
Abstract
A wide range of complex in vitro models (CIVMs) are being developed for scientific research and preclinical drug efficacy and safety testing. The hope is that these CIVMs will mimic human physiology and pathology and predict clinical responses more accurately than the current cellular models. The integration of these CIVMs into the drug discovery and development pipeline requires rigorous scientific validation, including cellular, morphological, and functional characterization; benchmarking of clinical biomarkers; and operationalization as robust and reproducible screening platforms. It will be critical to establish the degree of physiological complexity that is needed in each CIVM to accurately reproduce native-like homeostasis and disease phenotypes, as well as clinical pharmacological responses. Choosing which CIVM to use at each stage of the drug discovery and development pipeline will be driven by a fit-for-purpose approach, based on the specific disease pathomechanism to model and screening throughput needed. Among the different CIVMs, biofabricated tissue equivalents are emerging as robust and versatile cellular assay platforms. Biofabrication technologies, including bioprinting approaches with hydrogels and biomaterials, have enabled the production of tissues with a range of physiological complexity and controlled spatial arrangements in multiwell plate platforms, which make them amenable for medium-throughput screening. However, operationalization of such 3D biofabricated models using existing automation screening platforms comes with a unique set of challenges. These challenges will be discussed in this perspective, including examples and thoughts coming from a laboratory dedicated to designing and developing assays for automated screening.
Keywords
Introduction
Despite significant scientific and technological advances throughout the last few decades, the attrition rate during drug development has remained around 90%, 1 mainly due to lack of efficacy or unexpected toxicity in humans. 2 Modern molecular and cellular biology has allowed the development of cellular and animal models with technical robustness. However, such models are physiologically too simplistic and do not faithfully reproduce the complexity of human physiology under homeostatic and disease conditions, including models that reflect the diversity of the patient population. The lack of mimicry of diverse human physiology makes the current in vitro and in vivo models not predictive of drugs’ clinical efficacy and safety in humans. Capturing the complex cellular pathways in cellular backgrounds that are tissue and disease relevant, together with parenchymal and systemic cell–cell interactions, and cellular–microenvironment interactions, including mechanical factors in tissues, is therefore a critical factor to consider when developing cellular models that faithfully mimic native tissues and reproduce clinically relevant drug efficacy and toxicity in an in vitro setting.3,4
Over the past decades, complex in vitro models (CVIMs) have been described for many tissues and organs of the human body, produced by utilizing a wide variety of technologies. These CIVMs provide a platform of assays with increased physiological relevance for scientific research and drug efficacy and safety assessment to bridge the predictability gap between current in vitro, in vivo assays and clinical trials. 5 For these CIVMs to deliver on their promise of improved clinical predictability, their scientific validity, defined as the models’ physiological and clinical relevance, must be thoroughly established.6–8 Beyond their scientific validation and clinical benchmarking, these CIVMs must be operationalized as robust assay platforms for use in research and drug screening. By doing so, the hope is that, ultimately, the need for animal testing will be reduced and eventually eliminated, while producing data that will lower the attrition rate in the overall drug development process, to save costs and expand treatment options for patients. 9
