous nonreproducible results. Bacteria and yeast are probably the most common and obvious forms of contamination of cell culture. Overgrowth of these organisms often causes a turbid appearance of culture medium and can be accompanied by a distinctive odor. Use of antibiotics such as a combination of penicillin and streptomycin can help control contamination when preparing primary cultures from furry animals. Although the use of antibiotics has become routine for many cell culture protocols, if good cell culture practices are followed, antibiotics are generally not necessary. Although antibiotics can depress or eliminate bacteria, they can affect the biological responsiveness of eukaryotic cells as well as hide poor culture technique.2,3
Mycoplasma
Mycoplasma are small parasitic bacteria without a cell wall that are resistant to many common antibiotics such as penicillin and streptomycin. Because mycoplasma are challenging to observe during routine culture of animal cells, contamination often goes unnoticed. Although there is general awareness that problems associated with mycoplasma contamination exist, recent studies have documented that it remains a significant problem that is known to affect assay results and waste valuable resources.19,20 Routine testing every 1–2 months is recommended to identify mycoplasma contamination. Testing for mycoplasma can be done using culture techniques with a medium optimized for mycoplasma growth, DNA staining to view mycoplasma nuclei using fluorescent microscopy, or detection of mycoplasma-specific DNA using a PCR assay that is capable of detecting most species. Young et al. 21 provide a detailed overview of the detection of mycoplasma in cell culture. Commercial kits are available or testing services are available from places such as the American Type Culture Collection (ATCC). Combining more than one assay method can be useful to confirm results. If a culture is identified to be contaminated with mycoplasma, the best approach is to sterilize and destroy it. If the culture is valuable and no backup is available, it may be able to be salvaged by treatment with mycoplasma-selective antibiotics, but the harsh treatment itself could cause phenotypic drift in the cell line.
