
Abstract
The variety and complexity of drug targets are expanding rapidly. At the same time, there is significant interest in exploring a larger chemical space to identify new candidates. Fragment-based screening (FBS) has emerged as a popular alternative to traditional high-throughput screening campaigns to identify such drug candidates. FBS identifies hit fragments that exhibit weak interactions with the target of interest, thereby enabling the rational design of small-molecule compounds from the identified hit fragments, which serve as building blocks. This strategy reduces the number of molecules to screen while also allowing the exploration of a greater chemical space.
Here we use temperature-related intensity change (TRIC) technology to perform FBS against the target MAPK/ERK kinase-1 (Mek1). TRIC describes the change in fluorescence intensity of a fluorescently labeled molecule upon a change in temperature. This intensity variation is dependent on the physicochemical environment in the vicinity of the dye and strongly affected by binding events. Thus, the detection of binding events is independent of mass, making TRIC an ideal tool for FBS.
Using only 150 pmol of labeled Mek1, the authors screened 193 fragments from a prescreened library in less than 1 h of measurement time, leading to 66 hits. Among those hits, they identified more than 80% of the published top hits found using orthogonal techniques. Furthermore, TRIC allowed the identification of fragments that were of poor solubility but could be mistaken as false-positive hits in other methods.
Introduction
Fragment-based screening (FBS) is a well-established strategy in small-molecule drug development, and its application has been expanding since it was first developed decades ago. It screens molecules of a reduced size that are composed of fewer than 20 heavy elements. These molecular building blocks, generally smaller than 0.5 kDa, are referred to as fragments. Combinations of different fragments are used to refine the lead synthesis and generate larger molecules, 1 allowing for tighter affinities. There are compelling advantages to the application of FBS in the drug discovery field. The use of fragments allows the exploration of a large chemical space, while at the same time significantly reducing the number of required molecules to screen.
Although FBS is widely used in drug discovery, some challenges remain when it comes to building a steady and reliable workflow. 2 Fragments are low-molecular-weight compounds and thus require a detection method that is independent of mass. Since fragments show weak affinities and sometimes low solubility at high concentration, instruments need to be able to tolerate compound precipitation or great care is needed during experimentation. These and other parameters of FBS elicit the need for orthogonal and parallel detection technologies to yield reliable hit confirmation.2,3 For targets without biochemical activity, researchers look for biophysical methods as alternatives, but even with smaller library sizes, fast methods are needed, often a weak spot of biophysical techniques.
Here, we evaluated a biophysical method referred to as temperature-related intensity change (TRIC), which is derived from microscale thermophoresis (MST). 4 TRIC technology relies on a target molecule that is labeled with a single fluorophore for the detection of binding events. Any fluorescent molecule displays a correlation between brightness and temperature, a property that is dependent on its physicochemical environment. 5 If the temperature modification is highly controlled, a given labeled molecule will always display the same change in intensity over the course of the heating process. For TRIC, an infrared laser is used to induce a highly controlled temperature change by excitation of water molecules.4,6,7 The correlation between the brightness of the fluorescent probe and the temperature of the solvent is strongly linked to the solvation of the labeled molecule. Therefore, any modification of the physicochemical environment of the fluorophore, for example, a binding event, will modify the observed fluorescence temperature correlation.4,5 Hence, TRIC is a mass-independent detection technology that is highly suited for FBS. The high sensitivity of the fluorescence-based assay design works with low target concentrations. Not only does this allow the screening of a wide range of affinities (from pM to mM), but it also requires less protein sample. In comparison to the related MST technology, TRIC promises higher throughput by enabling a readout of measurement of a 384-well plate in 30 min. To adapt MST to a higher throughput, the sample format had to be changed to agree with SLAS standards. Measurements in a multiwell plate, in contrast to measurements in thin glass capillaries, do not allow the generation of a steep temperature gradient. Hence, the Dianthus instrument line relies on TRIC alone, not thermophoresis. The excellent comparability to MST as a method, as also demonstrated in this publication, results from TRIC also being a major component of the signal measured in MST instrumentation. 7
To test the capability of this emerging technology, we conducted a single-point screening campaign against the well-established target protein mitogen/extracellular signal-regulated kinase (Mek1) from the MAPK signaling cascade. Mek1 is a regulator of the ERK1/2 pathway that regulates proliferation, differentiation, transcription, and development. Aberrant signaling in this pathway can lead to unregulated cell growth, and targeting this cascade is promising to develop anticancer drugs. 8 We used a library of 193 fragments that had been identified in an initial virtual screen and previously tested for binding to Mek1 using various methods. 9 Selecting this library−target combination would allow a direct side-by-side comparison of TRIC technology and established biophysical methods such as MST, surface plasmon resonance (SPR), co-crystallization, and thermal shift assay (TSA) using differential scanning fluorimetry.
