
Abstract
K+ channels play a critical role in maintaining the normal electrical activity of excitable cells by setting the cell resting membrane potential and by determining the shape and duration of the action potential. In nonexcitable cells, K+ channels establish electrochemical gradients necessary for maintaining salt and volume homeostasis of body fluids. Inward rectifier K+ (Kir) channels typically conduct larger inward currents than outward currents, resulting in an inwardly rectifying current versus voltage relationship. This property of inward rectification results from the voltage-dependent block of the channels by intracellular polyvalent cations and makes these channels uniquely designed for maintaining the resting potential near the K+ equilibrium potential (EK). The Kir family of channels consist of seven subfamilies of channels (Kir1.x through Kir7.x) that include the classic inward rectifier (Kir2.x) channel, the G-protein-gated inward rectifier K+ (GIRK) (Kir3.x), and the adenosine triphosphate (ATP)-sensitive (KATP) (Kir 6.x) channels as well as the renal Kir1.1 (ROMK), Kir4.1, and Kir7.1 channels. These channels not only function to regulate electrical/electrolyte transport activity, but also serve as effector molecules for G-protein-coupled receptors (GPCRs) and as molecular sensors for cell metabolism. Of significance, Kir channels represent promising pharmacological targets for treating a number of clinical conditions, including cardiac arrhythmias, anxiety, chronic pain, and hypertension. This review provides a brief background on the structure, function, and pharmacology of Kir channels and then focuses on describing and evaluating current high-throughput screening (HTS) technologies, such as membrane potential-sensitive fluorescent dye assays, ion flux measurements, and automated patch clamp systems used for Kir channel drug discovery.
Introduction
K+ channels are transmembrane proteins that provide a high conductance pathway for the movement of potassium ions down their electrochemical gradient. In excitable cells of the heart and brain, these channels play a critical role in maintaining normal electrical activity by setting the cell resting membrane potential and by determining the shape and duration of the action potential.1–4 In nonexcitable cells of the kidney, these channels maintain ionic gradients required for transepithelial transport.5,6 K+ channels have traditionally been divided into two major categories defined as voltage-gated (Kv) and inward rectifier (Kir) channels.1–4 Kir channels conduct larger inward currents at membrane voltages negative to the K+ equilibrium potential (EK) than outward currents at voltages positive to EK. This property of the channels results in an inwardly rectifying current versus voltage relationship. This inward rectification is believed to result from the voltage-dependent block of the channels by intracellular magnesium or organic polyamines at potentials positive to EK. The Kir family of channels consists of seven subfamilies of channels (Kir1.x through Kir7.x) (KCNJ1–16 genes), which include the classic cardiac and skeletal muscle inward rectifier (Kir2.x), the G-protein-gated inward rectifier K+ (GIRK) (Kir3.x), and the adenosine triphosphate (ATP)-sensitive (KATP) (Kir6.x) and renal (Kir1.1, Kir4.1, and Kir7.1) channels.2,7,8 GIRK channels are found in cardiac atrial tissue, as well as throughout the peripheral and central nervous systems, where they mediate cellular actions of drugs and neurotransmitters that bind to muscarinic, opioid, dopamine, serotonin, and other G-protein-coupled receptors (GPCRs). Opening of the GIRK channel results from the binding of the Giβγ subunit to the cytoplasmic domain of the channel. KATP channels, expressed in the heart, brain, and pancreas, are inhibited during elevations in intracellular levels of ATP and therefore function to couple metabolism to cell excitability. Finally, renal Kir channels control the transepithelial potential in the kidney and thereby act to regulate electrolyte homeostasis of the blood and urine.