To achieve such a vision for CIVMs, there is a general agreement in the scientific community that guidelines for their validation must be clearly defined. 8 These guidelines will ensure that any CIVMs that are produced will be useful as tools for basic science research and drug screening. There is little consensus within the field as to how one can systematically define the models’ physiological relevance and clinical predictability. In addition to assessing what native-like molecular and cellular features of a tissue in homeostatic conditions are captured in a CIVM, there needs to be clear guidelines as to what clinical biomarkers are needed to be demonstrated experimentally for each disease CIVM. 7 Omics technologies such as single-cell transcriptomics, as well as microscopy and histological analysis, should be used to establish how faithfully the molecular and cellular complexity of these CIVMs mimics native human tissues.
There are many critical questions to address regarding the clinical relevance of these CIVMs, including, but not limited to, the following: How much better are 3D tissue models than 2D models in predicting drug outcomes in the clinic? How much cellular complexity is required to effectively model diseases? Can one reproduce changes in the levels of relevant clinical biomarkers that can then be used to discriminate disease versus normal tissues? Lastly, how much biological variability will be observed when using cells from different donors, and can this variation be used to capture patient heterogenicity and predict patient-specific responses to therapeutics? Experimental designs to answer these questions will be paramount in establishing the use of CIVMs in research and drug discovery and development disciplines.
Overview of CIVMs: Spheroids, Organoids, Bioengineered Tissues, and Organ-on-Chip Systems
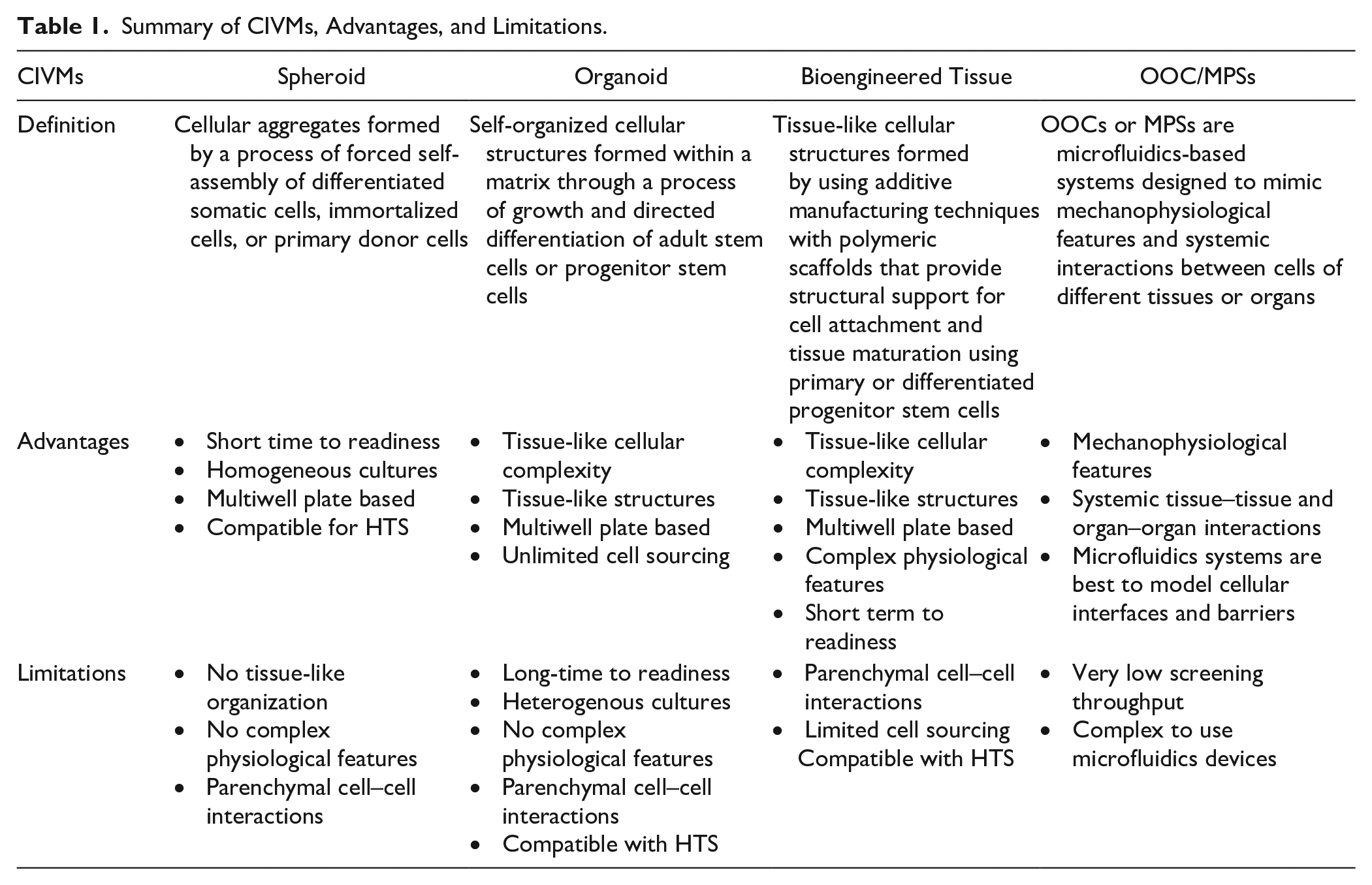
There is now a wide range of CIVMs with various levels of physiological complexity and throughput that can be applied toward preclinical research and drug testing ( Fig. 1 ). Spheroids are cellular aggregates produced in ultra-low-attachment (ULA) round-bottom plates or by other techniques, such as hanging drop and/or aggregating hydrogels like Matrigel. In most cases, spheroids are formed with differentiated somatic cells, including immortalized cell lines, but also with progenitor stem cell-derived cells or primary donor cells. Spheroid formation relies on spontaneous aggregation, and in most cases, these spheroids do not form a native-like tissue architecture.10,11 Spheroids can be produced in a homogenous size and cellular composition, and such reproducibility makes them very amenable as an assay platform for drug screening. Organoids are self-organized cellular structures formed within a matrix through a process of growth and directed differentiation of adult stem cells or progenitor stem cells. 12 For organoids, tissue-relevant cell types and native tissue-like spatial organization are formed after the extended period of differentiation, unlike spheroids that rely on spontaneous aggregation and short maturation.11,13 Although these organoids include physiologically relevant cellular complexity and a tissue-like structure, it is difficult to introduce other important tissue features like vascularization and cell types of different germ layers. 14 In addition, current protocols tend to produce organoids that are heterogenous in size, cellular composition, and tissue-like features, which make them more difficult to use as a robust assay platform for drug testing.
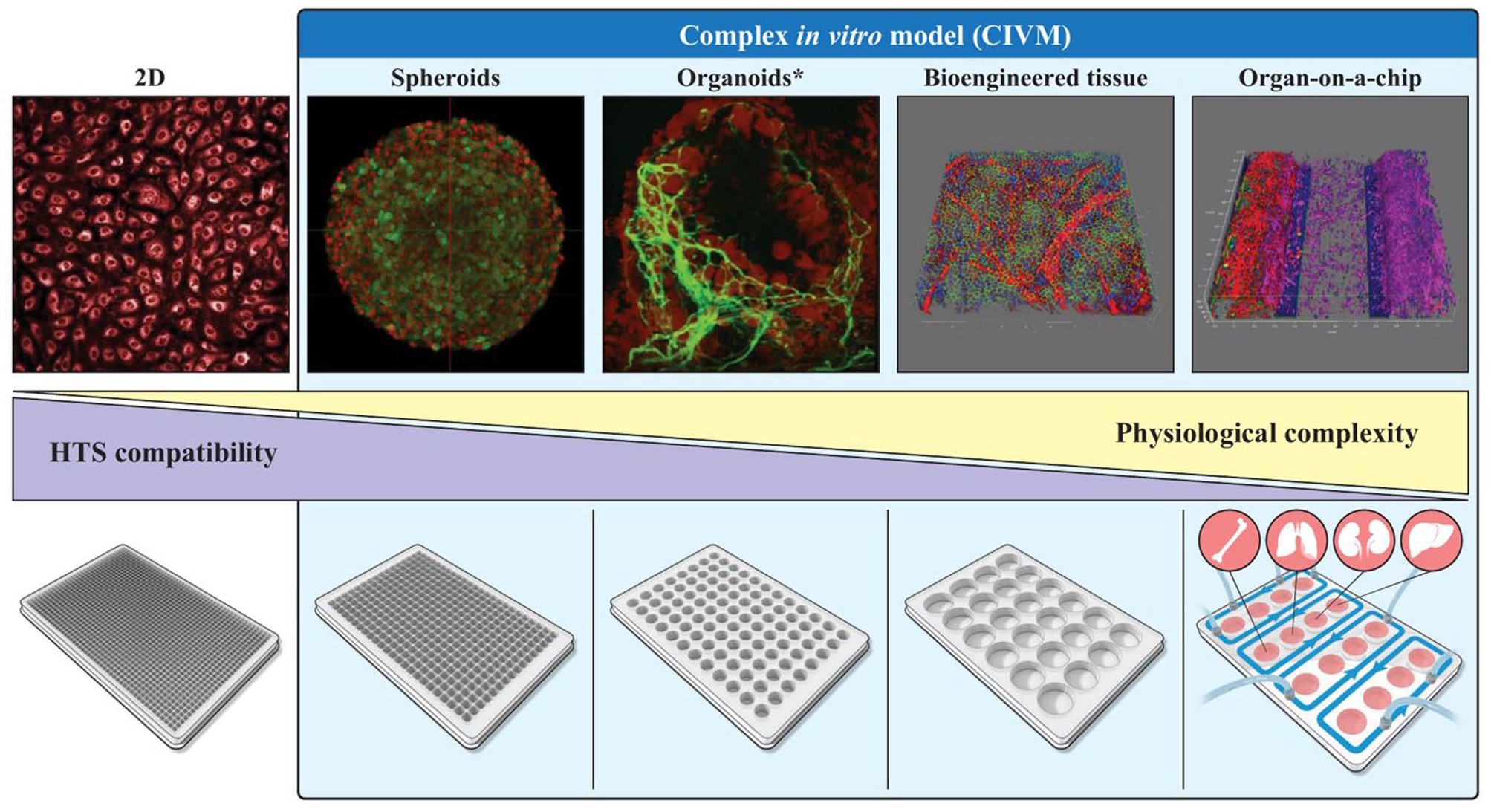
Balancing HTS compatibility and physiological complexity of CIVMs. 2D, primary human brain endothelial cells; spheroid, designer brain spheroid; organoid, inner ear organoid; bioengineered tissue, ocular tissue; organ-on-chip (OOC), blood–brain barrier. The organoid image is a screen capture of the S1 movie. 70 Figure generated by NIH Medical Arts.