Authentication of Cell Lines
One of the absolute requirements to achieve reproducibility of cell-based assays is to have the same type of cells available for each experiment. There is a growing body of peer-reviewed literature documenting the instances and problems associated with misidentified or contaminated cell lines.22–24 The use of misidentified or contaminated cell lines occurs in a significant number of laboratories and leads to invalidation of data, retraction of publications, and wasted time, money, and effort. Various literature reports indicate that approximately one in five cell lines tested are contaminated or misidentified, but in some cases, the contamination rate is much higher.25,26 A recent study of cell lines from institutes in China revealed that 46.0% (128/278) were cross-contaminated or misidentified. 27 Among the easiest practices to implement to help avoid cross-contamination include working with one cell line at a time in the laminar flow hood and using separate containers of culture medium for each cell line.
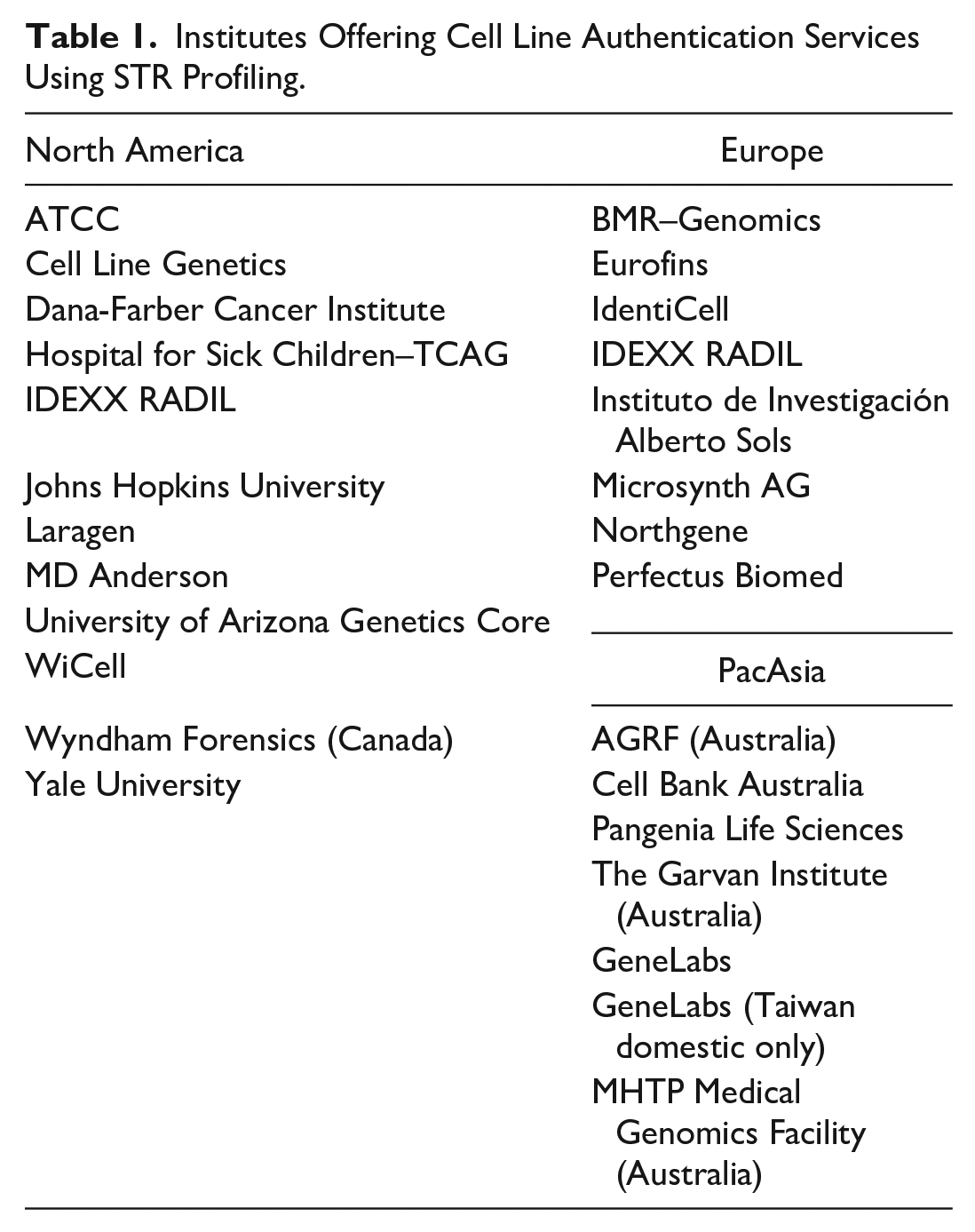
Cell line authentication can be accomplished easily with routine testing. The International Cell Line Authentication Committee (ICLAC) has prepared a Guide to Human Cell Line Authentication 28 that includes recommendations to perform authentication based on using short tandem repeat (STR) genotyping. That STR genotyping method to test for authenticity of cell lines is widely available and relatively inexpensive. Table 1 shows a growing list of service providers and core facilities offering cell authentication services.
Institutes Offering Cell Line Authentication Services Using STR Profiling.
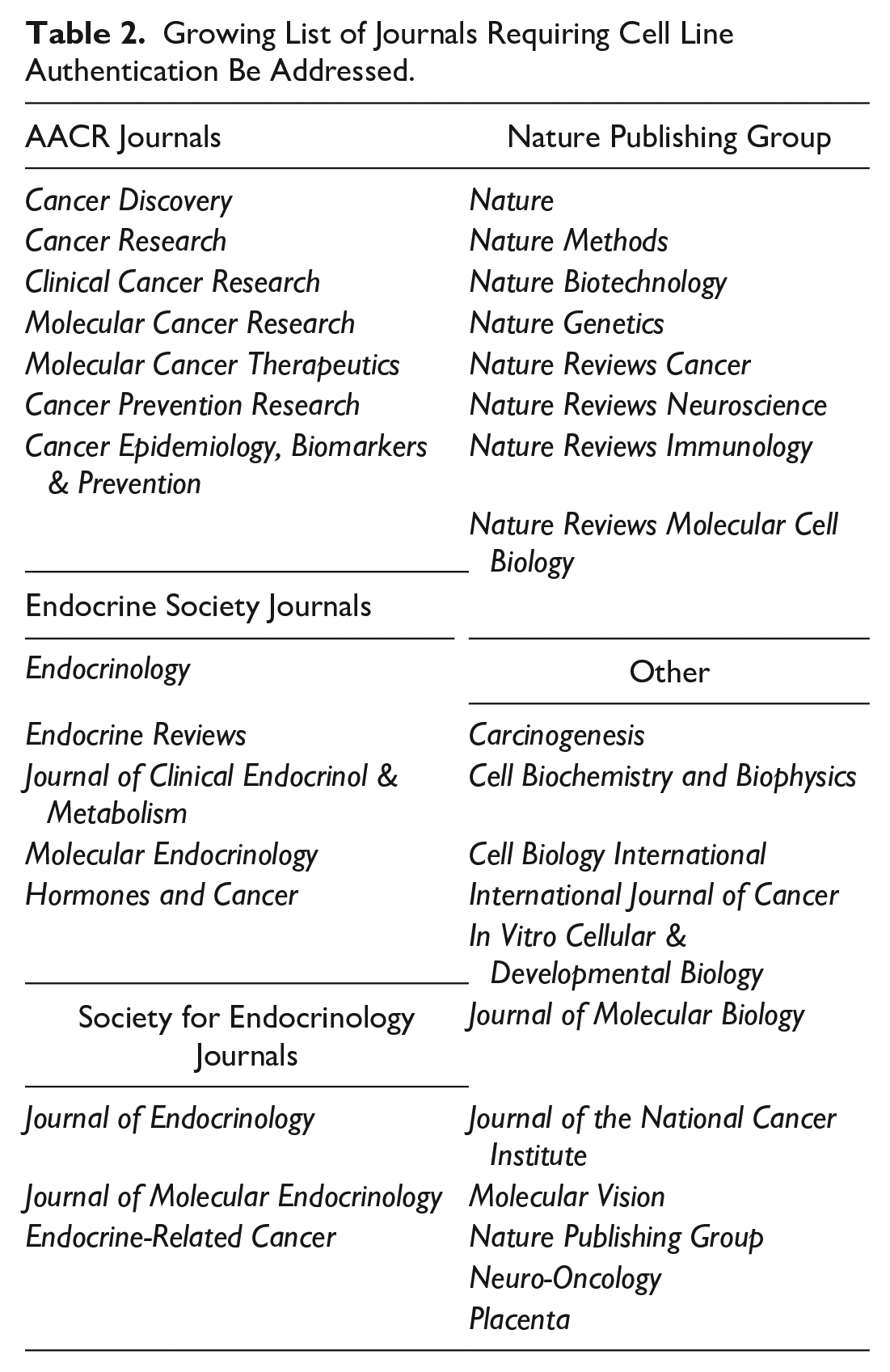
Providing evidence that the issue of cell line authentication has been addressed is now required by a long and growing list of peer-reviewed journals ( Table 2 ) that require authors to document cell line authentication as part of the process of submitting a manuscript for publication. However, there is an unmet need for journal editors to provide training and guidance for peer reviewers to identify unacceptable authentication practices.