Given the role of Kir channels in stabilizing the cell resting membrane potential and serving as downstream effectors for GPCRs (GIRK channels) and as metabolic sensors (KATP channels), it is not surprising that mutations in these channels give rise to a number of channelopathies. For example, loss-of-function mutations in the Kir2.1 channel cause Andersen–Tawil syndrome, which is characterized by cardiac arrhythmias and skeletal muscle myotonia. 9 Patients with Andersen–Tawil syndrome can develop cardiac long QT syndrome, leading to ventricular tachycardia and sudden death. 10 A point mutation in the pore-forming region of the Kir3.2 channel is found in the weaver mouse and causes a loss of K+ selectivity.11,12 Activation of the mutated channel results in neuronal depolarization (from Na+ influx through the channel) with cell death and gives rise to the ataxic gait and “weaving” motion observed in these mice. Finally, cardiac KATP channelopathies are associated with atrial fibrillation (AF), dilated cardiomyopathy, and sudden infant death. 13 In addition, gain-of-function mutations in the Kir6.1 channel and the associated sulfonylurea receptor (SUR1) cause Cantu syndrome, a rare disorder with clinical symptoms including hypertrichosis, heart valve abnormalities, and congenital hypertrophic cardiomyopathy.14,15
This review provides a brief background on the structure, function, and pharmacology of Kir channels with an emphasis on Kir1.1, Kir2.x, Kir3.x, Kir4.1, and Kir6.x channels. This is followed by a description and evaluation of current high-throughput screening (HTS) technologies used for Kir channel drug discovery. Readers wishing for a more in-depth description of the molecular physiology of Kir channels are directed to a number of excellent reviews.2,7,8,16
Structure, Function, and Pharmacology of Kir Channels
The first Kir channels to be cloned were named ROMK1, IRK1, and GIRK, corresponding to the Kir1.1, Kir2.1, and Kir3.1 channels.17–19 As is the case for Kv channels, Kir channels consist of a pore-forming α subunit ( Fig. 1A ). The α subunits are composed of a membrane-spanning ion pore that provides a conductive pathway for K+ movement across the cell membrane and a selectivity filter that allows K+ passage through the channel while excluding other ions. Kir channel α subunits consist of two transmembrane domains (TM1 and TM2) connected by a pore-forming loop region (P) ( Fig. 1A ). The pore region contains the glycine–tyrosine (phenylalanine)–glycine signature sequence that confers K+ selectivity to the channel. Kir channels are formed through a tetrameric arrangement of subunits with the TM2 helix from each of the subunits defining the pore region. Channels can be composed of four identical (homotetramer) or different (heterotetramer) subunits, with heterotetramer formation normally restricted to subunits of the same Kir subfamily. For example, Kir3.1 subunits will form heterotetramers with Kir3.2, Kir3.3, and Kir3.4 subunits. Heterotetramer formation can confer distinct kinetic properties to the channel and may enhance the activity of the channel. Co-expression of Kir3.1 subunits with Kir3.2 subunits greatly increases the size of the resulting inward rectifier currents when compared with currents recorded from the expression of Kir3.2 alone. 20
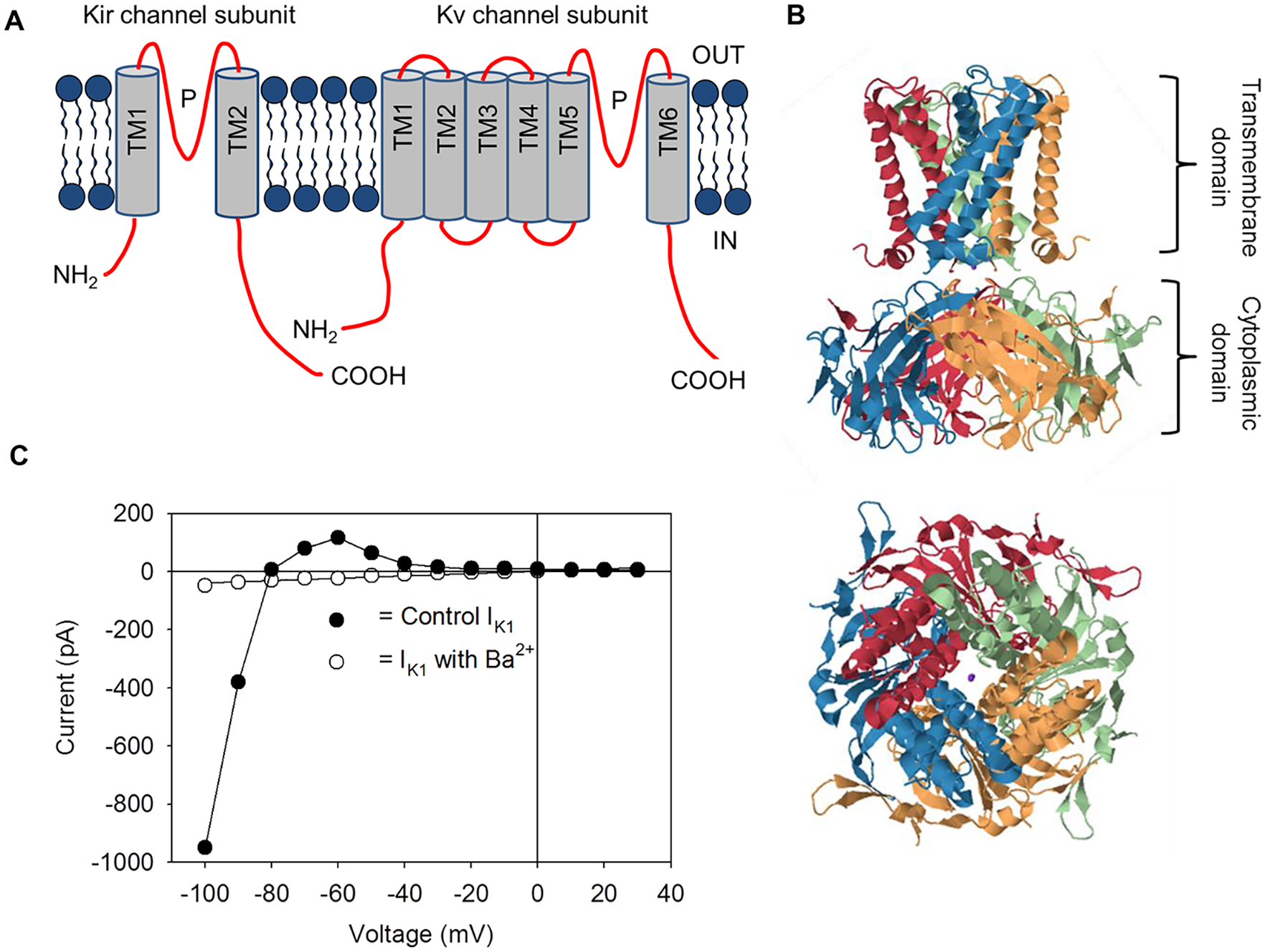
Structural and functional properties of Kir channels. (
Over the past 20 years a number of important structural versus functional details of Kir channels have been provided through x-ray crystallography ( Fig. 1B ). Both the TM2 and TM1 α-helices play critical roles in the gating of Kir channels.21–23 Comparisons between the GPCR-activated Kir3.x channel and the constitutively active Kir2.x channel indicate that mobility of the TM2 helix around a hinge point contributes to Kir channel gating. 24 Structural differences between the Kir3.x channel and Kir2.x channel suggest that activation of the Kir3.x channel, following the binding of the Giβγ subunit, causes a rotation in each of the four TM2 helices, bringing the channel into an open conformation equivalent to that of the active Kir2.x channel. 25 In addition to the transmembrane regions, putative cytoplasmic binding sites for the Giβγ subunit, nucleotides, and phosphatidylinositol 4,5-bisphosphate (PIP2) have been identified.24,25 Binding of PIP2 is postulated to stimulate the engagement of the cytoplasmic domain of the channel with the transmembrane domain, resulting in the opening of the pore. This interaction of PIP2 with the channel is of special significance since the presence of PIP2 on the intracellular surface of the plasma membrane is necessary for the activation of all Kir channels.26,27 Recent cyro-electron microscopy (Cyro-EM) has revealed putative PIP2-binding residues located at the ends of the TM1 and TM2 helices of the Kir6.2 channel.28,29 Interestingly, these residues are close to the inhibitory ATP binding site on the Kir6.2 subunit. Binding of one molecule (e.g., PIP2) might therefore constrain the binding site of the other molecule and result in antagonistic effects of the two molecules on channel opening (as previously observed 30 ).
