
Abstract
The secretin receptor (SCTR), a prototypical class B G protein-coupled receptor (GPCR), exerts its effects mainly by activating Gαs proteins upon binding of its endogenous peptide ligand secretin. SCTRs can be found in a variety of tissues and organs across species, including the pancreas, stomach, liver, heart, lung, colon, kidney, and brain. Beyond that, modulation of SCTR-mediated signaling has therapeutic potential for the treatment of multiple diseases, such as heart failure, obesity, and diabetes. However, no ligands other than secretin and its peptide analogs have been described to regulate SCTRs, probably due to inherent challenges in family B GPCR drug discovery. Here we report creation of a testing funnel that allowed targeted detection of SCTR small-molecule activators. Pursuing the strategy to identify positive allosteric modulators (PAMs), we established a unique primary screening assay employing a mixture of three orthosteric stimulators that was compared in a screening campaign testing 12,000 small-molecule compounds. Beyond that, we developed a comprehensive set of secondary assays, such as a radiolabel-free target engagement assay and a NanoBiT (NanoLuc Binary Technology)-based approach to detect β-arrestin-2 recruitment, all feasible in a high-throughput environment as well as capable of profiling ligands and hits regarding their effect on binding and receptor function. This combination of methods enabled the discovery of five promising scaffolds, four of which have been validated and further characterized with respect to their allosteric activities. We propose that our results may serve as starting points for developing the first in vivo active small molecules targeting SCTRs.
Keywords
Introduction
In 1902, Bayliss and Starling discovered secretin (Sec-FL [full-length]), a 27-amino acid hormone, in the duodenal mucosa stimulating the secretion of bicarbonate, enzymes, and potassium ions from the pancreas. 1 Sec-FL exerts its physiological effects by activating the secretin receptor (SCTR), which was cloned in 1991 as the first member of the class B family of G protein-coupled receptors (GPCRs). 2 Thus, secretin and its receptor formed the backbone of this highly important family of therapeutic targets, which are crucially involved in hormonal homeostasis. 3 Beyond its secretory effect, Sec-FL is implicated in a number of physiological and pathological conditions involving the heart, lung, liver, brain, and gastrointestinal (GI) system. 4 According to Grossini et al., 5 intracoronary infusion of secretin in animal models or human patients demonstrated beneficial cardiac responses, such as positive inotropic, chronotropic, and vasodilating effects, without changing the blood pressure. Furthermore, SCTRs have been shown to stimulate meal-induced brown fat thermogenesis resulting in satiation and short-term reduction of food intake. 6 A recent report highlighted elevated postprandial secretin plasma concentrations after Roux-en-Y gastric bypass (RYGB) surgery, which unveiled glucose-sensitive S (secretin) cells in the distal small intestine. 7 In addition, SCTR activation might be of therapeutic value for functional dyspepsia as indicated in a small clinical study in humans. 8 Altogether, modulating SCTR signaling outlines a unique strategy to develop novel therapeutics with potential benefits for the treatment of comorbid conditions, such as obesity, diabetes, abnormal gastric accommodation, and heart failure. However, to date, no ligands other than secretin peptide and closely related analogs have been identified to interact with SCTRs.9–11 Beyond that, synthetic Sec-FL represents the only clinically utilized SCTR ligand for diagnostic purposes.12,13 This might be due to the practical clinical limitations of Sec-FL, such as its short half-life (~2–4 min) and the need for intravenous administration. 8 Moreover, the development of potent small-molecule ligands for class B GPCRs remains challenging, likely due to the complex binding mechanism of orthosteric endogenous ligands within the N-domain and the highly open seven-transmembrane region of these receptors.3,14 Nonetheless, recent diabetes research has led the way by identifying a vast number of small-molecule modulators targeting the glucagon-like peptide-1 receptor (GLP-1R),15–20 a close relative to the SCTR. Although no orally available GLP-1R drug has been approved for human use, 20 the pace of emerging small-molecule GLP-1R ligands expands drug discovery for class B GPCRs. The quest for ligands that interact via allosteric binding sites contributed largely to this breakthrough. This class of compounds, which binds to structurally distinct receptor pockets, offers an opportunity to selectively modulate GPCR function in a diverse and rich way. 21 Positive allosteric modulators (PAMs) are able to elevate the natural effect of hormones and endogenous ligands, either by increasing potency or by maximizing efficacy, and may additionally display intrinsic activity (ago-PAMs). 21 Furthermore, PAMs may induce functional selectivity for signaling pathways not inherent to the natural ligand. 21 Another characteristic of allosteric modulators is their saturable effect, which has the potential to fine-tune receptor signaling, as well as minimizing risks such as drug overdosing. 21 Thus, the search for allosteric scaffolds provides a novel opportunity to identify the first orally bioavailable drug candidates modulating SCTRs.
Here, we outline the development of a four-stage testing funnel with the capability to detect, validate, and characterize biologically relevant SCTR small-molecule modulators. Our approach includes the design of a diverse selection of PAM primary screening assays differing not only in their detection method but also in their orthosteric stimulus response, which have been compared by screening a 12,000-compound small-molecule library. We also established a set of secondary assays to validate and characterize promising scaffolds regarding target engagement and functional selectivity. The results demonstrate how the combination of assays arranged in the testing funnel led to the discovery of the first low-molecular-weight, nonpeptidyl SCTR-specific PAMs.
Materials and Methods
Peptides and Ligands
Sec-FL (full-length human secretin [1–27]; cat. 4031250), GLP-1 (GLP-1 trifluoroacetate salt; cat. 4030663), and AVP ([Arg 8 ]-vasopressin trifluoroacetate salt; cat. 4012215) were obtained from Bachem AG (Bubendorf, Switzerland). Secretin 1–23 (Sec(1–23); HSDGTFTSELSRLREGARLQRLL-OH) and secretin 3–27 (Sec(3–27; DGTFTSELSRLREGARLQRLLQGLV-NH2) were custom synthesized by Biopeptide (San Diego, CA). GLP-1(9–36) (cat. AS-65070) was obtained from Anaspec (Fremont, CA). BETP (4-(3-(benzyloxy)phenyl)-2-(ethylsulfinyl)-6-(trifluoromethyl)pyrimidine) was purchased from Sigma-Aldrich (St. Louis, MO; cat. SML0558; purity ≥98% [high-performance liquid chromatography]).
Cells and Culture Reagents
Chinese Hamster ovary (CHO-K1) cells and human embryonic kidney (HEK)-293(T) cells were obtained from ATCC (Manassas, VA). CHO-K1 cells were maintained in CHO cell growth media (Ham’s F-12K [Kaighn’s modification]; Corning Life Sciences, Tewksbury, MA; Cellgro cat. 10-025-CV), 5% fetal bovine serum (FBS) clone II (GE Healthcare Life Sciences, Marlborough, MA; Hyclone cat. SH30066.03), 1% penicillin (10,000 units)/streptomycin (10 mg) (Pen/Strep; Thermo Fisher Scientific, Waltham, MA; Gibco cat. 15140122), and 1%
The chemical library was obtained from BioAscent Discovery Ltd. (Newhouse, Lanarkshire, UK), consisting of 12,000 cluster centroids providing diverse representatives of an expanded 125,000 small-molecule compound BioAscent collection. Each of the scaffolds in the expanded library contains 10–30 analogs that help to rapidly validate scaffolds and establish nascent structure–activity relationships (SARs) through cherry-picked orders of hits and analogs as liquid stocks. The library was stored as 2 mM stocks in 100% DMSO.
All assays were performed at the Conrad Prebys Center for Chemical Genomics (CPCCG) High-Throughput Screening Facility.
cAMP Accumulation Assays
Cyclic adenosine monophosphate (cAMP) assays were performed using frozen stocks of parental CHO-K1 cells, SCTR- or AVP2R-overexpressing CHO-K1 cells, or GLP-1R-overexpressing HEK-293T cells, all derived from a single cell clone at a low passage number. After reaching 80%–90% confluency, cells were harvested using TrypLE Express, centrifuged, and resuspended in freeze media (10% DMSO in growth media). The final concentration of the cell stocks was 20 million cells/mL. Corresponding ligand standard curves were recorded for each cell batch to obtain adequate EC20 and EC95 concentrations.