Growing List of Journals Requiring Cell Line Authentication Be Addressed.
The National Institutes of Health (NIH) has posted notices addressing reproducibility that includes urging the documentation of authenticity of cell lines and key biological reagents including antibodies (e.g., “Enhancing Reproducibility through Rigor and Transparency”). 29 The NIH has also provided guidelines for addressing cell line authentication in new grant applications. 30
If you do not already routinely test your cell lines for contamination, a good place to start is to check the cell line name against the surprisingly large database of misidentified cell lines. 31 Regardless of whether the cell lines you are using are on the list, it is recommended to use the STR genotyping testing method on a routine basis to confirm they are authentic and have not become contaminated.
The authentication process is simple. Details of the process vary depending on the testing service, but in general, a sample of cells (spotted on a provided piece of filter paper) or extracted DNA is sent to an STR profile testing service, the STR profile is compared with a reference (usually provided by the testing service), and a report is generated.
STR testing is recommended when receiving a cell line from an unreliable source, after 10 passages, and when preparing a cell bank. Documentation that a working bank is authentic eliminates the need for repeat testing if the “thaw-and-use” of cryovials approach is used for initiating cell-based assays. The cost is minimal, and the risk of contamination justifies investing in incorporating authentication as a routine procedure in the SOPs for handling cells in your laboratory. Many of these recommendations are applicable to established cell lines but do not apply to primary cells obtained directly from living tissue. Although reagents for authenticating rodent and other cell lines are becoming available, most reagents to date have been developed for human cell lines. A collection of resources describing the steps involved in cell line authentication is readily available online. 32
Establish a QC Performance Assay to Determine Reproducibility
Many times, the methods used to test and quantify the responsiveness of cells to a known positive control are absent in the Materials and Methods section of peer-reviewed papers. Potency values may be reported relative to a control compound, but changes over time or with increasing cell line passage number are seldom reported.
Measuring the responsiveness of different batches of cells treated with a range of concentrations of a validated control compound with a known activity is one way to monitor the variability of assay results. Evaluation of EC50 data recorded on different days from different users and different batches of cryopreserved cells will provide insight into the reproducibility of a cell-based assay. The minimum significant ratio (MSR) is a statistical parameter that can be used to describe the reproducibility of potency estimates from in vitro concentration–response assays. The MSR is described by Haas et al., in a chapter in the Assay Guidance Manual. 33 The MSR provides a quantitative expression of the reproducibility of potency values from cell-based assays. It is a valuable statistical tool for assessing the reliability of cell-based assays over time.
In addition to reporting potency, documenting microscopic observations of the morphology of cells (with photographs rather than written descriptions, if possible) can provide evidence to explain variability among assay results. Photographs can be included in the routine passage data template described above. Subtle deviations in morphology may be an indication of phenotypic drift.
Cryopreserved Cells
One approach to reduce variability associated with phenotypic drift in stock cultures is to use cells within a narrow range of passage number for each experiment. 34 For example, vials from a cryopreserved bank can be thawed on a routine schedule and used for approximately 10 passages over a 1-month period before discarding and starting over with a fresh vial from the bank. The useful upper limit of passage number will differ with cell type. Considerable investment may be required to establish the passage number when a substantial phenotypic drift occurs. Alternatively, if you have validated a procedure to demonstrate that cryopreserved cells are fit for purpose for directly seeding into an assay plate, then a working bank containing a large number of vials with the identical passage number can serve as a source to set up assays and replace the need for continuous culturing of that cell line.