All members of the Kir channel family display the property of inward rectification.2,7 Figure 1C shows the current versus voltage relationship for the Kir2.1 channel (IK1) recorded from a cardiac ventricular myocyte using the whole-cell arrangement of the patch clamp technique. The current “reverses” direction from an inward to an outward current at approximately −82 mV; this is close to EK (−95 mV) under the conditions of the experiment. The reversal of the current at a potential close to EK indicates that the channels underlying the whole-cell current are selective for K+. At potentials negative to the reversal potential, the Kir channels pass a large amount of inward current. In contrast, at potentials positive to the reversal potential, the channels pass only small outward currents. Therefore, small hyperpolarizations from the cell resting membrane potential will result in inward Kir currents, while small depolarizations from this potential will induce outward currents. In both situations, the flow of current will shift the membrane potential back toward the reversal potential, thus functioning to maintain the cell resting potential close to EK. Of course, under physiological conditions the resting potential for most cells is positive to EK, and thus outward current through the Kir channels will predominate.
Early studies concluded that strong inward rectification of Kir channels results from a combination of intracellular block by Mg2+ and an “intrinsic gating” mechanism. Insight into the nature of this intrinsic process first came from macropatch experiments in which patches of membranes containing multiple Kir2.1 channels were excised from cells.31,32 Before excision of the macropatch the Kir2.1 channels showed strong rectification. However, removal of the patch into a divalent cation-free solution resulted in the gradual loss of rectification. Interestingly, strong rectification was restored when the macropatch was brought back to the cell membrane or exposed to small organic amines. It was subsequently discovered that intracellular polyamines such as spermine, spermidine, and putrescine are responsible for the intrinsic gating mechanism of inward rectification.31,32 Thus, the affinity of Mg2+ and polyamines for binding sites in the TM2 helix and cytoplasmic domain determines the degree of inward rectification.16,23 This gives rise to strong inward rectifiers (such as the Kir2.x and Kir3.x channels) and weak inward rectifiers (such as the Kir1.1 and Kir6.x channels).
While the biophysical properties of Kir channels have been extensively studied, we currently possess only an elementary knowledge of Kir channel pharmacology. Classical K+ channel blockers used experimentally to characterize Kv and Kir channels include inorganic cations, quaternary ammonium compounds, and aminopyridines. In general, Kv channels are sensitive to 4-aminopyridine (4-AP) and relatively insensitive to tetraethylammonium (TEA) salts and Ba2+. In comparison, Kir channels are marked by their sensitivity to Ba2+ (see Fig. 1C ) and Cs+, but are insensitive to 4-AP. Some Kir channels are blocked by TEA, but only at millimolar concentrations. Several peptide toxins are also effective in blocking Kir channels.33,34 The most potent of these is tertiapin, a peptide isolated from the venom of the bee Apis mellifera. Tertiapin has been widely used as an inhibitor of Kir3.x and Kir1.1 channels.33,34 The following section highlights our current knowledge of Kir2.x, Kir3.x, Kir6.x, and renal Kir channel function, tissue distribution, and pharmacology (see also Table 1 ).
Structure, Tissue Expression, and Pharmacology of Kir Channels in Excitable Tissues.