General Procedure
The time-resolved fluorescence resonance energy transfer (TR-FRET)-based cAMP accumulation assays were performed according to the manufacturer’s instructions with a few modifications. In brief, orthosteric stimulator dilutions were prepared freshly in DMSO and transferred to Echo Qualified 384-well, low-dead-volume (384LDV) microplates (Labcyte, San Jose, CA) via a CAPP 16-channel pipette (CAPP, Nordhausen, Germany). Compounds and ligands were dispensed onto dry microplates with an Echo liquid handler (Labcyte). The final assay compound concentration was 10 µM for primary screening and hit confirmation (in triplicate) or 12.5 µM for 1025 hits and analogs (in triplicate). Frozen cell stocks were thawed quickly in a 37 °C water bath and diluted in stimulation buffer (HBSS [Hank’s Balanced Salt Solution with Ca2+ and Mg2+]; Gibco; cat. 24020117), 5 mM HEPES (hydroxyethyl piperazineethanesulfonic acid), 0.075% BSA (7.5% DTPA-purified bovine serum albumin; PerkinElmer Inc., Waltham, MA; cat. CR84-100), and 0.5 mM IBMX (3-isobutyl-1-methylxanthine; Sigma-Aldrich; cat. I5879) to obtain the desired cell density. Cells were dispensed using a Multidrop Combi dispenser (Thermo Fisher Scientific), centrifuged at 1000 rpm for 1 min, covered with a lid, and kept at room temperature (RT) for 30 min. cAMP standard dilutions (4-fold, 0–1 µM final) were prepared in the stimulation buffer and transferred to designated wells using a CAPP 16-channel pipette. Antifoam SE-15 (0.1%; Sigma-Aldrich; cat. A8582) was added to the cAMP detection buffer, which was subsequently filtered through a 40 µm cell strainer. Detection reagents were diluted according to the manufacturer’s manual (specific dilutions in supplementary information) and dispensed using a Combi dispenser. The plates were centrifuged at 1000 rpm for 1 min, covered with a lid, and read on a Pherastar Plus microplate reader (BMG Labtech, Ortenberg, Germany) with the HTRF (homogeneous time-resolved fluorescence) module after 30–60 min at RT. Data were uploaded and analyzed on CBIS (Chemical and Biology Information System software; ChemInnovation Software Inc., San Diego, CA). The EC20 of the ligand was set as the negative control and EC95 as the positive control. Further characterization was conducted using the TIBCO Spotfire software (PerkinElmer). Compounds with high fluorescence in the TR-FRET donor (reference) channel in comparison with the negative control wells were eliminated from further studies. Specific procedures are described in the Supplementary Information.
CRE-Luc2P Reporter Assay
Luciferase reporter assays were performed as described previously 22 with a few modifications. In brief, HEK-293 SCTR CRELuc cells were seeded in two T225 cell culture flasks and grown in HEK cell growth media (DMEM, 10% FBS, 1% Pen/Strep, and 1% L-glutamine). After 2 days, cells were detached using TrypLE Express, resuspended in phosphate-buffered saline (PBS), and centrifuged at 300g for 4 min. The cell pellet was resuspended in DMEM + 10% FBS. A cell suspension of 0.25 million cells/mL was seeded into tissue culture-treated 384-well microplates (Greiner Bio-One small volume 784080) via a Multidrop Combi dispenser at 5 µL/well. Plates were centrifuged at 500 rpm for 15 s and incubated overnight at 37 °C and 5% CO2. The next day, orthosteric stimulator dilutions were freshly prepared in DMSO (positive control: 156 pM Sec-FL final; negative control and compound wells: 5 pM Sec-FL final) and transferred to Echo Qualified 384LDV microplates via a CAPP 16-channel pipette. Compounds (25 nL/well) and ligands (5 nL/well) were dispensed onto microplates with Labcyte Echo, followed by incubation at 37 °C and 5% CO2. The final DMSO concentration was 0.60%. After 4 h, plates and Steady-Glo (Promega, Madison, WI) detection reagent were brought to RT for 15 min. Detection reagent was added via a Multidrop Combi dispenser (5 µL/well) and plates were centrifuged at 500 rpm for 15 s. Thereafter, assay plates were kept at RT protected from light for 15 min and luminescence was detected via a ViewLux ultra HTS Microplate Imager (PerkinElmer; 5 s read). Data were uploaded and analyzed using CBIS. The EC20 of the ligand was set as the negative control and EC95 as the positive control. Further characterization was conducted via TIBCO Spotfire. Cell line generation and clonal selection of HEK-293 SCTR CRELuc cells are described in the Supplementary Information.
TR-FRET SNAP-SCTR Binding Assay
Binding experiments were performed as previously described,23–26 with the following modifications.
Selected compounds A1, A9, B1, C1, and D1 were stored in 384LDV microplates in a desiccator as 16-point twofold dilutions in DMSO. Stock concentrations ranged from 0 to 10 mM. Fluorescein-labeled secretin (Fluo-Sec) and Sec-FL were diluted in DMSO and dispensed into a 384LDV plate. Ligand titrations were prepared in DMSO in adjacent wells. Using Labcyte Echo, Fluo-Sec was transferred (25 nL, 6 nM final) into all test wells of a 1536-well plate (Corning; cat. 3725), DMSO (25 nL, positive control), and Sec-FL (25 nL, 5 µM final, negative control) or ligand/compound titrations (25 nL, varying concentrations) were dispensed on top. Using a dounce homogenizer, thawed HEK-293 SNAP-SCTR membranes labeled with Lumi-4 Terbium cryptate (Cisbio Tag-lite) were diluted in binding buffer (10 mM HEPES, pH 7.4, 100 mM NaCl, 10 mM MgCl2, 1 mM ascorbic acid, and 0.2% BSA) to a final concentration of 5 µg/mL. Membrane solution was added via a Multidrop Combi dispenser at 5 µL/well. The plate was centrifuged 1000 rpm for 1 min and incubated at RT for 2 h. Competition binding/allosteric modulator titration was recorded by Pherastar FSX (LanthaScreen 520/490 module). Data were uploaded and analyzed via CBIS as well as via GraphPad Prism 8.4.0 (GraphPad Software Inc., San Diego, CA) by applying the equation “One site – Fit Ki” to determine equilibrium dissociation constants Ki of orthosteric ligands. Allosteric modulators were analyzed using the equation “Allosteric modulator titration” to obtain equilibrium dissociation constants Kb and cooperativity factors α. α =1 indicates neutral cooperativity, 0 < α < 1 indicates negative modulation, and α > 1 supports positive cooperativity. Experiments were performed in duplicate in at least three independent experiments. Procedures for cell line generation, clonal selection, and membrane preparation of HEK-293 SNAP-SCTR, as well as for saturation and dissociation binding experiments, are described in the Supplementary Information.
Calcium Flux Assay with FLIPR Calcium 6 Dye
SCTR-CHO-K1 cells were grown in the CHO cell growth media. After reaching 80%–90% confluency, cells were harvested using TrypLE Express, resuspended in growth media, and seeded into a 384-well plate (5000 cells/50 µL/well; Greiner Bio-One; cat. 781091). Plates were covered with lids, centrifuged at 500 rpm for 1 min, and incubated at 37 °C and 5% CO2 overnight. The next day, growth media was removed by an upside-down spin (300 rpm for 5 s). Immediately, 20 µL of FLIPR Calcium 6 dye (membrane-permeable Ca2+ indicator; Molecular Devices, San Jose, CA) in assay buffer (HBSS with Ca2+ and Mg2+ containing 20 mM HEPES, 0.1% BSA, and 2.5 mM probenecid [Sigma-Aldrich; cat. P-7861]) was added per well. The plate was centrifuged at 500 rpm 5 s and subsequently incubated at 37 °C and 5% CO2 for 2 h. Test ligand titrations were prepared on a 384LDV plate and then transferred to a 384-well NBS plate (Non-Binding Surface; Corning; cat. 3655). Assay buffer was added to the compound source plate using a Multidrop Combi dispenser to a final volume of 40 µL/well. Cell plates were equilibrated at RT for 30 min. Hamamatsu FDSS 7000 (Functional Drug Screening System; Hamamatsu Photonics, Hamamatsu City, Japan) was used for liquid dispenses and fast kinetic reads. Ten microliters of ligand solution was added to 20 µL of cells in dye media while monitoring fluorescence (3 min read, addition at 10th read interval, normal exposure FLUO3/4). All experiments were performed in duplicate in at least three independent experiments. Curve-fitting analysis was conducted by GraphPad Prism 8.4.0.