Upon initial receipt of an authenticated and contamination-free cell line from a reputable source, it is advisable to create a master cell bank of cryopreserved vials to ensure long-term future access to a consistent source of cells. Vials from the master bank can be used to create consistent batches (working banks) of cryopreserved vials. Some of the critical steps that must be validated for creating hundreds of vials of frozen assay-ready cells as a working bank include large-scale expansion, transfer to a cryoprotectant medium (usually containing DMSO), automated dispensing, controlled-rate freezing (typically around 1° per minute), and transfer to vapor-phase storage above liquid nitrogen. The formulation of the cryoprotectant, the number of cells per vial, and the freezing and thawing protocols all need to be optimized and validated to ensure each frozen vial can provide consistent results. General guidelines for cryopreservation and storage of cells are available from the ATCC website. 35
There are examples of vendors that provide frozen assay-ready cells as a custom product (acCELLerate GmbH, Hamburg, Germany) or as part of convenient assay kits (Promega Corporation, Fitchburg, WI). Some commercial vendors have products that contain frozen cells as a component that are “thaw and use” and throw-away at the end of the day, thus not requiring stock culture. 36
The main advantages of using assay-ready frozen cells include long-term access to a consistent resource, more flexible scheduling, and cost savings. Since properly stored cryopreserved vials of cells are stable for several years, a working bank of cells can contribute to providing consistent results throughout the duration of a research program. An assay can be initiated at any time by thawing a vial of cells (e.g., when new compounds arrive for testing) since the process for maintaining a stock culture in an acceptable condition is uncoupled from setting up an assay. Using cells directly from a frozen bank also has the advantage of saving cost because the time spent to maintain cell lines in culture, the quantity of culture reagents and disposables (pipets, T-flasks, etc.), and use of cell culture facilities are all reduced.
The deleterious effect from exposure to cryoprotectant needs to be considered when creating large working banks. Long-term exposure to cryoprotectant medium may become cytotoxic. If a long time is required to dispense hundreds of vials in a working bank, it may be necessary to verify percent viability from the beginning, middle, and end of a dispensing run. The number of vials that can be processed simultaneously for freezing needs to be considered and will likely limit the batch size.
Thawing is a critical step in the workflow when cells are fragile. A standard procedure should be followed each time, typically using gentle swirling of the cryovial in a 37 °C water bath to completely thaw the medium, followed by wiping the vial with 70% ethanol to reduce the possibility of contamination. If the established method removes the cryoprotectant to avoid cytotoxic effects or interference with subsequent assay performance, care should be taken to not damage the fragile cells during centrifugation and addition of fresh culture medium. Once the protocol for freezing and using a working bank is validated, each batch of cells can be QC performance tested and treated as a reagent for future use.
Cell Number Controls
There are several cell number-related controls that are of value when performing cell-based assays, and there are many options for surrogate markers available for obtaining those data.37,38 Data from measuring the number of live cells, the number of dead cells, or the total number of cells as controls can provide insight into the precision and reproducibility of the methods used as well as the overall value of the assay. Use of appropriate cell number controls for identifying potential problem areas during assay development can lead to improved reproducibility.
One of the recommended controls for any cell-based assay is to determine if there is a consistent number of viable cells dispensed into each assay well. That value will help identify if there are errors introduced by the dispensing process. For example, cells that tend to clump into aggregates can lead to variability among replicate wells, especially if small volumes of a heterogeneous suspension are being dispensed into high-density 384- or 1536-well assay plates. It is also important to know if handling the cells during the assay protocol is having an adverse effect on cell viability. If dispensing the cells is reducing viability, it can influence the physiological relevance of the assay model system.
The health and number of cells in individual assay wells can change over the course of the experiment, especially if incubation conditions are suboptimal or if the duration of incubation lasts for several days. It is important to know how the viable cell number in no-treatment (vehicle-only) control wells changes during the experiment. That information can help identify edge effects that occur across multiwell assay plates during the incubation period. There can be rapid cell proliferation or significant cell death that occurs in untreated controls during the assay incubation period. Those changes in viable cell number are often undetected or overlooked when using endpoint assays that record a single measurement at the end of the experiment.
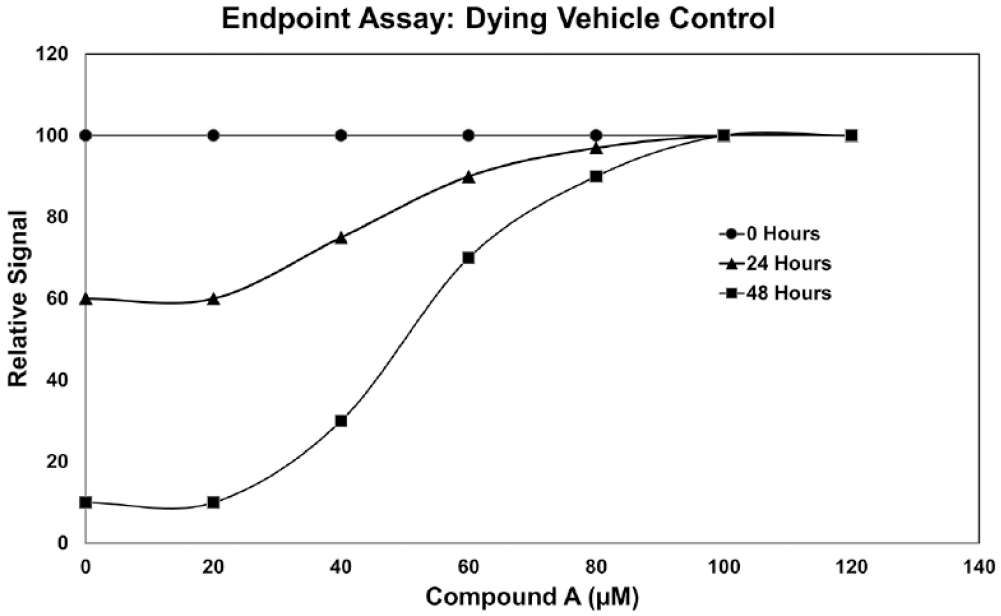
Hypothetical endpoint cell viability assay data representing different incubation times (0, 24, and 48 h) with compound A. Independent analysis of the sigmoid-shaped curve representing 48 h of incubation could be misinterpreted to suggest that the higher concentrations of compound A are stimulating cell proliferation. Considering data collected from all three incubation periods suggests that the untreated control cells (0 µM compound A) are dying over time and there is no change at the highest concentrations of compound A, consistent with compound A being required for survival, but not stimulating cell proliferation.