The comparative analysis of a single-dose screen of 193 fragment molecules establishes TRIC technology as a promising candidate for FBS, with sufficiently high throughput, mass-independent detection of binding events, high sensitivity for a broad affinity range, and the added advantage of robustness against and detection of poorly soluble fragments.
Materials and Methods
Mek1 Production
The recombinant protein Mek1 kinase domain was produced following a previously published method. 9 Mek1 was stored at 270 µM in 20 mM HEPES, 300 mM NaCl, 2 mM DTT, pH 7.5.
Mek1 Labeling
Free cysteine residues in Mek1 were labeled as per instructions in the RED-MALEIMIDE 2nd Generation labeling kit (NanoTemper Technologies, Munich, Germany). Ninety microliters of 10 µM Mek1 was mixed with 10 µL of 300 µM RED-MALEIMIDE 2nd Generation dye diluted from a stock solution resuspended in DMSO to 600 µM. This led to a final mix of 9 µM Mek1 and 30 µM dye at 5% DMSO in 25 mM Tris, 150 mM NaCl, pH 7.4. Note that the labeling buffer composition deviated from instructions in the kit manual. Labeling was performed for 30 min at room temperature in the dark. Unreacted dye was then removed using a gravity flow size-exclusion column provided by the manufacturer to separate free, unreacted dye molecules below 5 kDa from labeled protein material. The size-exclusion column was equilibrated with the assay buffer: 25 mM Tris, 150 mM NaCl, pH 7.4, supplemented with 0.1% Pluronic F-127. Labeled protein was aliquoted and stored at −80 °C to minimize the number of freeze/thaw cycles.
Fragment Library
Fragments were provided by Sanofi and are described by Linke et al. 9 Chemical structures remain confidential.
Affinity Determination for Selected Binders
The stability and function of labeled Mek1 were confirmed by an interaction test with a known reference molecule, EP1 (Sanofi). Ten microliters of a serial dilution series of EP1 in 25 mM Tris, 150 mM NaCl, pH 7.4, 0.1% Pluronic F-127 was transferred to each of eight wells. Ten microliters of labeled Mek1 was added to each well, resulting in a final concentration range from 625 nM to 4.9 nM of EP1 and a final concentration of 10 nM labeled Mek1.The titration of fragment 139 was done in the same way as described for EP1, with the only difference being that the final concentration of the fragment ranged from 1 mM to 488.3 nM.
The acquisition was performed on a Dianthus NT.23PicoDuo instrument from NanoTemper Technologies using 25% LED excitation power, picomolar detector sensitivity off, and a laser on-time of 5 s. TRIC dose−response plots show Fnorm (as for MST data) displayed semilogarithmically against the titrated ligand concentration. Fnorm is calculated by computing the ratio of the mean fluorescence recorded within the time window of 4−5 s after the infrared laser was turned on, by the mean initial fluorescence measured (before turning on the infrared laser).4,7 The analysis was carried out using the DI.Screening Analysis software (NanoTemper Technologies) to merge the data for the final output of the Kd value, as well as to estimate the response area expected for the interaction of EP1 and Mek1 in single-dose assays.