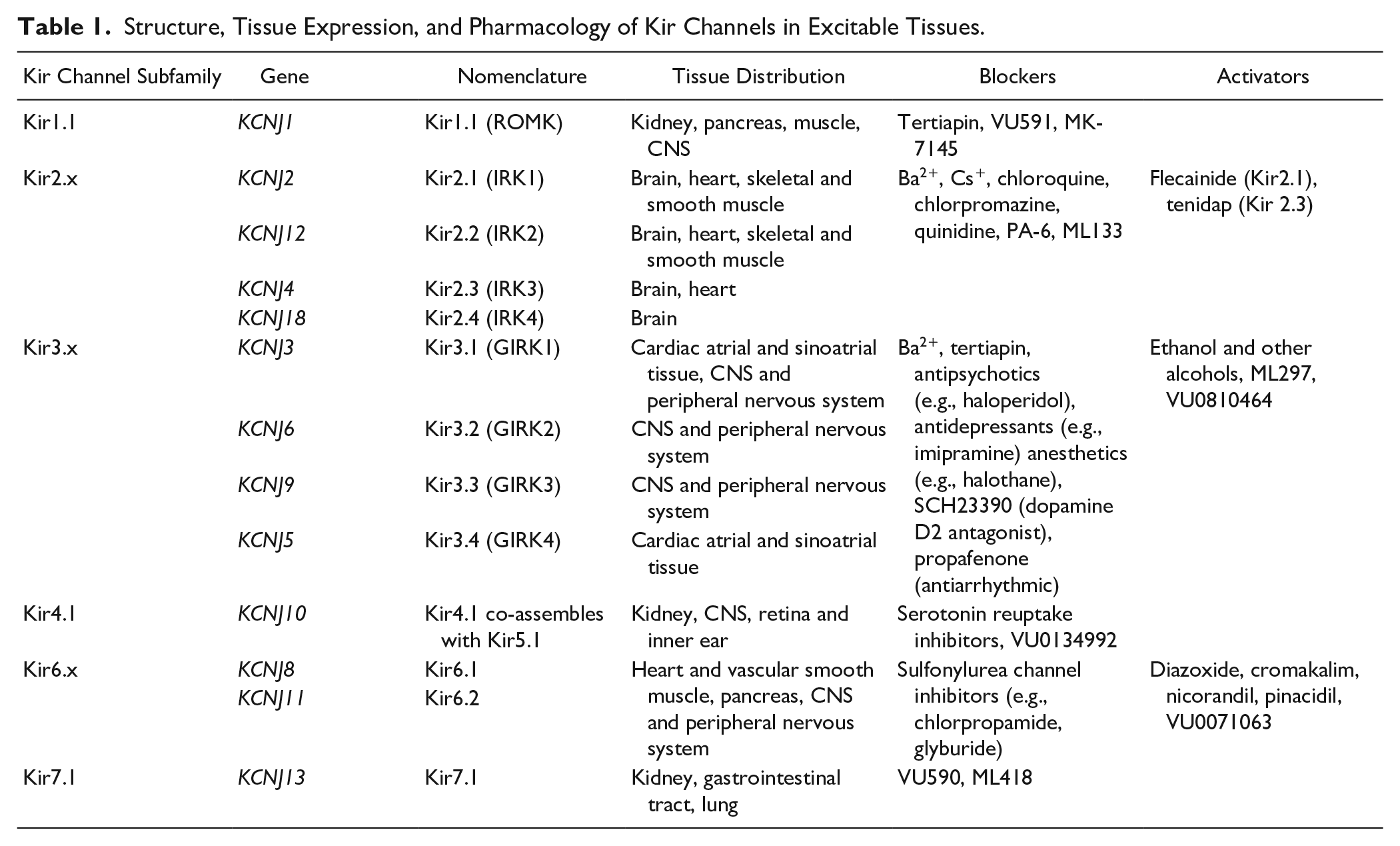
Kir2.x Channels
Kir2.1 and Kir2.2 channels underlie the classic cardiac inward rectifier current (IK1) that is expressed in ventricular and atrial myocytes. In addition to maintaining the resting membrane potential (see above), these channels contribute to the late repolarization phase of the cardiac action potential. Kir2.x channels are also expressed in skeletal muscle and in regions of the brain including the forebrain and cerebellum. In addition to their presence in brain neuronal tissues, Kir2.1 channels are expressed at high levels in brain astrocytes. 35 It is postulated that astrocytic Kir channels function to buffer increases in extracellular K+, which occur during periods of neuronal hyperexcitability and therefore may reduce the risk of seizure activity. 36
Several drugs are known to modulate Kir2.x channels. Block of the IK1 by the antimalarial drug chloroquine is associated with the development of fatal ventricular arrhythmias. Block of IK1 by chloroquine occurs in a voltage-dependent manner with an IC50 of approximately 9 µM.37,38 The antiarrhythmic drugs quinidine and flecainide also interact with the ventricular IK1, with quinidine blocking the channel in the micromolar concentration range. Surprisingly, Caballero and colleagues 39 found that flecainide augments Kir2.1 currents (EC50 = 1 µM) by binding to a cysteine residue in the cytoplasmic domain and reducing polyamine-induced rectification. Although the magnitude of the flecainide augmentation of IK1 is small, drug regulation of polyamine binding might represent a novel approach for treating some types of cardiac arrhythmias. Takanari et al. 40 reported that the diamine pentamidine analogue 6 (PA-6) blocks IK1 in ventricular myocytes (IC50 in the 50–200 nM range) but is without effect on voltage-gated Na+ and Ca2+ currents. In addition, PA-6 produces small but nonsignificant decreases in the cardiac transient outward K+ current (Ito) and the fast (IKr) and slow (IKs) delayed rectifier channels. 40 Finally, ML133 was discovered as a Kir2.1 inhibitor (IC50 = 1.8 µM) in an HTS campaign using a thallium (Tl+) flux assay combined with automated patch clamp (APC) experiments (see below). 41 Of particular relevance, ML133 shows marked selectivity for Kir2.x channels compared with Kir1.1, Kir4.1, and Kir7.1 channels. 41 The potency of ML133 (pKa = 8.7) in blocking IK1 is enhanced at basic pH but reduced at acidic pH, indicating that ionization of ML133 limits access of the compound to its binding site. 41
Kir3.x Channels
The Kir channel subunits Kir3.1 and Kir3.4 (GIRK1/4) underlie the acetylcholine-activated inward rectifier current (IK,Ach) found in cardiac atrial and nodal myocytes. Binding of acetylcholine and other muscarinic agents to the muscarinic M2 receptor causes a dissociation of the Giβγ subunits (from the Giα subunit), which subsequently bind to and activate IK,Ach.42,43 In the central nervous system (CNS) and peripheral nervous system, Kir3.1, Kir3.2, and Kir3.3 subunits are expressed in areas including the amygdala, ventral tegmental area, cortex, hippocampus, cerebellum, and spinal cord.44,45 Kir3.1/Kir3.2 (GIRK1/2) heterotetramers are the most abundantly expressed GIRK channels in the CNS and are activated by a large number of neuromodulators including somatostatin, dopamine, endorphins, and endocannabinoids.8,46 Activation of the GIRK1/2 channels in the presynaptic nerve terminal following GPCR stimulation causes the presynaptic resting membrane potential to become more negative. As a result, there is a decrease in spontaneous action potential formation and an inhibition of excitatory neurotransmitters released.8,46
Both cardiac and neuronal GIRK channels have been explored as potential drug targets. NTC-801, a selective inhibitor of IK,Ach, was developed by Nissan Chemical and Teijin Pharmaceuticals (Tokyo, Japan) for the treatment of AF. 47 IK,Ach is constitutively active or upregulated in some patients with AF.48,49 The constitutively active Kir3.1/Kir3.4 channel causes the atrial action potential duration to shorten with a resulting increase in cardiac excitability. NTC-801 (IC50 = 10 nM for inhibiting IK,Ach) and other IK,Ach inhibitors decrease atrial excitability and reduce the incidence of AF when tested in canine models of AF.47,49 Although NTC-801 (renamed BMS 914392) did not advance beyond phase 2 trials, recent studies suggest that drugs that bind to the Kir3.4 subunit may be more effective in treating AF. 49