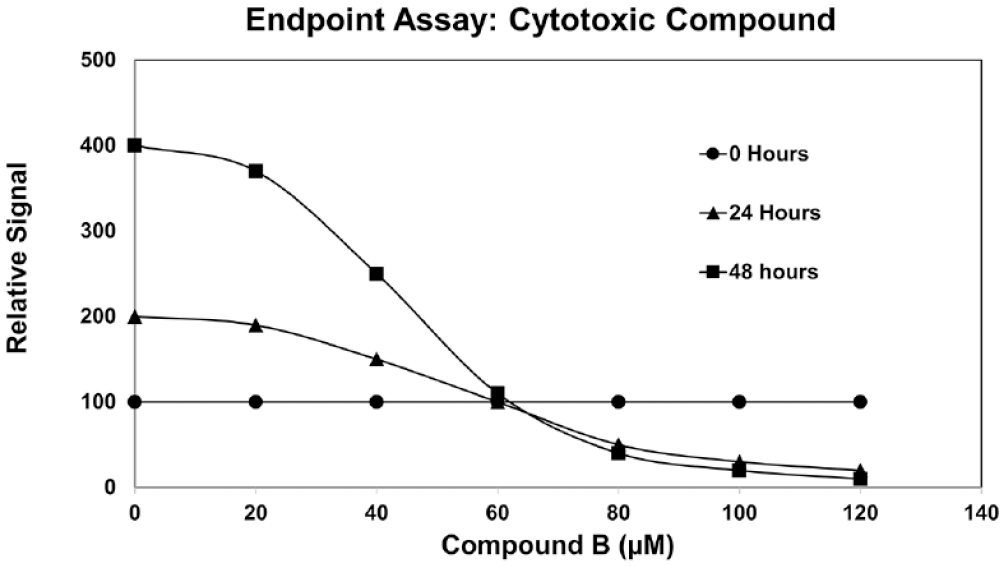
Hypothetical endpoint cell viability assay data representing different incubation times (0, 24, and 48 h) with compound B. Independent analysis of the sigmoid-shaped curve representing 48 h of incubation suggests that compound B is cytotoxic. Data from the untreated control samples (0 µM compound B) at 0, 24, and 48 h suggest that those cells continued to grow, resulting in a doubling of the relative signal each day. Data from incubation with 80–120 µM compound B for 24 and 48 h show a lower relative signal compared with that for 0 h, consistent with compound B being cytotoxic. Because endpoint viability assays typically kill the cells, a separate assay plate is required for each time point.
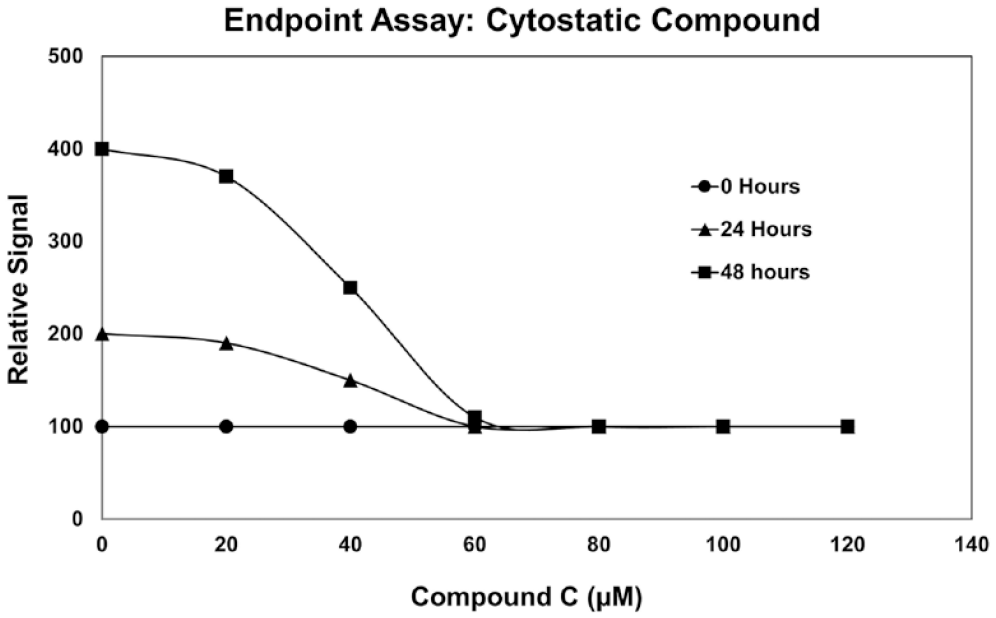
Hypothetical endpoint cell viability assay data representing different incubation times (0, 24, and 48 h) with compound C. If the 48 h data were all that were available, analysis of the sigmoid-shaped curve could be misinterpreted to suggest that compound C is cytotoxic. Similar to compound B shown in Figure 3, data depicting the untreated control samples (0 µM compound B) at 0, 24, and 48 h suggest that those cells continued to grow, resulting in a doubling of the relative signal each day. However, the relative signals are the same for all compound C concentrations from 60 to 120 µM at 0, 24, and 48 h of incubation, suggesting that compound C is cytostatic.