Biofabricated cellular models are produced using various tissue engineering techniques, including 3D bioprinting, hydrogels, and biomaterials.3,15,16 Unlike spheroids and organoids in which cell aggregation provides its own scaffold-free 3D support, bioengineering enables the assembly of tissue-like structures using additive manufacturing techniques with polymeric scaffolds that provide structural support for cell attachment and tissue maturation. Tissue engineering enables the production of tissues with tailored cell composition, and 3D bioprinting can be further used for spatially controlled cell deposition that mimics native-like tissue architecture. The spatial control and modular features allow for the introduction of complex physiological features into these biofabricated CIVMs, including vascularization or innervation, as well as the introduction of normal and disease cells to study the effects of disease and nondisease cell types in the development of a disease phenotype. 17 The biomaterials used range from natural biopolymers such as collagen, gelatin, fibrin, and alginate, to chemically modified biopolymers like gelatin methacrylate (GelMA), to synthetic polymers with designed scaffolding properties. The choice of each hydrogel will depend on the nature of the tissue, including cell attachment and maturation, material stiffness, and, in some cases, degradation properties to mature tissues with native-like extracellular matrices (ECMs). The use of primary cells and/or stem cell-derived cells to produce biofabricated tissues has enabled the incorporation of complex physiological features with a relatively short maturation period of culture, leading to better reproducibility. However, the sourcing/expansion of primary cells and maturation/differentiation of induced pluripotent stem cell (iPSC)-derived cells are critical logistical and cost issues to consider when creating these tissues, especially because this approach requires the use of a large number of cells that are not easy to expand and might therefore limit their scaling up for large-scale drug testing.
Spheroids, organoids, and biofabricated tissues mostly capture parenchymal or stromal cell–cell interactions and cell–microenvironment interactions. Most of them do not recapitulate the systemic tissue–tissue and organ–organ interactions, or the spatiotemporal gradients of chemotaxis or the mechanical shear flow that is produced by the in vivo circulatory system. To address the latter unmet need, microfluidics-based systems like organs-on-chip (OOCs) 18 or microphysiological systems (MPSs)—OOCs perhaps mostly being single-organ assay platforms, for example, lung-on-chip, while MPSs are assay systems that include cells from multiple organs/tissues—are designed to comprehensively mimic the organs’ biological activities, dynamic mechanical properties, and biochemical functionalities by incorporating flow. These microfluidics assay platforms enable a more accurate modeling of physiological transport and distribution of nutrients as well as other biomolecular cues ranging from hormones to neurotransmitter release and circulation in the 3D tissue constructs.19,20
One should view these different CIVMs not as exclusive but as complementary models, and really think about what the best use is for each one of these 3D assay platforms, depending on the desired application. In fact, there is an emerging trend of combining different aspects of each platform in one. For example, spheroids and organoids can be used as building blocks and be integrated into tissue-relevant ECMs, scaffold-based cellular tissue-like microenvironments, or OOC platforms.21–23 The application of bioengineering technologies to induce more physiologically relevant tissue patterns and heterogenous production is now being pursued.24–26 Choosing the appropriate CIVM assay platform for scientific research or a specific phase of the drug discovery and development process needs to be established through a fit-for-purpose approach: 7 (1) the disease to model, (2) the disease stage, (3) the physiological complexity needed to model the overall disease physiology or a specific stage, and (4) the desired sample throughput, whether it is a low number (tens) for research, lead optimization, and preclinical development, or larger numbers (hundreds to thousands) for lead discovery ( Fig. 2 ). There are now commercial sources of CIVMs, mostly spheroidal models of cancer tumors and liver. Liver is perhaps the organ for which CIVMs have been further developed and characterized, driven by their use in drug toxicity studies and early assessment of their predictability over hepatic cell cultures and animal models. There have been coordinated efforts providing guidelines as to their validation and qualification. 27 Tumor models based on aggregated cells have been developed for decades, and the acceleration of their use has occurred with the commercial availability of ULA round-bottom well plates and other techniques to speed up the process of spheroid formation.