NanoBiT β-Arrestin-2 Recruitment Assay
β-Arrestin-2 recruitment assays were developed and performed as reported previously, 27 with a few modifications.
HEK-293 cells were seeded into a 6-well plate at a cell density of 0.3 M/well and were incubated overnight at 37 °C and 5% CO2. Following the manufacturer’s manual, pFC220K-SCTR-SmBiT and LgBiT-ARRB2, pFC220K-SCTR-SmBiT and ARRB2-LgBiT, or pFC220K-AVP2R-SmBiT and LgBiT-ARRB2 were transfected using TransIT-LT1 transfection reagent (Mirusbio, Madison, WI; cat. MIR2300) delivering 0.5 µg of DNA of each construct per well. After 24 h, cells were harvested using TrypLE Express, resuspended in growth media, centrifuged at 300g for 3 min, and resuspended in assay buffer (HBSS with Mg2+ and Ca2+, 5 mM HEPES, 0.1% BSA) to a final cell density of 0.4 million/mL. Cell suspension was dispensed in an AlphaPlate-384 (light gray, shallow well; PerkinElmer; cat. 6008350) using a Multidrop Combi dispenser. After centrifugation at 500 rpm for 15 s, 3 µL of NanoBiT detection reagent was added per well via Multidrop Combi. The plate was covered, centrifuged at 1000 rpm for 1 min, and monitored with Pherastar FSX (luminescence, kinetic mode, 0.2 s read) for 2 h or until the baseline appeared to stabilize. Ligand (Sec-FL, Sec(1–23), Sec(3–27), or AVP) titrations were prepared in DMSO in 384LDV plates. Utilizing an Echo liquid dispenser, ligand titrations (40 nL/well) were transferred into cell plates. In the case of characterization of PAMs, DMSO (20 nL) or compound A1 or B1 (20 nL, 12.5 µM final) was dispensed into sample wells before Sec-FL titration. The plate was covered and centrifuged at 1000 rpm for 1 min, and β-arrestin-2 recruitment was recorded via Pherastar FSX (luminescence, kinetic mode, 0.2 s read) for 30 min. Experiments were performed in duplicate or triplicate in at least three independent experiments and curves were fitted using GraphPad Prism 8.4.0. Plasmids, construct generation, and the clonal selection of HEK-293 SCTR-SmBiT LgBiT-ARRB2 are described in the Supplementary Information.
Results
Development of a Four-Stage Testing Funnel to Detect PAMs Targeting SCTRs
To enhance our chances for discovering orally bioavailable drug candidates targeting SCTRs, we focused our efforts on the identification of PAMs, which may not only potentiate the effect of secretin and related peptides, but also selectively channel activation of signaling pathways ( Fig. 1A ). We fortified our drug discovery program by developing a panel of diverse assays that build four essential stages for our screening campaign. Starting with high-throughput screening (HTS) and hit confirmation ( Fig. 1B ), it was our aim to develop a robust and efficient primary screening assay based on Gαs protein signaling, which represents the dominant physiological effect upon SCTR activation and leads to increased cAMP levels. It is well known in the HTS field that different assays detecting the same cellular processes would inadvertently produce distinct sets of hits; some of them may be enhanced by the detection approach used in the assay, while others may be enhanced by unique settings specific to each of these assays. 28 Therefore, we compared three different cAMP detection methods (PerkinElmer LANCE Ultra, Cisbio Gs Dynamic [Cisbio GsD], and Promega CRELuc2P reporter [CRELuc] technology 22 ) by performing pilot screens of a small-molecule library consisting of 12,000 compounds. Since PAMs may display reduced or no activity without an orthosteric ligand present,29–31 we developed PAM screening assays using a fixed concentration (EC10 to EC20; based on TR-FRET ratios or relative luminescence units [RLUs]) of agonist as the basal orthosteric stimulator response in compound and negative control wells. Since PAM assays are able to detect both PAMs and (allosteric) agonists, we additionally characterized compounds in agonist mode assays performed in the absence of peptide ligands to determine their intrinsic activity. The EC95 concentration of the peptide agonist served as a positive control in both cases. Similar approaches have been described for recent PAM screening efforts against other GPCRs.20,32
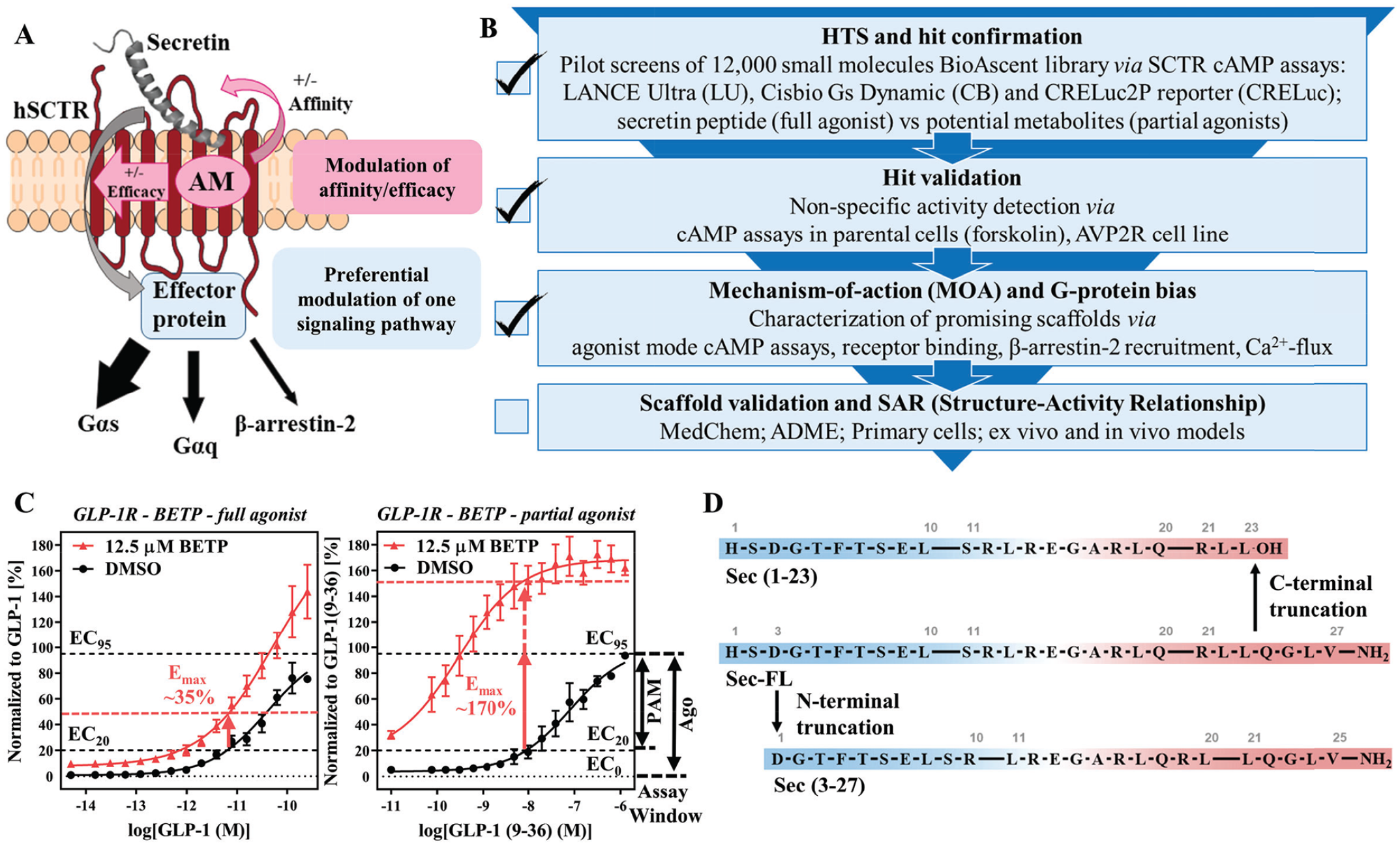
Strategies for the development of a testing funnel to detect PAMs targeting SCTRs. (