Figure 3 shows hypothetical viability assay data to illustrate a further limitation of using an endpoint assay at a single time point. The 48 h data (squares) show a sigmoid curve with the higher concentrations of compound B resulting in a lower signal; however, the vehicle control (0 µM compound B) shows a doubling of the signal every 24 h, suggesting that the cell number is increasing in control wells. At higher concentrations of compound B, the decrease in signal with increasing time of incubation suggests a cytotoxic effect.
In Figure 4 , the 48 h endpoint viability assay data show a typical sigmoid curve, suggesting that compound C is cytotoxic. However, by including additional data from 0 and 24 h of incubations, it becomes apparent that the relative signal for all concentrations above 60 µM is the same as that for the 0 h data. Those data suggest that compound C is cytostatic (i.e., cell proliferation does not occur), but it does not kill the cells. The assays and additional information from 0 and 24 h require separate samples when using an endpoint assay approach.
Live cell kinetic assays provide flexibility in designing cell number controls. In contrast to the endpoint assays in
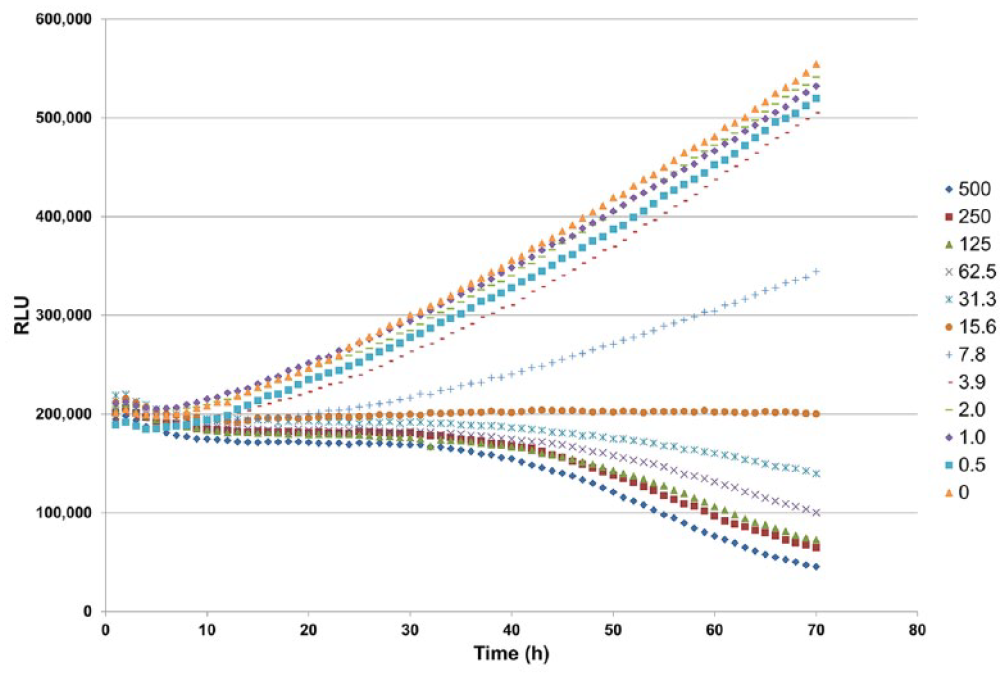
Example of a live cell kinetic assay to measure viable cell number using the RealTime-Glo MT Cell Viability Assay (Promega Corporation). A549 cells (500/well) were plated in 40 µL of medium containing 2× RealTime-Glo reagents. A thapsigargin titration was prepared in medium at 2× concentrations and added to the plate at an equal volume. The final concentrations of thapsigargin ranged from 0.5 to 500 nM. The vehicle control was 0.1% DMSO. Luminescence was monitored every hour for 72 h using a Tecan Infinite 200 Multimode Reader with Gas Control Module (37 °C and 5% CO2). The vehicle-only controls and the lowest concentrations of thapsigargin continued to grow over the 3-day period and displayed increasing luminescent signal over time. The highest concentrations of toxin resulted in a decrease in luminescent signal over time, indicating cell death. Adapted with permission from Riss TL, Moravec RA, Niles AL, et al. Cell Viability Assays. Assay Guidance Manual [Internet]; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, 2013. Published in 2013 under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported license (CC BY-NC-SA 3.0).