In contrast to cardiac IK,Ach inhibitors, neuronal Kir channel drug discovery has concentrated on identifying GIRK channel openers. Investigators at Vanderbilt University synthesized a number of asymmetrical urea compounds displaying neuronal Kir3.x subunit selectivity. One such compound, ML297, shows moderate selectivity as a Kir3.1/3.2 channel opener (EC50 = 0.3–1 µM) and possesses anxiolytic and antiepileptic activity in rodent behavioral models.50,51 Recently, nonurea GIRK channel openers, including VU0810464, have been developed that show greater Kir3.1/3.2 channel selectivity. 52 Accordingly, VU0810464 is selective in activating GIRK channels in neurons but not cardiomyocytes. 52
Kir6.x Channels
KATP channels were discovered in cardiomyocytes as K+-selective channels that are activated under conditions that cause an increase in intracellular ADP, but are inhibited during increases in ATP. 53 KATP channels consist of four Kir6.2 or four Kir6.1 subunits and regulatory sulfonylurea receptor (SUR) subunits.2,13 The SUR subunits are members of the ATP-binding cassette (ABC) transporter family of proteins that contain nucleotide-binding domains (NBDs). The SUR subunits confer sulfonylurea and K+ channel opener (KCO) sensitivity to the channels.2,13 The Kir6.2 channel, along with the SUR1 subunit, is expressed in β-pancreatic cells where they play a critical regulatory role in insulin secretion (see below). 54 KATP channels are also expressed throughout the CNS including the pituitary gland, basal ganglia, cerebral cortex, hippocampus, and basal forebrain.55,56 Both the Kir6.1 and Kir6.2 subunits are expressed in the brain. In the hypothalamus KATP channels play an important role in regulating the release of hormones such as glucagon and epinephrine.57,58 When glucose levels fall in the brain, hypothalamic neurons experience a decrease in intracellular ATP levels with a subsequent activation of KATP channels.57,58 The resulting neuronal hyperpolarization stimulates the autonomic nervous system, causing the release of glucagon and epinephrine that serve as counterregulatory hormones to insulin and increase blood glucose levels.
The availability of both KCOs and sulfonylurea inhibitors has provided a valuable tool for studying the function of KATP channels in a variety of tissues.2,13 KCOs include diazoxide, cromakalim, nicorandil, and pinacidil, which activate the channel upon binding to the SUR subunit. The ability of these drugs to open KATP channels is regulated by intracellular nucleotide levels. For example, nicorandil requires the presence of ADP in order to open the cardiac channel. 59 In addition, KCOs can reduce the inhibition of the channel caused by intracellular ATP. 60 Sulfonylureas include first-generation (chlorpropamide, tolbutamide, etc.), second-generation (glyburide, glipizide, etc.), and third-generation (glimepiride) chemicals. Unlike KCOs, binding of sulfonylureas to the SUR subunits inhibits the opening of the channels. Using Cyro-EM, Martin et al. 61 identified a structural binding pocket for sulfonylureas (e.g., glyburide) present in the SUR1 subunit. Residues from TM7, TM8, and TM11 of the SUR1 transmembrane domain 1 (TMD1) and residues from TM15 and TM17 (of TMD2) contribute in forming the glyburide binding pocket. 61 Binding of sulfonylureas is predicted to stabilize the SUR1 subunit in a conformation that prevents its structural interaction with Kir6.2 and thus inhibits channel opening.
Sulfonylurea KATP channel inhibitors are widely prescribed in the treatment of type 2 diabetes mellitus.
54
Inhibition of the pancreatic β-cell KATP channel by sulfonylureas results in membrane depolarization and the opening of
Renal Kir1.1, Kir4.1, and Kir7.1 Channels
Renal epithelial Kir channels play an essential part in maintaining electrolyte levels and fluid volume in the body.5,6 Kir1.1 (ROMK) channels are expressed in the apical surface of epithelial cells found in the thick ascending loop of Henle (TAL) and the cortical collecting duct (CCD) of the nephron. In the TAL, the Kir1.1 channels provide the K+ concentration gradient across the apical membrane needed for the effective functioning of the Na+/K+/2Cl– co-transporter. In the CCD, the Kir1.1 channels provide a major secretory pathway for K+ elimination from the body. In addition, by providing a favorable electrochemical gradient for operation of the Na+/K+-ATPase, K+ efflux through the Kir1.1 channel is coupled to the activity of the amiloride-sensitive epithelial Na+ channel. Loss-of-function mutations in the Kir1.1 channel result in Bartter’s syndrome, a life-threatening disorder associated with salt wasting, metabolic alkalosis, and hypokalemia. Of significance, patients who are heterozygote carriers of these loss-of-function Kir1.1 mutations have a reduced blood pressure and a decreased risk of developing hypertension. 64 Thus, it has been hypothesized that pharmacological inhibitors of the Kir1.1 channel might represent a new class of diuretic and antihypertensive agents.