Liquid Handling Sample Preparation for Screening
The final assay condition in the plate was a volume of 20 µL per well at 10 nM Mek1. The assay buffer composition was 10 mM Tris, 150 mM NaCl, pH 7.4, supplemented with 0.1% Pluronic F-127 and 1% DMSO to ensure fragment solubility. A STARlet system (Hamilton) was used for liquid handling. First, fragments were diluted from the 100 mM stock solution into assay buffer to 4 mM and 4% DMSO. The fragments were further diluted fourfold into the Mek1 target stock, to a final concentration of 1 mM and 1% DMSO. The first compound dilution into assay buffer occurred in a conventional microwell plate (Nunc), while the second dilution step into Mek1-containing assay buffer occurred in the Dianthus 384-well microplate (NanoTemper Technologies, cat. DI-P001), where the measurements took place. Each sample was prepared in duplicate and positioned side by side. At regular intervals (18 times in duplicate over the entire screen), a negative control of labeled Mek1-target alone was included in duplicate to ensure robot pipetting accuracy and act as a reference sample for nonbinding conditions. A duplicate for EP1 (five times in duplicate over the entire screen), also pipetted at regular intervals, was used as a positive control to certify Mek1 stability by probing the response area as established in initial assay development experiments. We prepared four rows on five different 384-well plates to make best use of the four-channel STARlet system. Once prepared, each plate was shaken for 6 min at 2400 rpm, incubated for 20 min, and centrifuged for 30 s at 1000g before beginning the measurement.
Fragment Screening with TRIC
The prepared 384-well plates were placed in the instrument immediately after centrifugation. Data acquisition was done at a 25 °C set temperature using 40% LED excitation power along with a sequence of 5 s of infrared laser irradiation.
Acquired data were analyzed using the DI.Screening Analysis software, using a Z-score threshold of 3 and plate-wise referencing of ligands to the corresponding negative controls on the same plate. Subsequent analysis and comparison of the data with orthogonal techniques were done manually using data tables in Microsoft Excel.
Results and Discussion
Assay Development and Optimization
To demonstrate the capability of TRIC technology for FBS, we selected Mek1, a target that has already been extensively screened in the literature and for which data sets from various established technologies were readily available. 9 Since TRIC requires a fluorescent target molecule, we coupled Mek1 to a far-red fluorescent dye (RED-MALEIMIDE 2nd Generation from NanoTemper Technologies). Far-red fluorophores are ideal probes for target labeling since they rarely show a background signal in fragment- and small-molecule libraries.
To start, we established the robustness and stability of the system using a known and well-characterized compound, EP1. We measured the area response signal of Mek1 against EP1 in various buffer compositions using a Buffer Exploration Kit (NanoTemper Technologies; data not shown). The best buffer was selected based on the response in amplitude (
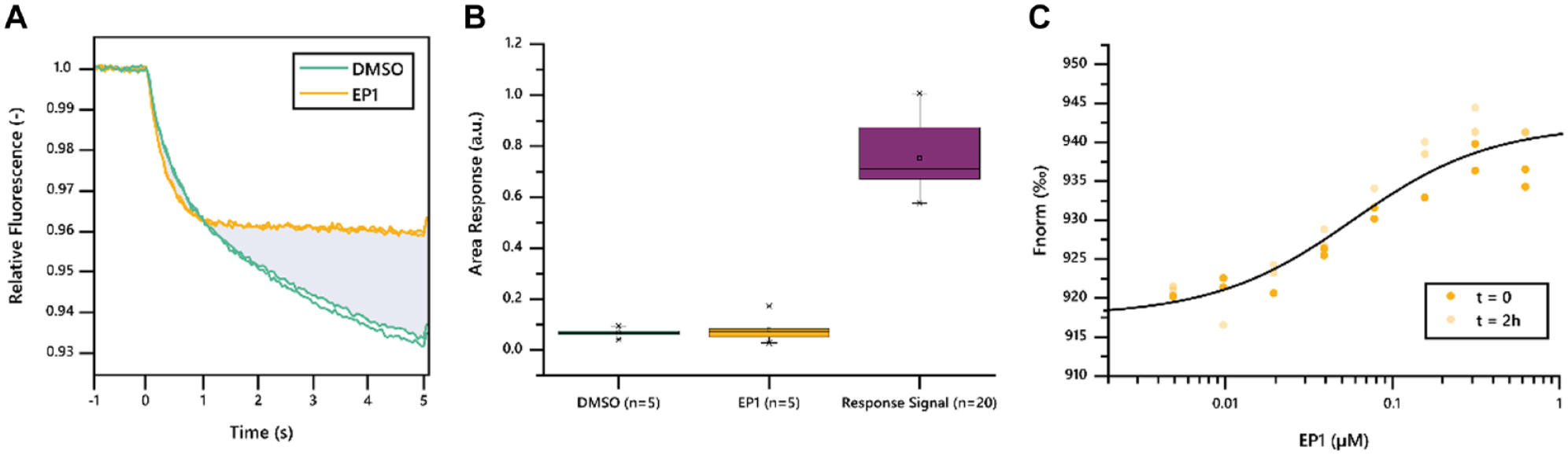
(
Overall, maleimide labeling of Mek1 in combination with buffer screening quickly resulted in excellent assay quality as determined by titration of EP1, a known binder. The established conditions were used in a single-point screening of 193 fragment molecules with a low sample consumption of just 150 pmol of labeled protein.