It is also of value to know the number and rate of accumulation of dead cells present in assay wells. The accumulation of a significant number of dead cells in the untreated (vehicle-only) control wells may indicate a problem with the culture environment and a poor representation of physiological relevance of the assay. The number of dead cells can be estimated by using a vital dye that does not penetrate viable cells but can enter and stain dead cells with a compromised cell membrane. There are numerous fluorogenic vital dyes available.38,39 There are also numerous assay methods to measure apoptosis or oxidative stress that can provide insight into mechanisms leading to cell death.40,41
Figure 6
shows an example of a kinetic assay to detect dead cells. This real-time assay is based on using a fluorogenic DNA binding dye that is not permeable to live cells but can penetrate dead cells and stain DNA. Because the dye is not permeable to live cells, it is nontoxic and can remain in the culture medium for days, enabling the recording of fluorescence repeatedly from the same sample to provide a real-time live cell kinetic assay. In contrast to the endpoint assays that typically lyse cells and provide data from a snapshot in time when the assay was performed, the kinetic assay methods shown in
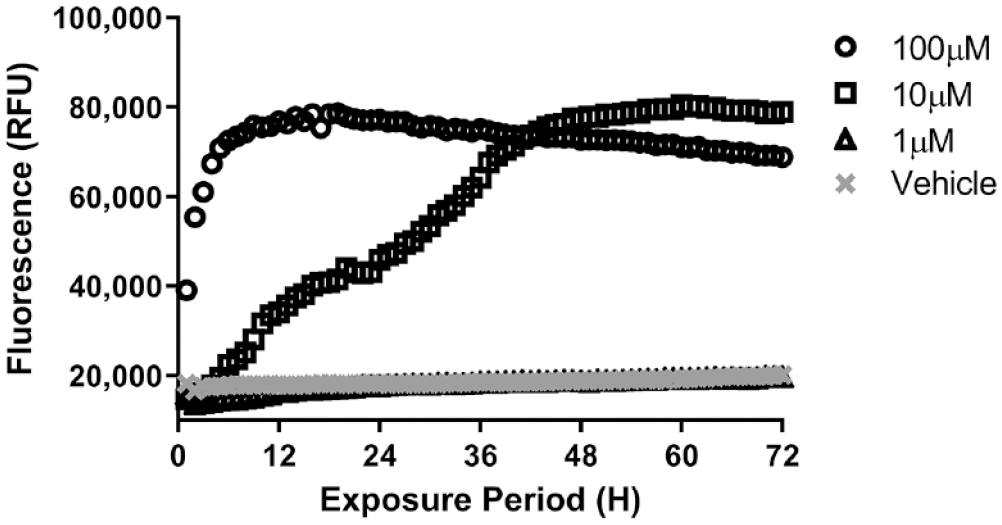
Example of a kinetic assay to measure dead cell number repeatedly from the same samples for 72 h using the fluorescent CellTox Green Cytotoxicity Assay (Promega Corporation). HepG2 cells were treated with various doses of terfenadine. CellTox Green Dye was added at time of terfenadine dosing. Fluorescence was measured every hour for 3 days using a plate reader with an environmental chamber to control temperature at 37 °C and CO2 at 5%. Increasing fluorescence indicates an increase in the number of dead cells in the culture well. Adapted with permission from Riss TL, Niles AL, Moravec RA, et al. Cytotoxicity Assays: In Vitro Methods to Measure Dead Cells. Assay Guidance Manual [Internet]; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, 2013. Published in 2013 under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported license (CC BY-NC-SA 3.0).
Live cell kinetic assays not only provide cell number controls but also enable more flexibility for multiplexing with other assays. For example, the Incucyte SX5 Live-Cell Analysis System is essentially an automated microscope built into an instrument that can fit inside an incubator and record images repeatedly from the same location using up to five fluorescent channels to measure various surrogate markers. Separate staining of live and dead cells with different fluorophores is possible. Another example providing kinetic cell viability and cytotoxicity data is the multiplex combination of the RealTime-Glo MT Cell Viability Assay ( Fig. 5 ) and the CellTox Green Cytotoxicity Assay ( Fig. 6 ).37,38,42 The RealTime-Glo assay emits a constant luminescent signal that is proportional to the number of viable cells, and the CellTox Green assay contains a vital fluorogenic dye that stains only the dead cells with a compromised membrane. The reagents are added once, and then the signals can be recorded repeatedly over a period of days using a multimode plate reader. The kinetic assay methods provide an advantage by enabling repeated multiplexed measurement of live or dead cells simultaneously in the same sample in real time using reagents that do not kill the cells.37,38
Live cell kinetic assay methods provide insight into the rate of cell proliferation or death during the incubation period of the experiment. The results from kinetic assays can help eliminate the type of misinterpretations described in examples shown in
Each assay approach has its own set of advantages and limitations. It is difficult to identify artifacts and assay chemistry interference using only one approach to measure a surrogate marker. Using an orthogonal approach to combine more than one assay chemistry and measure separate markers of the same parameter (such as live and dead cells) can help confirm results and identify artifacts.38,43 Figure 7 illustrates an example of multiplexing orthogonal assays measuring viable cells and dead cells from the same sample wells. At high concentrations of toxin, the increased fluorescent staining of dead cells by the vital dye correlates with a decrease in ATP content from the luminescent assay, indicating a lower number of viable cells. Many such examples of multiplexing orthogonal assays exist and can be implemented if the assay chemistries are compatible. Multiplexing orthogonal assays can be a valuable method for confirming a cell number control. The data from a second assay run on the same sample can be used to help identify artifacts and confirm the performance of a primary assay.