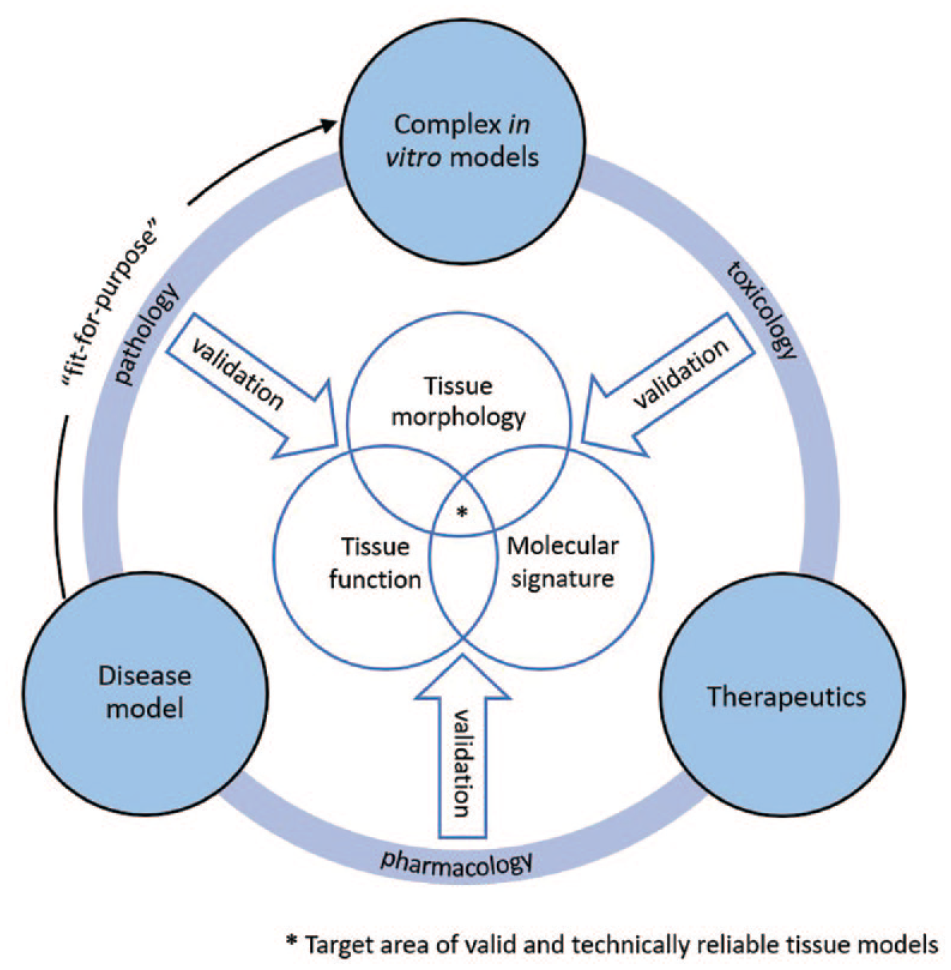
Determining domains of validity for a CIVM using clinically relevant data as benchmarks. The fit-for-purpose approach allows for systematic evaluation of how a CIVM can be applied in a drug R&D pipeline.
The field is moving fast toward the development of CIVMs. However, data are still lagging to provide a thorough scientific validation of these models to assess cell composition, whether molecular and cellular pathways are regulated like in native tissues, and whether pharmacological responses are clinically relevant. Techniques such as immunofluorescence, proteomics, metabolomics, single-cell sequencing (scRNA), and tissue-relevant functional studies will be crucial to benchmark these cellular models and define their clinical predictability.
Biofabricated Tissues: A Versatile Approach to Assemble CIVMs with Modular Complexity
Bioengineering techniques have emerged as powerful tools to create tissues of tailored cellular composition and spatial arrangements. The use of polymeric scaffolds, micropatterning, and bioprinting additive manufacturing techniques has allowed for precise spatial controls and rapid prototyping of cell-laden tissue-like structures. The key advantage of this technology over other CIVMs is to enable simultaneous control of both cellular and structural complexity, demonstrating most of the strengths and complementing weaknesses of other CIVMs: (1) spatial organization of multi-cell-type tissue-like structures using cell-laden scaffolds, (2) controlled cellular complexity using both primary and iPSC-derived differentiated cells, (3) controlled cellular complexity that can be easily modulated using normal or disease cells, (4) controlled structural complexity in tissue physiology (e.g., design of vascularization patterns to better quantitate vasculogenesis and angiogenesis), (5) shorter time to assay readouts unlike organoid systems because of the use of differentiated cells, (6) higher reproducibility due to the automation process used in bioprinting, and (7) compatibility with medium-throughput automated screening (up to a 96-well platform) with multiwell plate platforms. The versatility of biofabrication approaches provides major opportunities for disease-specific assay development by using protocols established for normal bioprinted tissues. The challenges remain in creating such complex biofabricated tissues in a multiwell plate format for drug testing, expansion of relevant cells, and development of disease-relevant quantitative readouts that can be used for drug testing.
For example, a protocol developed for a full-thickness skin equivalent can be easily adapted to build different skin disease models such as atopic dermatitis (AD) 28 or cutaneous squamous cell carcinoma. 29 Such models can then be used as screening platforms to evaluate drug efficacy and toxicity or to study the effect of skin microbiota imbalance on integumentary homeostasis. Additionally, biofabricated vascular networks containing endothelial cells, pericytes, and fibroblasts can easily be customized to vascularize bioprinted tissues of any organ systems, including the skin, retina, or lung. These biofabrication methods allow for stepwise modularity in the physiological complexity of the tissue by the choice and availability of cell types that can be printed. Such versatility in controlling physiological complexity provides a unique opportunity to assess the effects of a particular cell type on a disease phenotype and the effect of compounds on specific cell types, tissue, or disease pathology.
Establishing Domain of Validity with Bioengineered 3D Tissues: Case Study with Skin Models of AD
To compare in vitro to in vivo biology, one must thoroughly assess the native-like cellular composition and function in CIVMs through immunohistological studies and single-cell transcriptomics analysis. However, there also needs to be a similar level of benchmarking when establishing disease relevance by identifying disease biomarkers commonly seen in clinical settings. As increased cellular complexity comes at a high technical cost when assembling these bioengineered tissues, a progressive and iterative approach of increasing cellular complexity, tissue validation, and disease relevance with preestablished disease biomarker benchmarks is needed. The concept of domain of validity, referring to the determination of parameters in a tissue model to establish what disease stage the cellular assay system will replicate, 30 is a guiding principle that should be used to develop and optimize any CIVM ( Fig. 2 ).