Beyond that, we evaluated the screening efficiency comparing full agonists and partial agonists. This was based on the observation of substantial probe dependency of GLP-1R PAMs, such as BETP,17,33 toward the partial agonist GLP-1(9–36), which is generated via N-terminal truncation of GLP-1 by DPP4 (dipeptidyl-peptidase-4). 34 Figure 1C illustrates benefits of using a partial agonist in the PAM assay. The extent of response from BETP in the presence of the EC20 concentration of GLP-1 ( Fig. 1C , left panel) is significantly lower than that observed with the partial agonist GLP-1(9–36) ( Fig. 1C , right panel). Intriguingly, BETP would be considered a weak hit with a 35% response using the full agonist as the basal stimulator, but would reach activity well beyond 100% when used with the partial agonist. Inspired by this increase of sensitivity, we explored potential secretin peptide metabolites. Sec-FL, like other peptide hormones, is known to be metabolized and cleared rapidly (half-life of ~2–4 min 8 ). Although there is no clear description of physiologic Sec-FL degradation products, we mimicked metabolites after known truncated versions of endogenous ligands targeting other class B GPCRs, which could serve as useful tools to evaluate the assay sensitivity and probe dependency of potential PAMs. For example, Sec-FL was shown to be degraded by NEP (neutral endopeptidase) 24.11, but no specific cleavage products were reported. 35 Another in vitro study suggested the formation of Sec(1–23) ( Fig. 1D , top) by VIP-degrading endoprotease through C-terminal cleavage of Sec-FL ( Fig. 1D , middle). 36 To explore effects of C- and N-terminal truncation of Sec-FL, we acquired Sec(1–23) and Sec(3–27) ( Fig. 1D , bottom). In stage 2 of our testing funnel ( Fig. 1B ) we validated hits by eliminating nonspecific activators. To that end, we translated our developed cAMP detection methods to cell lines not expressing SCTRs, such as parental cells and type 2 arginine vasopressin receptor (AVP2R)-bearing cells. AVP2R was selected, since it also couples to Gαs proteins while exerting physiologic effects contradictory to SCTR activation. 37 Besides being a class A GPCR, AVP2R should not be affected by SCTR targeting hits.
Development of Secondary Assays to Enable Broader Scaffold Validation and Compound Profiling
To strengthen our ability to validate and profile promising scaffolds regarding their mechanism of action (MOA), we developed and optimized secondary assays to evaluate the target engagement and functional selectivity of potential PAMs. Inspired by previously reported TR-FRET-based binding assays,23,24,38 we inserted a SNAP-tag to the N-domain of SCTR and created stably expressing HEK-293 cell clones. The construct was compared with a wildtype (WT) receptor by monitoring the agonist response in cAMP accumulation assays to ensure functional integrity (
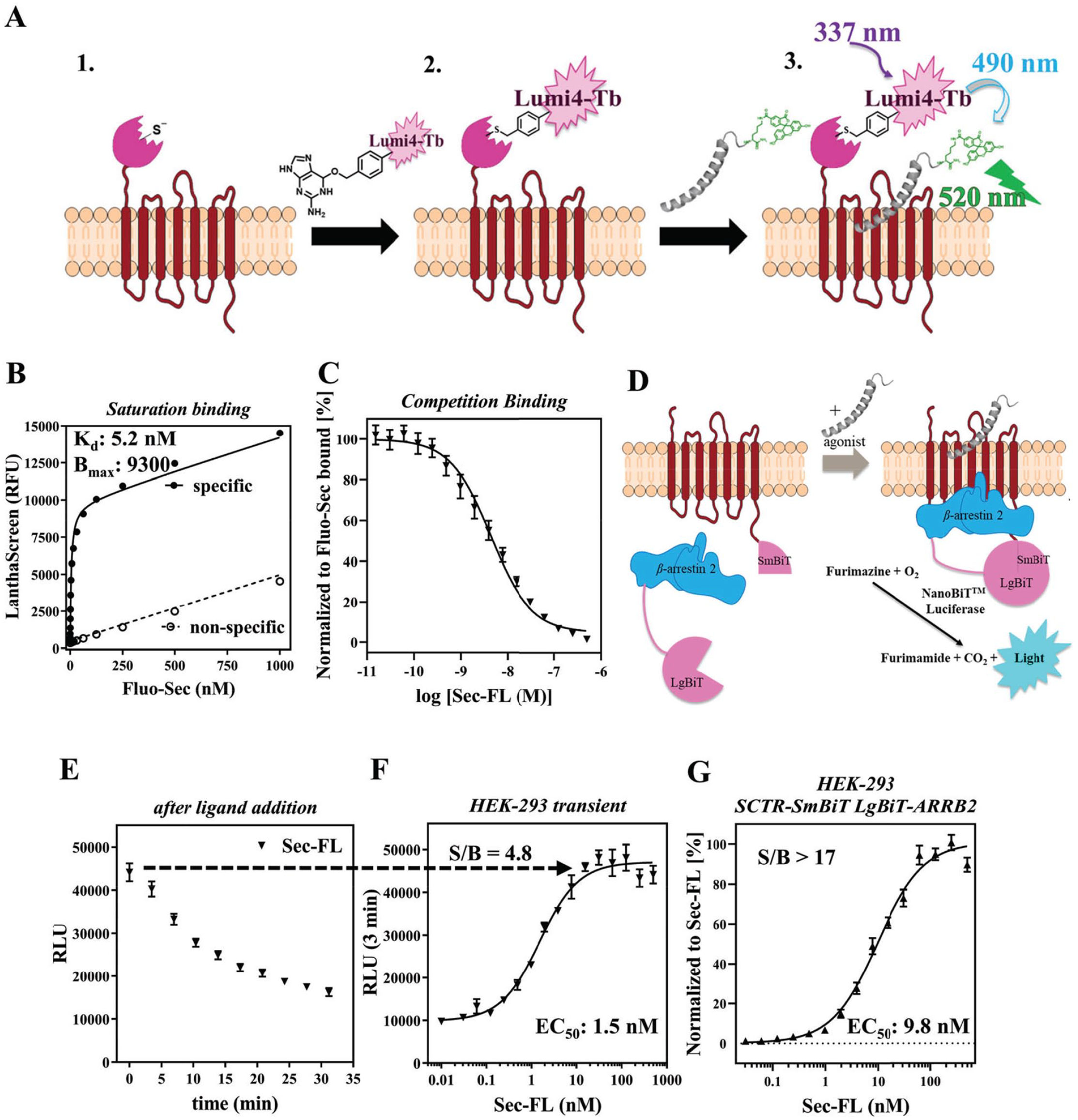
Development of GPCR-specific secondary assays to enable immediate scaffold validation and compound characterization. (
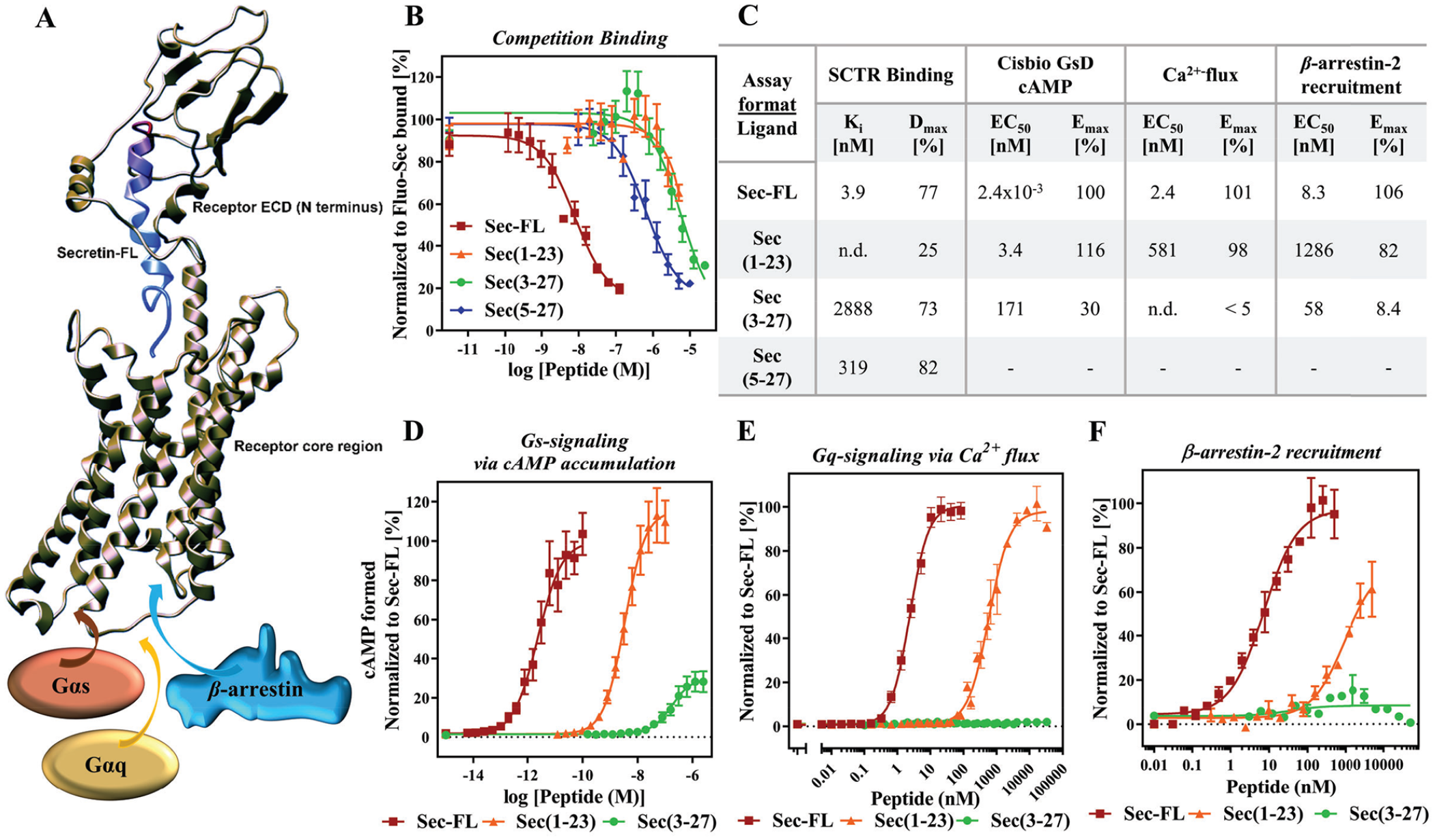
C-terminal truncation of secretin leads to a low-affinity, low-potency but fully efficacious agonist, whereas N-terminal cleavage drastically reduces efficacy among investigated signaling pathways. (
Since functional selectivity has been shown to provide potential therapeutic benefits relevant for GPCRs,39–43 we designed a β-arrestin-2 recruitment assay applying Promega’s NanoBiT technology.
44
As reported in a similar study on cannabinoid receptors,
30
we made use of an 11-amino acid peptide (Small BiT [SmBiT]) attached to the C-terminal end of the GPCR and the 17.6 kDa Large BiT (LgBiT) tag introduced either on the N- or C-terminal end of β-arrestin-2 (ARRB2). To validate our procedure, we generated and tested constructs for SCTR and AVP2R (
C-Terminal Cleavage of Secretin Leads to Low-Affinity, Low-Potency, but Fully Efficacious Agonist, whereas N-Terminal Truncation Drastically Reduces Efficacy among Investigated Signaling Pathways
After establishing a strong panel of assays, we validated the developed assays by characterizing our truncated secretin analogs Sec(1–23) and Sec(3–27). Several examples in recent studies revealed a two-site binding mode of hormones to class B GPCRs, which is also depicted in
Figure 3A
, illustrating a three-dimensional (3D) model
45