Single-Dose Screening and Evaluation
We screened 193 fragments against Mek1 using one single high dose of 1 mM fragment concentration. We included the well-characterized reference compound EP1 at a saturating concentration of 500 nM every 24 fragments (n = 10) and a Mek1 target-alone negative control every 6 fragments (n = 36). The data were analyzed using the same area analysis approach as described above (
The analysis resulted in the identification of 66 hit fragments with a signal response exceeding the set threshold (
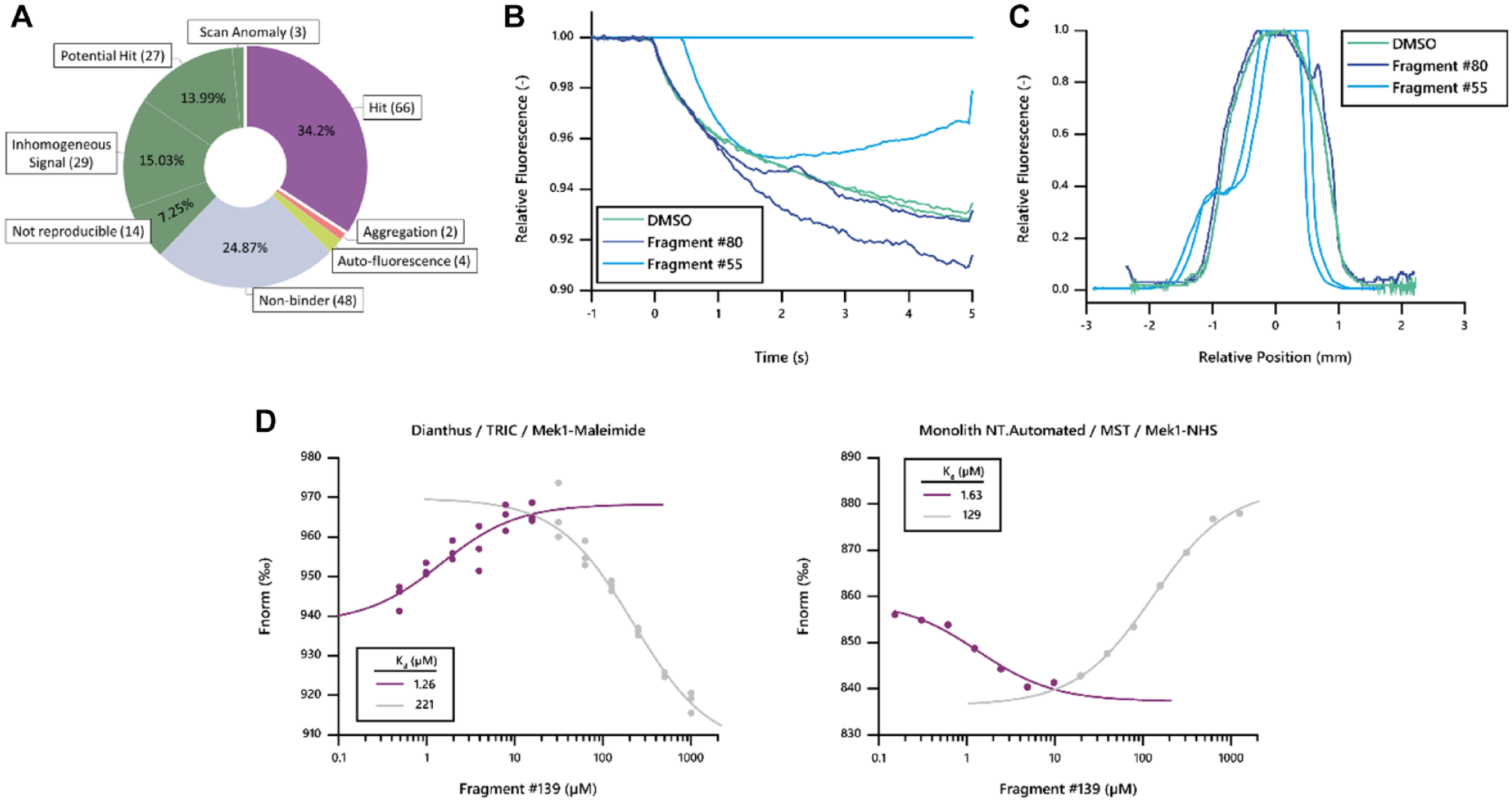
(
Ligand Categories Automatically Assigned by DI.Screening Analysis in Single-Dose Screens.

In a typical workflow, additional control experiments for the potential hit and autofluorescence categories would yield a final hit list that would then be used for a secondary Kd screening. While a dilution series of fragments requires more data points than a single-dose screening, it allows for a more detailed and thorough separation of nonspecific and specific binders. However, a secondary screen was beyond the scope of this study, which was designed to test for the comparability of hit identification between different biophysical techniques.
Comparison to Other Biophysical Techniques
For detailed data comparison and interpretation, we followed the approach of the previously published study and focused on the top 25 fragments identified by each orthogonal technique used to characterize Mek1. 9 We made use of the established ranking based on the resonance unit (RU) signal from SPR, Kd ranking from MST, ΔTm ranking from TSA, and co-crystallization successes.
From the top 25 fragments identified by either MST, TSA, or SPR, TRIC technology identified more than 80% as interactors, demonstrating an excellent rate of hit identification by TRIC with low rates of false-negative results. Interestingly, some of the SPR hits with a high RU response showed aberrant well scans in TRIC measurements and also TRIC traces, biased by aggregated target material. For example, fragment 80, which featured a high RU response in SPR, along with a ΔTm of −1 °C in TSA,