To illustrate the points above, we recently reported on a bioprinted skin tissue model of AD. 28 AD, also known as eczema, is an inflammatory disease of the skin that results in red and itchy rashes on the skin. AD is triggered by immunogenic sensitization to exposure of environmental allergens that promote a Th2 cell response. Physiological manifestations of AD include spongiosis and hyperplasia, early and terminal expression of epidermis differentiation proteins, and increases in levels of pro-inflammatory cytokines. We set out to experimentally assess what tissue complexity was needed to reproduce the various biomarkers of AD in skin. Skin models have been developed and extensively validated for safety assessment in the cosmetics industry.31,32 Based on these technologies, skin tissue equivalents of different cellular complexities were biofabricated: epidermis-only equivalents produced with keratinocytes, full-thickness skin equivalents with dermis made up of fibroblasts and epidermis with keratinocytes, and vascularized full-thickness equivalents in which microvasculature was developed in the dermis by adding endothelial cells and pericytes, in addition to fibroblasts. The tissues were biofabricated in 96-well transwell plates for reconstructed human epidermis skin tissues, and 24-well plates for full-thickness skin and vascularized full-thickness skin tissues. The different tissues were then validated by using a set of preestablished criteria based on clinical data, including skin tissue morphology by hematoxylin and eosin (H&E) staining, skin protein biomarkers by immunohistochemistry, reduced barrier function measured by electrical resistance, and secretion of a panel of cytokines. Using the different skin tissue variants, AD disease phenotypes were induced by interleukin 4 (IL-4) treatment, a cytokine secreted by Th2 cells, to mimic the T-cell response in clinical AD. 28
IL-4 treatment was used to recapitulate the acute inflammatory stage in AD, and the expectation was to reproduce clinical phenotypes such as hyperplastic skin morphology, loss of skin barrier function measured by transendothelial/epithelial electrical resistance (TEER), secretion of canonical inflammatory cytokines that were reported to be clinically relevant in AD, and observation of vasculogenesis and angiogenesis, since several clinical reports showed excessive angiogenesis in AD.33,34 By design, the models did not fully capture the whole AD disease pathomechanism, such as the initial nonlesional phase, Th2 secretion from the lymph nodes, and subsequent chronic phases of the AD postacute inflammatory stage. However, by using skin tissues with increasing complexity, we demonstrated that different disease pathologies, as measured by different phenotypes above, could be achieved with the different tissue variants. While epidermis tissues were able to show hyperplasia, they did not produce measurable changes in barrier function and secretion of relevant cytokines. When dermis was added to the tissue, the AD skin tissue showed not only hyperplasia, but also other AD clinically relevant effects, such as disruption of barrier function and increased secretion of inflammatory cytokines.
Although vasculature did not significantly affect the AD-specific responses as addition of a dermis to the skin tissue, it did provide additional insights into the unexpected effects of IL-4 in reducing angiogenesis and secretion of angiogenic factors, contrary to what is observed in clinical AD.
35
An important point was that none of the AD-relevant phenotypes were detected in 2D-based keratinocytes or other skin cell culture systems (
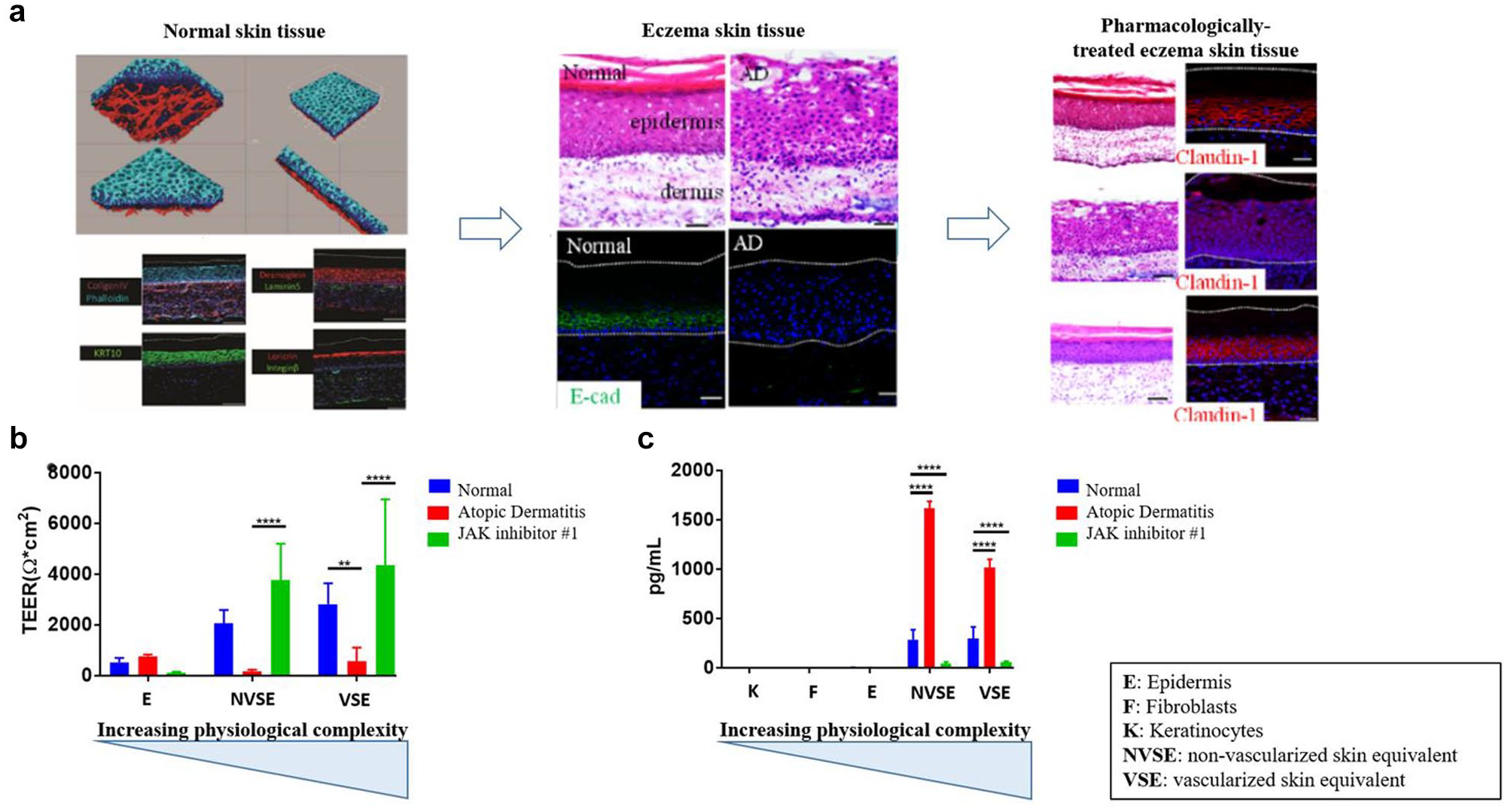
Reproduction of images from Liu et al.
28
(
In addition to identifying the domains of validity for the different skin tissue models, several JAK inhibitors that are currently in clinical trials for AD 36 were evaluated in vitro with the above skin models. The selected JAK inhibitors, which block the downstream signaling of IL-4, were able to reverse the AD disease phenotypes in each of the tissue variants ( Fig. 3A ). The models also showed that dexamethasone, a general immunosuppressor that is not expected to be therapeutically relevant in AD, did not alleviate any AD phenotypes, thereby establishing the pharmacological domain of validity for the bioprinted skin model. While IL-4 inhibited angiogenesis in vitro, JAK inhibitor surprisingly did not correct the IL-4-induced effect in vitro despite its known mechanism of action on IL-4 and, instead, downregulated vasculature formation even further. This observation highlights that while the vascularized model did not mimic clinical effects on angiogenesis for AD, it was a useful phenotype to assess specific pharmacodynamic effects of targeted therapeutics.