Tertiapin, isolated from the venom of the honeybee, was the first potent inhibitor of the Kir1.1 channel (Ki = 2 nM). 33 However, as noted above, tertiapin also blocks Kir3.x.channels (Ki = 9 nM). 33 HTS assays combined with medicinal chemistry have successfully been applied to develop new small-molecule inhibitors of the Kir1.1 channel. Bhave and colleagues identified VU591 as the first potent (IC50 = 240 nM) and selective small-molecule blocker of the Kir1.1 channel. 65 In silico modeling experiments suggested an energetically favorable interaction between VU591 and an arginine residue found within the pore of the channel. 66 The subsequent mutation of this positively charged arginine to a negatively charged residue was found to eliminate the blocking activity of VU591. Based on the structures of piperazine carboxamide and piperazine diamine compounds obtained from earlier screens of the Kir1.1 channel, Tang and coworkers at Merck developed MK-7145. 67 This compound displayed good Kir1.1 selectivity over other Kir channel subfamilies and selectivity over cardiac ion channels, including the ether-a-go-go-related gene (hERG) channel. In addition, MK-7145 displayed oral bioavailability and lowered the systolic blood pressure in spontaneously hypertensive rats. The efficacy of MK-7145 in lowering blood pressure was comparable to that of the diuretic hydrochlorothiazide.
In contrast to Kir1.1 channels, Kir4.1 and Kir7.1 channels are expressed in the basolateral surface of the distal convoluted tubule (DCT) and CCD of the nephron.5,6 Kir4.1 channels either form homomeric channels or assemble with Kir5.1 to form heteromeric Kir4.1/Kir5.1 channels. Kir5.1 channels do not form functional channels when expressed alone. However, co-assembled Kir4.1/Kir5.1 channels have a higher single-channel conductance and greater pH sensitivity than homomeric Kir4.1 channels. 68 In the DCT and CCD, Kir4.1 (and Kir4.1/Kir5.1) channels function to recycle K+ across the basolateral membrane and thus maintain a driving force for renal Na+ reabsorption via the basolateral Na+/K+-ATPase transporter. It is therefore not surprising that mutations in Kir4.1 (e.g., SeSAME disease) result in NaCl loss and plasma hypokalemia. In addition to their expression in the kidney, Kir4.1 channels are found in the CNS, retina, and inner ear. While less is known about the renal Kir7.1 channels, it is speculated that they serve a similar physiological function as Kir4.1 channels.
The selective serotonin reuptake inhibitor (SSRI) fluoxetine and the tricyclic antidepressant nortriptyline are among a number of CNS drugs that nonselectively block Kir4.1 channels. 69 Inhibition of the Kir4.1 channels by the drugs is concentration dependent and reversible with an IC50 of 76 μM for fluoxetine and 93 μM for nortriptyline. Block by fluoxetine occurs in a voltage-independent manner, while nortriptyline inhibition is voltage dependent. Both drugs are selective in blocking the Kir4.1 channels over Kir1.1 channels. Mutations of residues Thr128 and Glu158, located within the pore and TM2 helix of the channel, respectively, markedly reduce the inhibition by the drugs. 69 Of special note, Zaika et al. 70 found that dopamine produces a natriuretic action in the kidneys by inhibiting Kir4.1 (Kir4.1/Kir5.1) channels. As described in the following section, developments in HTS technologies have enabled the discovery of new Kir inhibitors and activators. The structure of some of these compounds is shown in Figure 2 .
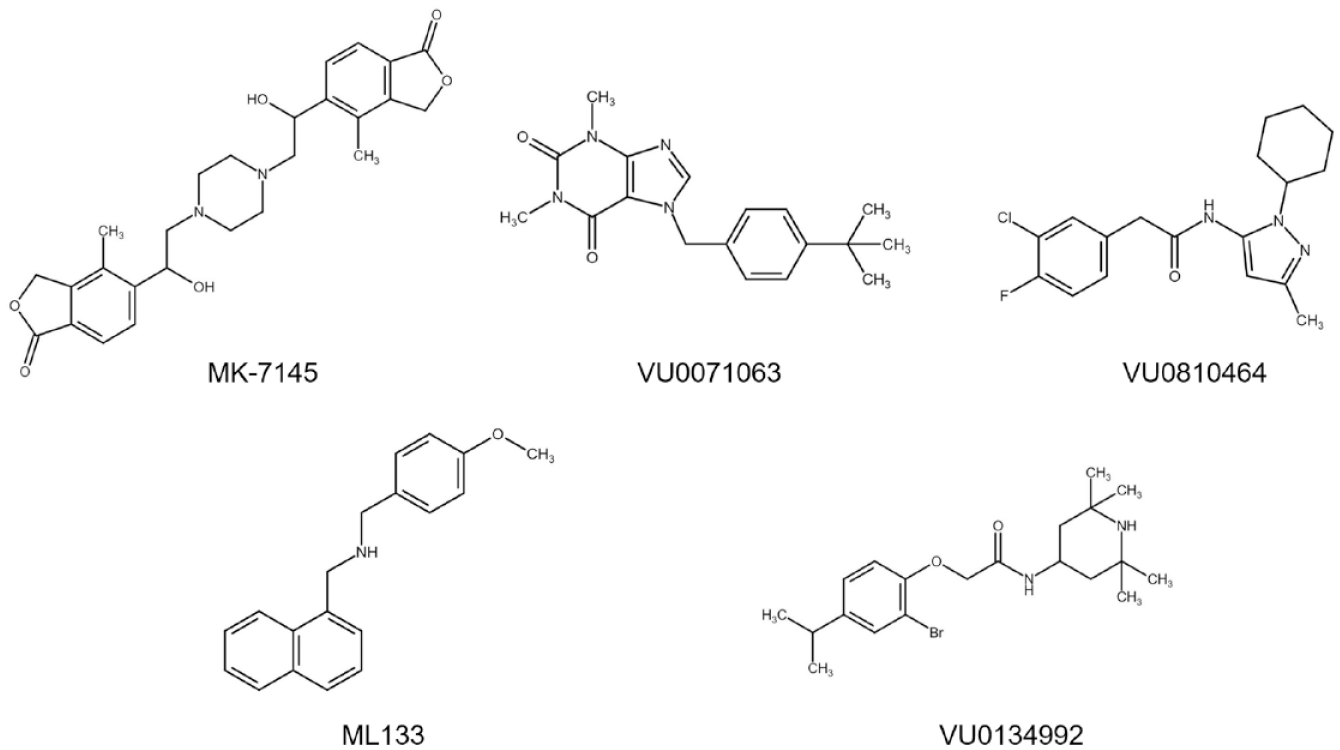
Kir channel inhibitors and activators. Chemical structures of MK-7145 (Kir1.1 inhibitor), VU0071063 (Kir6.2/SUR1 activator), VU0810464 (Kir3.1/Kir3.2 activator), ML133 (Kir2.1 inhibitor), and VU0134992 (Kir4.1 inhibitor).