9
was identified as aggregated and did not yield a reproducible TRIC response (
Among the 13 fragments that successfully co-crystallized in the original publication, 9 TRIC succeeded to identify all, except fragment 48. This fragment was highly ranked in the MST assay and TSA, but not in the SPR assay. We speculate that the corresponding fragment stock solution might have undergone degradation and thus did not yield a hit in the TRIC assay.
In summary, TRIC technology was used in a single-dose screening with duplicate measurements of one fixed high compound concentration. Nevertheless, a secondary Kd analysis would typically follow, also using TRIC, as illustrated during the assay optimization using EP1. A follow-up Kd screen would enable a more thorough distinction of binders and nonbinders but would increase the required number of data points to be screened.
Nevertheless, to further demonstrate the excellent comparability of TRIC measurements in a Dianthus instrument to MST measurements in a Monolith instrument (both NanoTemper Technologies), we performed a titration series experiment for fragment 139, which showed the highest affinity in the published MST screen.
9
Comparison of the titration data obtained on Dianthus (
The data presented here clearly demonstrate that TRIC is suited for FBS with some key advantages. The technique is mass-independent and highly sensitive for binding detection in a wide affinity range. Furthermore, the method is buffer-independent, suggesting that it can be used even on unpurified targets, if there is an option for labeling specificity, such as His-tagging.10,11 This may open the scope to more challenging targets, which are not easily purifiable. Finally, TRIC is highly comparable to and in agreement with other biophysical screening methods, especially MST.
Footnotes
Acknowledgements
Sanofi thanks Maryse Lowinski for fragments, compounds, and logistical support. We acknowledge Dr. Kwame Amaning for data management support. We thank Dr. Thomas Bertrand for the scientific discussion of the results.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.