The above example shows how domains of validity for a CIVM can be established by developing and validating models with different cellular complexities as well as using clinically available biomarkers and pharmacological interventions. It is therefore critical to establish such domains of validity for any model by (1) using available clinical biomarkers of cellular identity, tissue morphology, and different omics technologies; (2) testing tissues of modular cellular complexity, and inducing a quantifiable disease phenotype using patient cells or relevant biological and mechanical stressors; (3) developing different assay phenotypes with clinically relevant disease biomarkers; and (4) assessing whether the model responds to different clinical or cell signaling relevant pharmacological interventions.
Operationalizing the Use of 3D Biofabricated Tissues for Drug Screening
Once the domain of validity for a disease-based CIVM is established, technical robustness in a multiwell plate format must also be evaluated to increase the models’ applicability toward screening. Technical reproducibility not only enables the use of these models as screening assay platforms, but also provides opportunities to study the biological variability among patient populations by using different patient-derived cells, and thus investigate the roles of patient specificity in disease manifestation and therapeutic response in a laboratory-controlled tissue microenvironment.
Most automated screening platforms currently in place are not optimized or designed for CIVMs. Therefore, assay development for these complex assay systems requires further refinement to help operationalize the models for high-throughput screening (HTS). Examples of automated screening platforms have already been published, mostly applied to spheroids and organoids.37–39 First and foremost, the technical robustness and reproducibility of these assays must be assessed well to well, plate to plate, within a lot of cells, and among different cell lots used. Criteria for robustness normally used for 2D HTS assays must be included to assess assay quality (Z factor or equivalent) for 3D HTS assays. 40 Dose responses for key control compounds must also show proper assay pharmacology in these 3D cellular systems in HTS format. In our work with biofabricating full-thickness skin tissues in a 96-well plate to screen for skin irritants, we systematically obtained coefficient of variance (CV) values of 20% when measuring TEER for the wells in the sample field. 41 Additional unpublished in-house data demonstrate that assays with full-thickness skin models of melanoma and cutaneous squamous cell carcinoma assays in 96-well transwell plates, in which fluorescence signal from green fluorescent protein (GFP)-expressing cancer cells is measured by fluorescence microcopy, have Z factors of 0.4–0.6, which are very robust for 3D tissue models.
Operationalization of 3D biofabricated tissues for medium-throughput screening (MTS), for example, a 96-well plate, requires the integration and streamlining of several processes in an end-to-end pipeline ( Fig. 4 ). The process of cell procurement and expansion, reproducible and robust tissue biofabrication protocols, assay development with 3D in vitro tissues, and, lastly, the generation, processing, and analysis of large data sets are perhaps the four key challenges to operationalizing the use of biofabricated 3D tissues for drug screening. To biofabricate 3D tissues that faithfully mimic human tissues, it is critical to use cells that are as physiologically relevant as possible. Primary cells or iPSC-differentiated cells must be prioritized over other cell types, such as immortalized cell lines. However, the expansion of such cells is nontrivial and should not be underestimated, as it can be costly and resource-intensive. Primary cells are isolated directly from living tissue or organs, and therefore they closely resemble cells in in vivo tissues. However, their growth ex vivo requires complex specialty media conditions, and even in these optimized growth media, because they are terminally differentiated, they possess limited to no proliferative capacity, and some lose their mature phenotype and function when adapted to 2D cultures for propagation, which can limit their physiological relevance. There is also the issue of molecular and physiological variability in primary cells, depending on the donors, which, although making the use of these cells for assay development and reproducibility challenging, provides an opportunity to study personalized cellular models. Although iPSCs have the capacity for unlimited expansion, their production involves lengthy and complex differentiation protocols that require extensive automation to scale up production. 42 In many cases, iPSC-derived cells are not fully mature and resemble the differentiation stage of fetal primary cells rather than adult mature cells. In addition to their ability for unlimited expansion, iPSCs also have the advantage that they can be genetically modified using techniques such as CRISPR, enabling a syngeneic type of normal and disease cells that might reduce biological variability due to genomics variability between cells of different patient backgrounds. In our experience, the catalog of commercially available primary cells has increased substantially to enable production of tissues like liver, lung, skin, and retina, including endothelial cells and pericytes for vascularized tissues. For other tissue types like brain models, there is a growing catalog of human iPSC-derived neurons. Testing cells from different sources and batches is a common exercise in our lab, and the final choice of one human cell type over the other for tissue production will be given by their scientific relevance, but also by commercial availability in enough amounts, budget, and resources for internal large-scale differentiation of iPSC.