Screening Technologies for Kir Channels
A number of technologies including membrane potential-based fluorescent dye assays, ion flux analysis, and electrophysiology are used for HTS of Kir channels ( Fig. 3 ). While fluorescence-based measurement of Tl+ fluxes has become the most widely applied methodology for studying K+ channel pharmacology, APC systems are now reaching the high-throughput capacity needed for screening large chemical libraries. Other approaches, such as monitoring yeast cell growth and measuring potassium fluxes from Kir channel-containing liposomes, have also been utilized to study specific Kir channels.
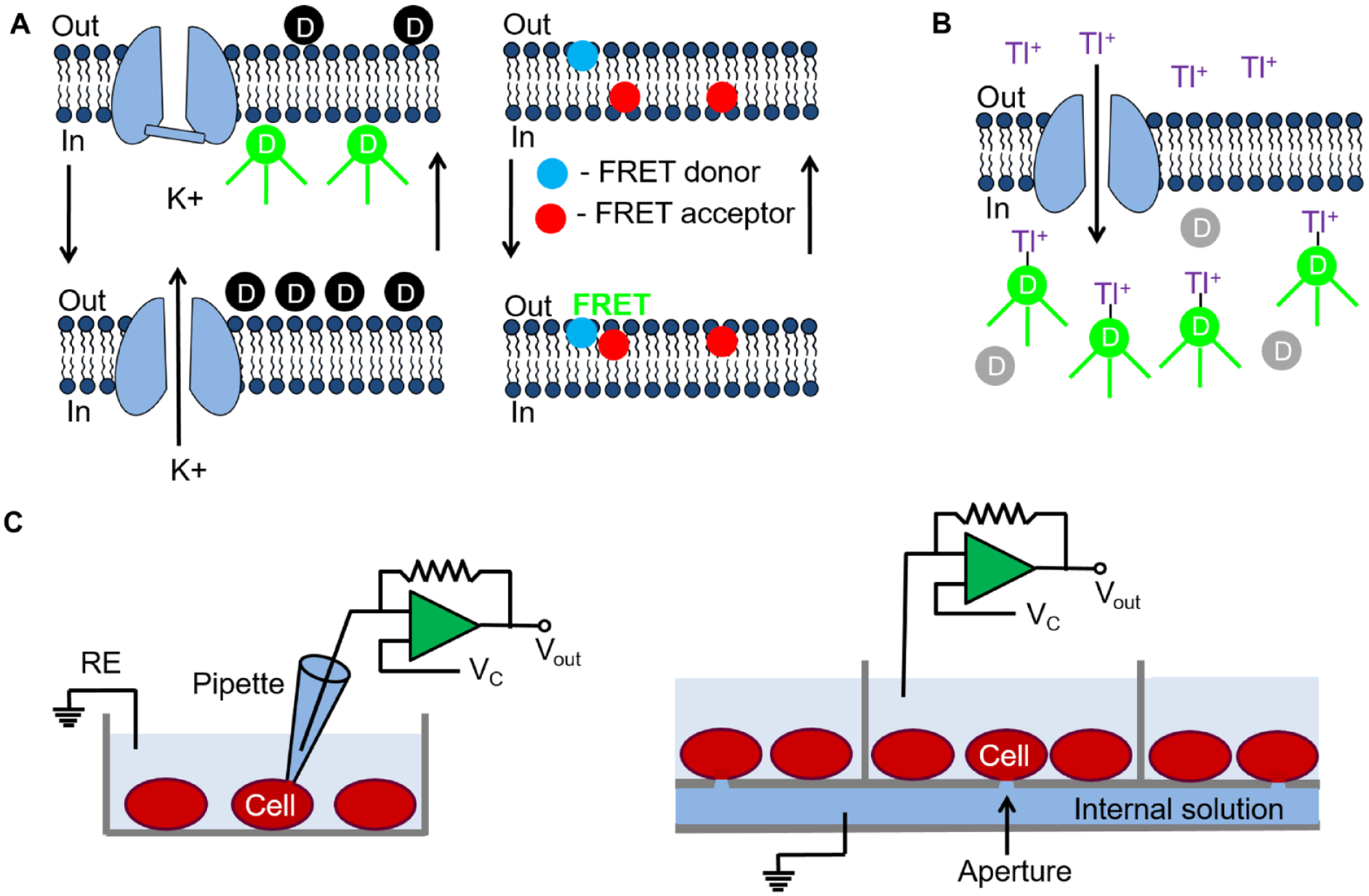
HTS technologies used for discovering Kir channel modulators. (
Fluorescent MPSD Assays
Membrane potential-sensitive dyes (MPSDs) have been utilized in a number of primary screening assays of Kir channels. Membrane potential can be an extremely sensitive indicator of K+ channel modulation since relatively small changes in K+ currents can produce relatively large changes in voltage. There are two types of MPSDs: fast-response and slow-response probes. Fast-response probes respond to a change in the membrane potential through a change in their electronic structure that produces a corresponding change in their fluorescent properties ( Fig. 3A ). This includes fluorescence resonance energy transfer (FRET) sensors. Fast-response probes detect fast (millisecond) changes in the membrane potential, such as during an action potential. However, the magnitude of the fluorescence change for the fast-response probes is usually small. Slow-response probes exhibit potential-dependent changes in their transmembrane distribution ( Fig. 3A ). While these probes detect slow (second) changes in the membrane potential, the change in the fluorescent signal is usually large. This make these MPSDs ideal for screening Kir channels expressed in clonal cell lines or in nonpolarized HEK293 and CHO cells.