of secretin-bound SCTR. As shown previously,14,45,46 the C-terminal end of secretin peptide binds tightly to the large extracellular domain (ECD) of the receptor while directing the N-terminal end of secretin toward the helical core bundle, inducing intramolecular arrangements that lead to the activation of effector proteins, such as Gαs and Gαq proteins or β-arrestins. Considering this binding model, we were not surprised that compared with Sec-FL (red, Ki = 3.9 nM), Sec(1–23) (orange, Ki not determined) suffered from a slightly greater loss of affinity than Sec(3–27) (green, Ki = 2.9 µM). Interestingly, further cleavage of the N-terminal tail resulting in Sec(5–27) led to higher binding affinity (Ki = 319 nM) compared with other truncated analogs (
Stage 1.1: Pilot Screen of BioAscent Library Using LANCE Ultra cAMP Assay Revealed Probe Dependency of Hits Toward Secretin or Its Truncated Forms
Our screening efforts began with a pilot screen utilizing the PerkinElmer LANCE Ultra cAMP detection kit ( Fig. 4A ) against the 12,000-compound BioAscent library at a 10 µM concentration. Deploying Sec-FL as the orthosteric basal stimulator in PAM mode, we obtained 361 hits (3% hit rate). Hit confirmation studies (EC20 Sec-FL, 10 µM compound in triplicate) led to 106 confirmed hits. Purchase of these 106 hits and 919 analogs (5–15 analogs/scaffold, 1025 compounds total), selected to provide a nascent SAR for identified hits as liquid stocks from BioAscent and subsequent evaluation in hit confirmation format (EC20 Sec-FL, 10 µM compound in triplicate), yielded 368 hits (90 reconfirmed original hits and 278 active analogs). Eleven of these hits were eliminated by screening in parental CHO-K1 cells using the same detection method. To investigate the effects of the truncated secretin analogs, we performed in parallel three screens of 1025 hits and analogs in the PAM format with either Sec-FL, Sec(1–23), or Sec(3–27) as the orthosteric basal stimulator (EC20 for each peptide). These efforts resulted in a pool of 34 confirmed hits, and by elucidating the chemical structures we were able to identify five common scaffolds (scaffolds A–E). In-depth analysis of 34 confirmed hits via scatterplot and pie chart ( Fig. 4B ) revealed a probe dependency of the hits toward certain peptides. Two of 34 molecules have been confirmed with all three secretin analogs, whereas 30 hits were obtained from screens with individual peptides. The pie chart illustrates the distribution of scaffolds among the individual peptides and indicates that scaffolds B and E could be found by employing either one of the peptides, whereas the confirmation of scaffolds A, C, and D was dependent on the orthosteric stimulator applied. It should be pointed out that scaffolds A, B, and C have related chemical structures, mainly differing from their R2 substituents at the pyrimidine ring ( Fig. 4C ), strengthening their credibility as hits. While implementing and performing these assays, we observed high day-to-day variability in the detected level of stimulation with the same concentration of agonist. For example, screening the same compound set of 1025 hits and analogs in the Sec-FL PAM mode resulted in two very different hit rates, 368 hits in the first run (indicated as squares in the scatterplot) and 21 hits in the second run. After in-depth attempts to troubleshoot, we established that minor experimental variances in cell density and the concentration of the agonist resulted in a disproportionally high variation of signal due to the high slope of the standard curve, resulting in a narrow dynamic range. We addressed this issue by comparing calibration curves of cAMP detected by either the LANCE Ultra or Cisbio GsD cAMP kit ( Fig. 4D ), which is advertised to perform with a greater dynamic range. The assay dynamic range was determined by a cAMP concentration range between IC10 and IC90 of the standard curve. Indeed, in contrast to LANCE Ultra (slope ~ 1.5; dynamic range = 0.18–3.52 nM cAMP), the CisBio GsD detection kit demonstrated a slope close to unity and a much broader dynamic range (slope ~ 0.9; dynamic range = 0.22–38.2 nM cAMP). Greater sensitivity and a steeper slope were also observed in the dose–response curves of secretin peptides detected with the LANCE Ultra cAMP kit ( Fig. 4E ). We decided that the small dynamic range of the LANCE Ultra kit is a major drawback for performing larger screens in the PAM mode, since it is crucial to ensure consistent baseline stimulation.