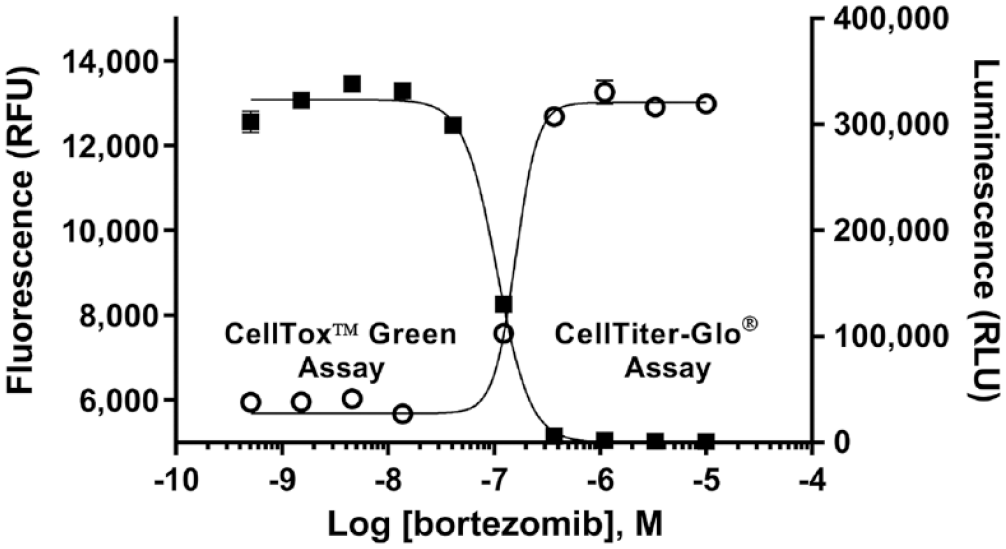
Example of sequential multiplexing of two orthogonal assays to measure the cytotoxicity of bortezomib, a known proteasome inhibitor chosen as a model toxin. The CellTox Green reagent (fluorogenic DNA binding dye) was applied to bortezomib-treated K562 cells after 48 h of incubation. Fluorescence was recorded first (open circles) to indicate the number of dead cells, and then the CellTiter-Glo Reagent (Promega Corporation) was added (which lyses the cells) and luminescence associated with the amount of ATP as a marker of viable cell number was measured (filled squares). At low bortezomib concentrations, the DNA staining of dead cells is low and the ATP levels are high. At high bortezomib concentrations, the ATP values are low and the DNA staining (indicating dead cells) is high. The inverse relationship of the two data sets can be used as a control to help confirm assay performance. Adapted with permission from Riss, TL, Niles AL, Moravec RA, et al. Cytotoxicity Assays: In Vitro Methods to Measure Dead Cells. Assay Guidance Manual [Internet]; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, 2013. Published in 2013 under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported license (CC BY-NC-SA 3.0).
Summary
Cell-based assays generally have a higher degree of variability than biochemical-based methods. The main reason is that cells are inherently variable in their response to a test substance, even within a clonal population. Variability in cell-based assays can result from cells being contaminated with bacteria or other eukaryotic cells, phenotypic drift that can change responsiveness compared with early passage cultures, changes in the procedures to handle cultures due to a lack of specific detailed instructions to follow, and variability in the number of viable cells per assay well. Variation can be reduced by creating and rigorously following an SOP describing detailed handling instructions that include routine testing for contamination and limiting the range of passage numbers of cells used to set up assays. Understanding that phenotypic drift occurs in cell lines and it is something that can be controlled by limiting passage number supports the practice of frequent replenishing of stock cultures from cryopreserved banks of cells that have been performance tested to ensure a consistent response. The number of cells per well can be verified using live cell kinetic or orthogonal endpoint assay methods to determine if proliferation or cytotoxicity has occurred during the assay and to normalize data to cell number. Taken together, these methods to treat cells as reagents can minimize variation among data from cell-based assays.
Footnotes
Acknowledgements
The authors thank members of Promega’s Cell Health Assay Development and Sample Analysis groups within Research and Development, members of the Strategic Portfolio Management for Cell Health and Functional Analysis, operational teams for the many years of effort to develop reproducible cell health assays, and Kristine Pitzen for assistance with formatting the manuscript. No funding was received outside of Promega’s normal departmental budgeting process.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors are employees of Promega Corporation, and their research and authorship of this article was completed within the scope of their employment with Promega.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.