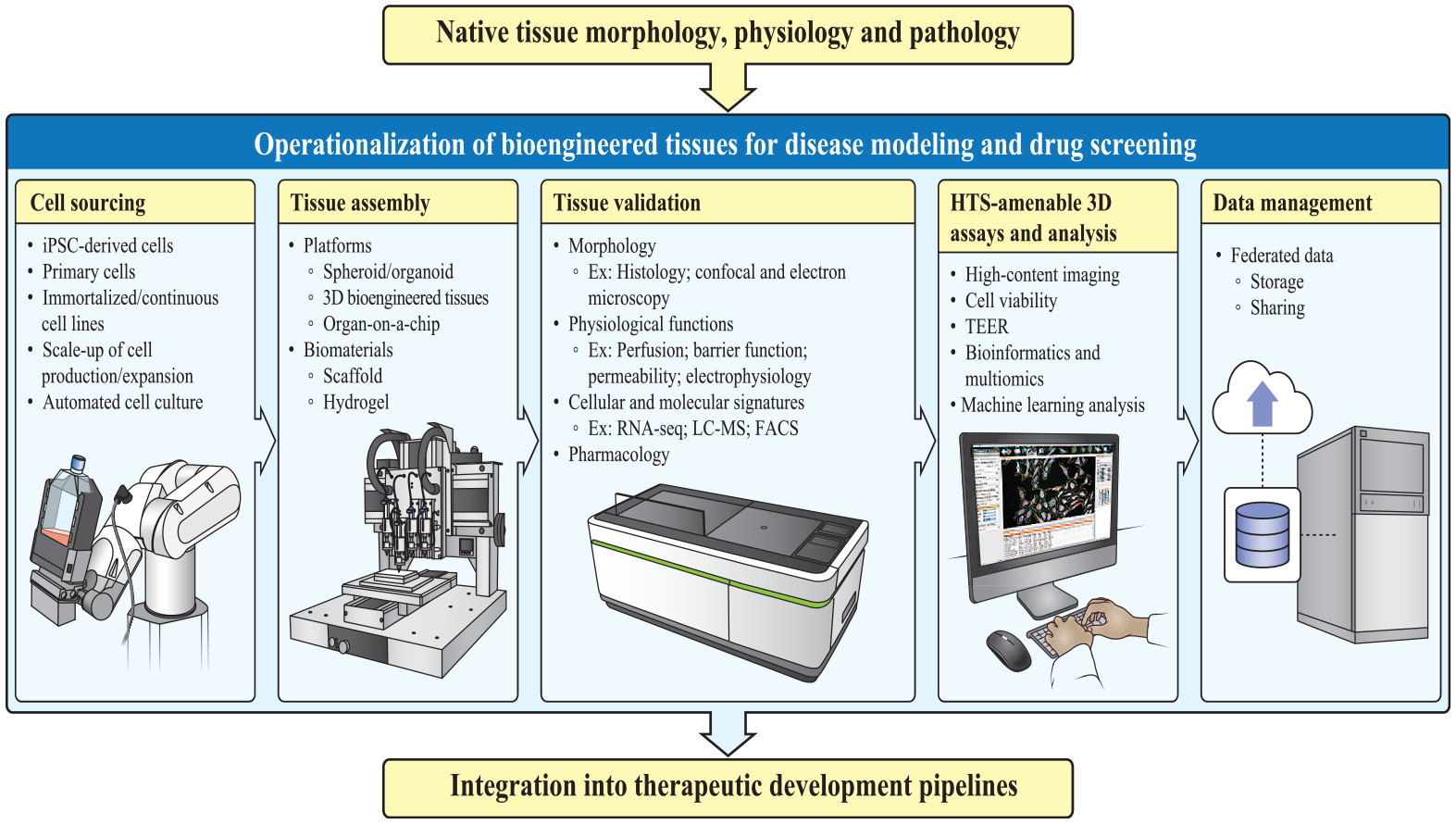
Entirety of drug R&D process in the context of CIVMs. Once the CIVM is developed and validated, there needs to be a streamlined assembly line-like process of scaling up and producing the models for HTS. Figure generated by NIH Medical Arts.
The choice of hydrogels to use to bioengineer each tissue will depend on the cell composition, as well as the end goal. For most of the tissues produced in our laboratory, gelatin or fibrin-based hydrogels have produced the desired results, but the choice of starting hydrogel to use really depends on the physiological features desired in the tissues being produced. 43 Additionally, types of transwell inserts, such as the material and pore size of the inserts, have increasingly become a critical parameter to consider, as they can determine the tissue and disease models to bioengineer. 44 A larger pore size might be needed to allow cell migration in models such as those for cancer metastasis, 45 but it might produce unwanted cell mobility in other cases. ECM coating with biomolecules such as collagen, fibronectin, and laminin, among many others, will be another important assay constraint to evaluate during assay development. Biodegradable membrane-based inserts are also becoming commercially available, 46 providing another option for a tissue to reach its parenchymal homeostasis while maintaining the desired layering, with the membrane separating the tissue depth during the initial fabrication.
Another critical aspect for the use of biofabricated tissue, or any other CIVM, in the drug screening pipeline is the adaptation of assay technologies that are commonly used in 2D-based HTS for 3D cellular systems. High-content fluorescence microscopy is widely used for 3D-based screening, but it must be acknowledged that sample processing, with currently available technology, is not straightforward from an automation perspective.47,48 Initial image acquisition and subsequent data transfer, storage, and analysis require robust IT infrastructure. In many laboratory settings, confocal immunofluorescence of whole or sliced microtissues is commonly used to validate cellular content and observe tissue morphology in depth. However, the imaging of tissue models might require laborious clearing techniques, 49 and it is too resource-intensive for large-scale screening. On the other hand, epifluorescence 50 or bright-field imaging51–53 coupled with machine learning can provide less time-intensive imaging alternatives for primary screening. Once lead candidates are identified, confocal imaging, which provides more biological information but quid pro quo on throughput due to temporal limitation, can be used at the hit validation stage, postprimary screens.
Alternatively, assays that have not been used for HTS due to high cost and lower throughput might be very amenable in the context of CIVMs. Such assays include enzyme-linked immunosorbent assay (ELISA)-like formats, which can detect biomolecules secreted in the supernatants and be easily multiplexed by using bead-based or other plate-based assay platforms. Additionally, many biofabricated tissues have barrier functions, which can be measured in a high-throughput manner using noninvasive TEER probes. 41 These functional TEER measurements can be a highly sensitive readout of tissue health and can be an effective primary, quantifiable output for disease models.
We believe that the power of using biofabricated tissues for drug testing will be in using multireadout approaches that provide rich phenotypic information on different aspects of a disease captured in a tissue model. Whether it is providing efficacy and/or safety information, quantitating different normal and disease-relevant features of a biofabricated tissue will allow us to assess the on- and off-target effects of the compounds tested, as well as the efficacy disease correction effects versus potential safety liabilities that a compound might produce, in both normal and disease cells of a tissue. The now more common use of artificial intelligence and machine learning methods to help quantitate multiparametric features of tissues measured using different technologies will be very powerful tools to be able to assess the effects of pharmacological interventions in the context of an engineered tissue model.
Lastly, the data produced from screenings must be stored and be accessible at later time points. As briefly mentioned in earlier sections, the large amount of data generated from screening using CIVMs poses a unique IT infrastructure challenge. This is especially the case for high-content imaging-based assays. Developing a robust IT infrastructure that enables storage in the cloud as well as a system that can facilitate faster processing and trouble-free data sharing will be critical in increasing practical usage of these models in the preclinical phases of the drug R&D pipeline.
Looking Ahead: Integration of CIVMs into the Drug Discovery and Development Pipeline
Although the interest in CIVMs for research and drug screening is increasing fast, there is a clear realization in the scientific community that for them to make a meaningful and long-lasting impact in research and the drug discovery process, there must be a standardized and harmonized path to their scientific validation to help establish their clinical relevance and predictability. These CIVMs must meet preestablished guidelines and markers for native-like tissue mimicry, clinical relevance, well-defined domain of validity, and measurable and screening amenable disease-relevant endpoints. 6 Their technical reproducibility in an MTS format is a critical prerequisite before their operationalization into the drug R&D process, and for these models to be instrumental in appropriate compound selection and evaluation of the structure–activity relationship for medicinal chemists and help prioritize compounds for in vivo evaluation for toxicologists and clinicians. In addition to testing for efficacy, these CIVMs will help elucidate clinically relevant data for toxicity and drug metabolism in disease conditions where pharmacokinetics are abnormal.