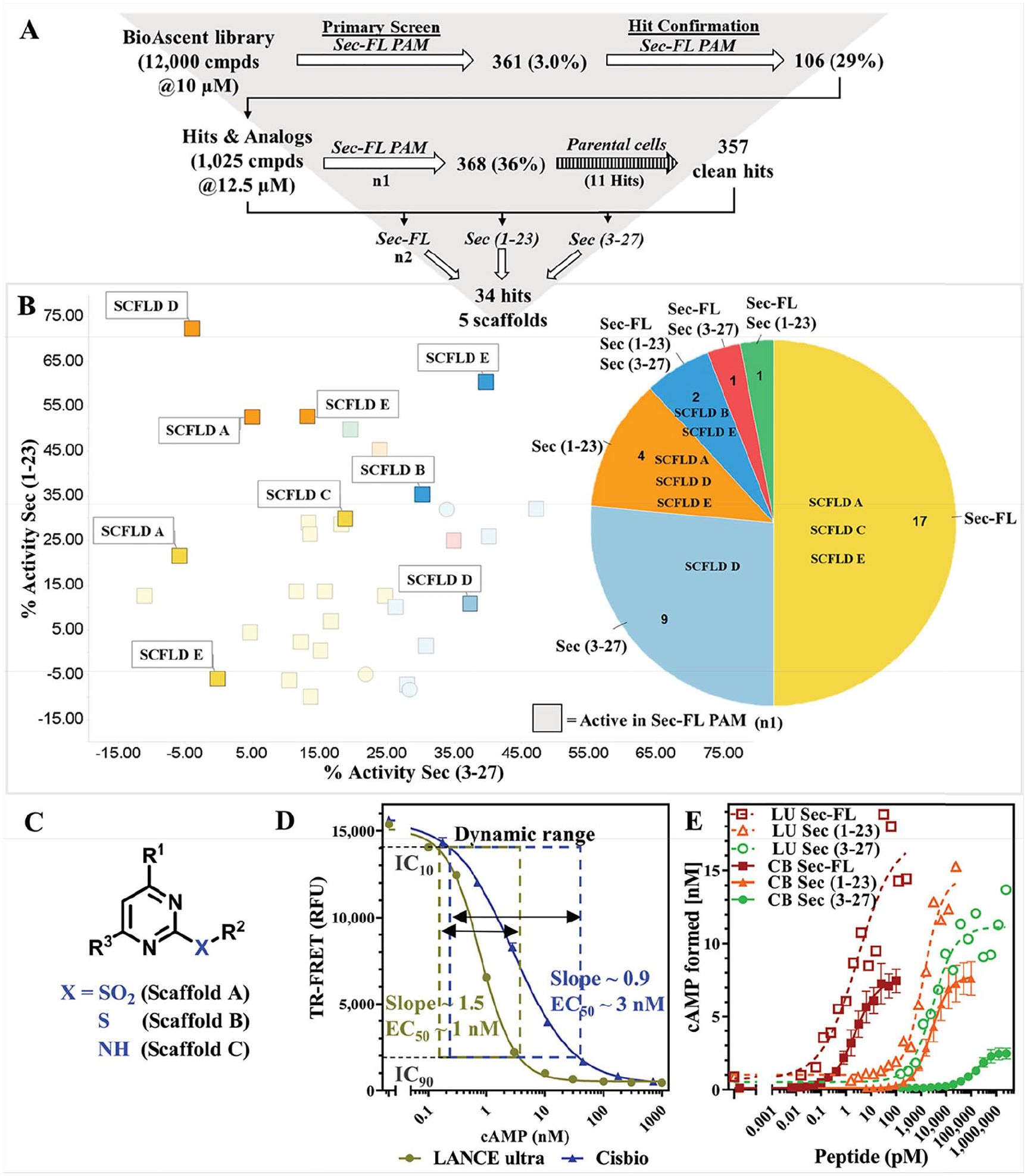
Pilot screen of BioAscent library employing the LANCE Ultra cAMP assay revealed theprobe dependency of hits toward secretin or its truncated forms. (
Stage 1.2: Pilot Screens Comparing Three Different Detection Methods and Two Sets of Ligand Probes Elucidate the Strength of Combining Sec-FL, Sec(1–23), and Sec(3–27) as the Orthosteric Stimulator
To explore further primary screening options, we tested the 12,000-compound BioAscent collection in three further formats (
Fig. 5A
). Taking into account the probe dependency and potentially increased sensitivity for stimulating with truncated peptides, we decided to perform in parallel a screen with full agonist Sec-FL and a screen with a mixture of EC10 concentrations of all three peptides (3-peptide mix), which resulted in a basal stimulation comparable to EC20 of Sec-FL alone. To obtain better reproducibility and a greater dynamic range, we used the Cisbio GsD cAMP kit as the detection method. We were also interested in applying a technology that is distinct from TR-FRET detection of intracellular cAMP. Hence, we selected the CRELuc system with a luminescence-based detection system for our third pilot screen employing Sec-FL as the basal stimulator. All four methods performed well in the primary screen, having decent to good Z′ factors (0.54–0.61) and reasonable S/B ratios (
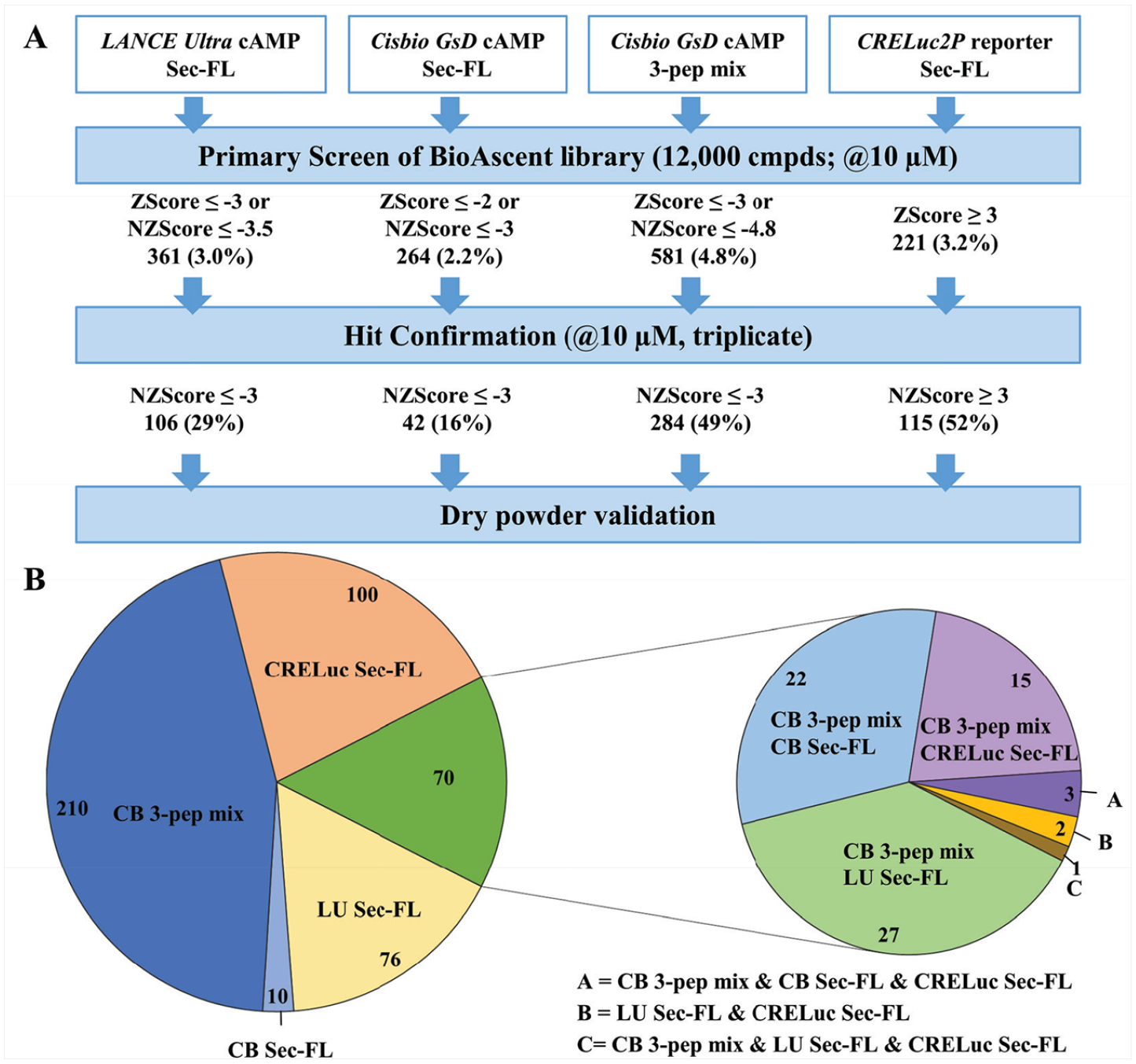
Pilot screens comparing three different detection methods and two sets of ligand probes elucidate the strength of combining Sec-FL, Sec(1–23), and Sec(3–27), that is, the 3-peptide mix as the orthosteric stimulator. (
Stages 2 and 3: Molecules with Scaffolds A–D Are SCTR-Selective PAMs
Moving into stages 2 and 3 of our testing funnel (
Fig. 1B
), we purchased dry powders of 110 compounds for hit validation. In the course of this study, we focused on two compounds (A1 and A9) containing scaffold A; two compounds (B1 and C1) with closely related structures, scaffold B and C, respectively; and one compound (D1) incorporating scaffold D. To explore intrinsic activity, we recorded dose–response curves in SCTR-CHO cells in the agonist mode (
Fig. 6A
, left panel). In this setting, we detected negligible activity for compounds A1, B1, and C1. Performing the same assay except stimulating cells with an EC20 concentration of Sec-FL (
Fig. 6A
, middle panel), we were able to measure dose–response curves for all five compounds with potencies in the single-digit micromolar range and efficacies ranging from 20% (D1, purple) to 40% (A1, dark green) (
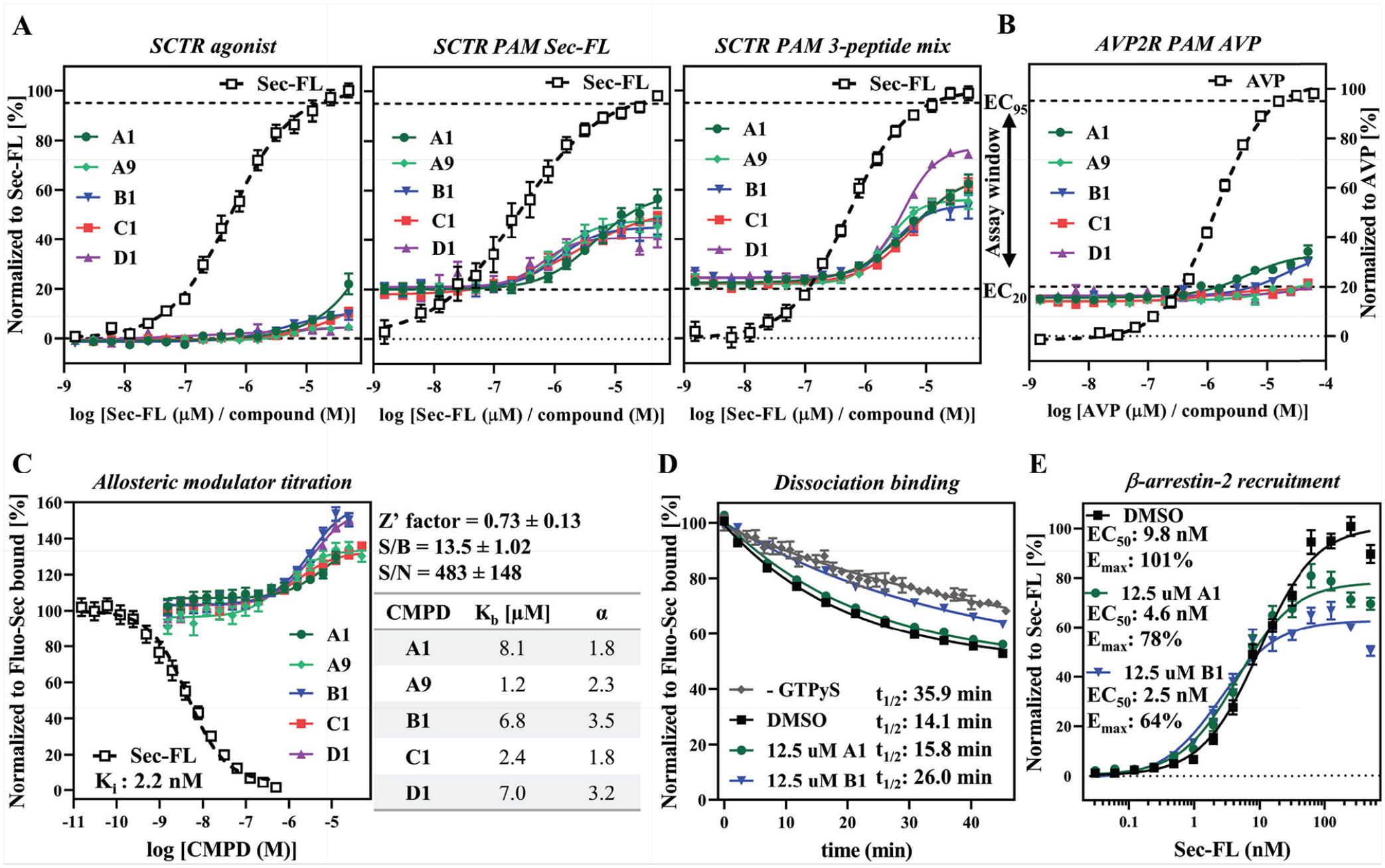
Validation and characterization of scaffold. (
Binding assays are useful tools not only to dissect orthosteric from allosteric ligands but also to determine allosteric activity parameters.
14
In a competition binding-type experiment we detected a dose-dependent increase of Fluo-Sec binding for all five compounds supporting an allosteric binding site on SCTRs (
Discussion
Comparison of Methods Reveals Key Features for a Successful Screen to Identify PAMs
Here we report the development and results of a testing funnel that incorporates a structured comprehensive toolbox to identify, validate, and characterize low-molecular-weight nonpeptidyl SCTR modulators. To date, no other small-molecule compounds have been described to interact with SCTRs. Hence, it was crucial to develop and select a robust and global primary screening method. By comparing not only three different detection methods (LANCE Ultra, Cisbio GsD, and CRELuc), but also the effect of individual as well as mixed full and partial peptide agonists deployed as orthosteric basal stimulators, we were able to evaluate the most critical aspects for successful implementation of an SCTR PAM screening assay based on pilot screens comprising 12,000 compounds. Our goal was to establish a primary screening assay with great sensitivity, reproducibility, and hit detection range. Initial efforts were conducted applying LANCE Ultra cAMP technology stimulating with Sec-FL. While demonstrating great assay sensitivity and good assay performance in the 1-day primary screen, the approach suffered from a lack of day-to-day reproducibility, likely due to a narrow dynamic range of the detection kit. Hence, we decided to test orthogonal approaches relying on intracellular cAMP accumulation, such as Cisbio GsD and CRELuc detection systems. Despite having a broader dynamic range, the Sec-FL Cisbio GsD assay demonstrated lower assay sensitivity, which led to a significant reduction in the number of confirmed hits. Utilizing the luminescence-based CRELuc reporter system resulted in a higher assay sensitivity, an improved S/B ratio, and a higher number of hits. These factors, beyond the lower cost of detection reagents, make it attractive for large-scale HTS campaigns. However, the overlap of CRELuc-derived hits and hits confirmed by TR-FRET-based methods was only around 30%, indicating substantial differences in the nature of hits.