Summary of CIVMs, Advantages, and Limitations.
Establishing the Domain of Validity for Tissue Skin Models of Atopic Dermatitis (AD).

JAK inhibitors and dexamethasone disrupt the vasculature network in the VFTS model.
Not Applicable
Currently, most in vitro, in vivo, and clinical drug metabolism and toxicity studies are carried out in homeostatic animals and humans. However, drugs are intended to treat patients with preexisting conditions. Most patients, in addition to their primary diagnosis, will have a higher likelihood of comorbidities that may affect drug metabolism and clearance. Therefore, more clinically relevant adsorption, distribution, metabolism, and elimination (ADME) studies must be established in order to evaluate the effects of disease pathology in altering the above kinetics. Alternatively, finding effective and safe dosage concentrations and regimens for new drugs depends on their bioavailability as well as hepatic and renal metabolism. As such, having preclinical, in vitro disease models that mimic epithelial barrier penetration, first-pass metabolism, and organ-specific enzymatic turnover of a drug candidate should provide time- and cost-effective predictive drug dosing in relevant patient populations. 10 Similarly, blood–brain barrier (BBB) penetration is a separate, major hurdle for many promising central nervous system drug candidates 54 —while early lead compounds may show high efficacy in vitro, some fail to show sufficient penetration in later stages of the drug development pipeline. Having tissue models to mimic the BBB physiology, under both normal and disease conditions, will provide permeability evaluation in a higher-throughput manner and allow groups to establish the basal permeability, accumulation, and toxicity of a compound in the brain.38,55
Establishing the clinical predictability of CIVMs for drug responses is perhaps the most challenging question to answer. There is likely to be an iterative process of benchmarking between data generated from in vitro tissue models with tailored cellular complexity and physiology and clinical data, both retroactively and proactively. Defining the domain of validity of each CIVM, regardless of the technology used, is critical, and as illustrated here, there must be a systematic process of increasing cellular complexity and physiological features along rigorous validation of tissue physiological function, omics validation of both normal and disease models, and pharmacological responses to a set of benchmarking therapeutical interventions. To affirm the applicability of these in vitro 3D systems, it will be paramount to show with a specific set of known therapeutics the correction in patient-specific tissues while simultaneously benchmarking clinical response in the same patients. For cancer, there have been studies published with patient-derived organoids to identify specific chemotherapeutics with maximal therapeutic benefit while patients are simultaneously followed in the clinics receiving the same chemotherapy.56,57 Such examples of parallel preclinical and clinical cross-checks of compound treatment must be performed on a larger scale, rigorously designed, and systematically implemented to prove that these in vitro models can be used for planning a patient-specific drug regimen either proactively, retroactively, or simultaneously in clinical settings.
Although the focus in CIVM development has traditionally been on human-based models, there is a wealth of in vivo data for animals that can be leveraged to establish the in vitro-to-in vivo translation for animal models. 58 Developing animal CIVMs and benchmarking with the already widely available in vivo animal data should provide a much clearer path to validating the physiological likeness to the whole organism, as well as to establishing the domains of validity for the in vitro system. 59 The resulting data similarity or discrepancy from a species-based CIVM to in vivo studies, as well as between animal and human CIVMs, should allow researchers to predict the clinical response of therapeutics more accurately in humans.
Lastly, if one can establish the technical reproducibility of these CIVMs, it will open the door to assessing how race, sex, age, and other predispositions can play a role in disease manifestation and tailor pharmacological intervention for specific patients. Although personalized medicine is now being developed quite effectively in cancer by using patient-derived models, 60 for nononcogenic diseases, such an approach is still at its early stages or nonexistent. 61 Although much of the discussion for the latter is focused on the biological technology (i.e., patient-derived iPSC differentiation to produce a target cell population for disease modeling), the logistical questions and hurdles will still be an inevitable bottleneck in the R&D process that must be addressed. As such, proactive collaboration with both private industries and academic laboratories will be crucial in resolving such issues.
Conclusion
There is growing evidence that CIVMs can produce relevant pathophysiological phenotypes and drug responses that are not reproduced in 2D cellular systems and are clinically more relevant.9,62 The challenge for those in the drug discovery and development field is to establish and validate the domain of validity of each CIVM by using available clinical biomarkers and drug responses, and operationalize toward robust HTS platforms. Fortunately, the discipline has seen an explosive growth in technology and interest in the practical application of the models for screening therapeutics. Over the last 10 years, we have seen the development of many key technologies, which helps to foster the use of CIVMs for various applications rather than focusing on technology development, for example, new biomaterials for 3D cell culture, so we expect the focus to be on scientific validation of the models to better establish their relevant use. As such, the challenge of scientific validation, clinical benchmarking, and pharmacological validation should be addressed with a sense of urgency and desire to change the current state of the drug discovery process.63–65 In doing so, it can reasonably be expected that, in the foreseeable future, such changes in the screening infrastructure will allow the scientific community to accelerate therapeutics development. Although there is now not much public information as to whether CIVMs have been used to gain approval for and bring a new drug to market, there is increasing evidence that data generated from CIVMs are being included in submissions for new drug approvals. 66 Data from reconstructed human epidermis are providing information on skin corrosivity and irritation for regulatory decisions,67,68 and pharmacological data from organoids have been included in study reports in investigational new drug (IND) filings. 69 Data generated using different types of CIVMs should continue to be included in study reports for IND, new drug application (NDA), and biologics license application (BLA) filings to provide additional pharmacological and toxicity information along with animal data. Eventually, as more clinical information on the therapeutic agents is obtained, it will be possible to do a retrospective analysis to assess the clinical predictability of each CIVM model, at each stage of R&D. Ultimately, by pushing the boundaries and current status quo in drug R&D, the scientific community may be able to create lasting and consequential socioeconomic impacts and provide medical, social equity for marginalized patients in the healthcare system.
Footnotes
Acknowledgements
We would like to thank Drs. Madhu Lal, Sharon Presnell, and Jens Kelm for their critical reading of this manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.