Novel Application of 3-Peptide Mix Results in Higher Assay Sensitivity and Allows Identification of SCTR PAMs with Distinct Probe Dependencies
The profiling of 1025 hits and analogs via LANCE Ultra cAMP employing Sec-FL or truncated secretin peptides Sec(1–23) and Sec(3–27) revealed that the response of potential PAMs is dependent on the nature of the orthosteric stimulator. Although this phenomenon has already been described for PAMs acting on other GPCRs, 33 there is no such pattern described for SCTRs. This might be due to the poorly understood metabolism of Sec-FL, which might vary depending on its site of expression. However, potential secretin metabolites are likely to be cleavage products from either end of the full-length peptide; thus, we focused our studies on one analog truncated at the C-terminal tail 36 (Sec(1–23)) and one analog designed as a hypothetical N-terminal degradation product (Sec(3–27)). Due to the development of a comprehensive set of SCTR binding and functional assays, we were able to thoroughly characterize our metabolite models, revealing that C-terminal peptide truncation leads to a lower affinity, but fully functional analog, whereas N-terminal cleavage results in an analog exerting only partial SCTR activation. These distinct activity profiles in combination with the modification of receptor occupancy and restricted binding space can be utilized to increase the sensitivity and effectiveness of PAM screening assays. 32 Looking ahead, PAMs able to enhance activity of truncated partially active versions of Sec-FL might be particularly useful for therapeutic applications by extending the functional response of the natural ligand after its degradation and inactivation.20,30,32 Our studies with individual secretin peptides in the LANCE Ultra PAM screening assay elucidated that each peptide finds distinct sets of compounds. Since the nature of physiological metabolites remains unclear, we decided to combine all three analogs into one orthosteric stimulator probe (3-peptide mix) to identify PAMs that are likely to work with any potential metabolite. Due to the broader dynamic range, we employed the Cisbio GsD detection kit to conduct the screen of the BioAscent library in 3-peptide mix format. The primary screen worked well with a good Z′ factor and decent S/B ratio of around 3. Moreover, Cisbio GsD 3-peptide mix was not only able to identify the largest number of hits but also empowered to detect 97% of overlapping hits, which were found utilizing Sec-FL in LANCE Ultra, Cisbio GsD, or CRELuc assays. Hence, the Cisbio GsD 3-peptide mix assay is a robust, reproducible, sensitive, and effective primary screening assay.
Secondary Assays Reveal That Identified Scaffolds Exert Different MOAs
The pool of overlapping hits provided five interesting scaffolds, four of which were investigated in detail. Dry powders of compounds A1, A9, B1, C1, and D1 were validated in dose–response studies on SCTRs regarding their ability to accumulate cAMP with no orthosteric ligand, Sec-FL, or 3-peptide mix basal stimulation. In general, compounds showed no to marginal intrinsic activity in agonist mode, but significant responses in PAM modes. Intriguingly, compounds A1 and A9 demonstrated comparable activities in both Sec-FL and 3-peptide mix cAMP assays, whereas compound D1 exerted greater effects with mixed agonists, therefore suggesting distinct MOAs. Compounds A1 and B1 appeared to produce slight responses in CHO-AVP2R cells stimulated with AVP; however, compound B1 displayed 18-fold selectivity toward SCTRs. Of note, structural analog A9 was inactive in the AVP2R-PAM cAMP assay, proposing that specificity is achievable through structural modification. The development of a fluorescence-based target engagement assay enabled the characterization of compounds on SCTR binding in a high-throughput environment. We were excited to see that all five compounds were able to increase Fluo-Sec binding in a dose-dependent manner. Resulting cooperativity factors α revealed further subtle differences between investigated scaffolds. Compound A1 displayed lower α values in the allosteric modulator titration and only slightly decreased Fluo-Sec dissociation rates in SNAP-SCTR dissociation binding experiments. However, compound B1, exhibiting stronger positive cooperativity to Fluo-Sec, was able to exert comparable deceleration of Fluo-Sec dissociation-like coupled G proteins, the most widely studied endogenous PAM targeting GPCRs. To explore potential signaling bias, we also subjected compounds A1 and B1 to Sec-FL-stimulated β-arrestin-2 recruitment studies. Both compounds reduced efficacy but were able to slightly enhance Sec-FL potency. Whether this phenomenon is due to functional selectivity of PAMs or a nonspecific interaction with the assay will be addressed in future studies.
Testing Funnel Identifies the First Small-Molecule Modulators Described for SCTRs
The combination of a diverse selection of primary screens, intensive comparison of their primary screen performance, design of GPCR-specific secondary assays, and consequential arrangement of funnel stages resulted in the discovery of PAMs targeting SCTRs. One of the main novelties of our testing funnel was the development of the 3-peptide mix stimulator that not only substantially enhanced assay sensitivity and effectiveness but also allowed the detection of probe-dependent hits. Another unique advantage was the development and implementation of GPCR-specific secondary assays, which led to immediate profiling and validation of distinct sets of hits with respect to target engagement and functional selectivity. This is the first report, to our knowledge, describing small-molecular-weight compounds that not only interact with SCTRs but also significantly enhance the binding and response of secretin peptides. Encouragingly, discovered hits cover effects on both natural peptide ligand and metabolite models, considerably increasing chances for potential therapeutic application.
Future Directions: Testing Funnel Stage 4
In this study we discovered bona fide SCTR PAM hits. We continue working with identified scaffolds in further hit validation and optimization studies by extending structural variety through analog-by-catalog and medicinal chemistry efforts. Like many HTS-suitable cell-based assays, the use of receptor overexpressing cell lines may result in overly amplified or distorted signaling. Thus, hits will be confirmed using cell models with endogenous SCTR expression and signaling. Hit-to-lead studies involving further optimization of MOA, pharmacokinetics, and toxicity profiles are expected to identify analogs suitable for in vivo testing. Beyond that, future work will include the expansion of GPCR selectivity screens not only to detect possible off-target effects but also to explore the potential of class B GPCR polypharmacology, 49 which might provide additional benefits for the treatment of metabolic disorders.
Supplemental Material
Supplementary_Information_Testing_Funnel_SCTR-PAMs_by_Dengler_et_alR – Supplemental material for Development of a Testing Funnel for Identification of Small-Molecule Modulators Targeting Secretin Receptors
Supplemental material, Supplementary_Information_Testing_Funnel_SCTR-PAMs_by_Dengler_et_alR for Development of a Testing Funnel for Identification of Small-Molecule Modulators Targeting Secretin Receptors by Daniela G. Dengler, Qing Sun, John Holleran, Sirkku Pollari, Jannis Beutel, Brock T. Brown, Aki Shinoki Iwaya, Robert Ardecky, Kaleeckal G. Harikumar, Laurence J. Miller and Eduard A. Sergienko in SLAS Discovery
Footnotes
Acknowledgements
We would like to thank personnel of the Conrad Prebys Center for Chemical Genomics (CPCCG) at the Sanford Burnham Prebys Medical Discovery Institute (SBP) for help in diverse aspects of this project.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We acknowledge support from the U.S. National Institutes of Health (NIH) National Heart, Lung, and Blood Institute (NHLBI) grant R01HL133501.
Supplemental material is available online with this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.