In a commonly used MPSD screening protocol, heterologous cells expressing a Kir channel of interest are incubated with a negatively charged fluorescent oxonol dye, such as bis-(1,3-dibutylbarbituric acid) trimethine oxonol DiBAC4(3), which distributes across the plasma membrane ( Fig. 3A ). The oxonol molecules inside the cells become strongly fluorescent upon binding to intracellular proteins and other cytoplasmic components. Activation of Kir channels with the subsequent K+ flux through the channel causes a change in the membrane potential. As a result, the MPSD molecules redistribute across the plasma membrane with a resulting increase or decrease in the fluorescent signal. Newer proprietary MPSDs produced by Molecular Devices (FLIPR Membrane Potential Assay kit, San Jose, CA) and Anaspec (HLB 021-152, Fremont, CA) provide faster response times and larger fluorescent signals. In addition, kits containing an MPSD along with a fluorescent quencher have been introduced that allow “no-wash” assays for HTS. This reduces well-to-well variations that can result from plate washing-induced loss or disruption of cell attachment in the wells.
Wolff et al. 71 demonstrated the feasibility of using MPSDs for measuring the drug block of an endogenous Kir2.1 channel in RBL-2H3 cells. A similar approach was used for examining the pharmacology of KCOs and sulfonylureas on KATP channels expressed in urinary bladder smooth muscle and neonatal rat cardiac cells.73,74 MPSDs have also been used for screening endogenous Kir3.1/Kir3.2 and Kir3.1/Kir3.4 channels expressed in immortalized pituitary AtT20 cells and atrial HL-1 cells. 75 In addition to screening Kir3.1/Kir3.2 channels, MPSD assay systems have been utilized in AtT20 and HEK293 cells to examine the efficacy of GPCR ligands such as somatostatin, opioids, and cannabinoids in activating GIRK channels.76–78
While fluorescent MPSDs provide a convenient and inexpensive approach for measuring changes in membrane potential, they represent an indirect measure of Kir channel activity. This method contrasts with ion flux analysis and patch clamp measurements that directly measure channel flux or current (see below). The lipophilic nature of oxonol dyes results in a lack of membrane selectivity, causing them to respond to membrane potential signals not only from the plasma membrane but also from other cellular membranes. In addition, MPSDs are sensitive to changes in temperature and can be perturbed by test compounds and compound solvents (DMSO, ethanol, etc.). This can make MPSD assays disposed to high rates of false-positive results. As a consequence, strong secondary screening of identified compounds is required. 79 Finally, FRET-based sensors may interfere with the change in membrane potential caused by current flow through the Kir channels.
Ion Flux Analysis
Radioactive 86Rb+ flux assays have long been successfully employed in the screening of KATP channels and in examining the properties of the Kir6.1 and Kir6.2 channels.80,81 In addition, the chemical screening of inhibitors of Kir1.1 channels has utilized 86Rb+ flux assays. 67 In this procedure, cells are first loaded with 86Rb+ and the cells and supernatant are collected following KATP channel activation for radioactive counting. Certainly, the major drawback to this procedure is the inconvenience and cost associated with the handling of radioactive reagents. A nonradioactive Rb+ flux assay was developed by Aurora Biosciences for the analysis of KATP channels. 82 In this procedure, Rb+ efflux through the KATP channels is measured using atomic absorbance spectrometry (AAS). One advantage of the Rb+ flux assays is that the signal-to-noise ratio is high when compared with most MPSD fluorescent assays. However, consideration must be given to the time required for cell Rb+ loading and efflux measurements. In addition, unlike fluorescent and electrophysiological assays, the technique does not provide real-time experimental analysis since the concentration of the Rb+ must be determined post hoc with either a scintillation counter or AAS.
A Tl+-sensitive, fluorescent-based assay for multiwell plate screening of K+ channels was first introduced by Weaver and colleagues ( Fig. 3B ).83,84 In this procedure, cells are first loaded with a Tl+-sensitive, membrane-permeant reporter dye such as the BTC-AM or FLUXOR. The cells are then incubated in assay buffer containing varying amounts of K+ and Tl+. Since many Kir channels are permeable to Tl+, K+ channel modulators can be rapidly screened by monitoring changes in the Tl+-induced fluorescent signal ( Fig. 3B ). The Tl+ assay has been utilized as a primary screen for discovering activators and inhibitors of Kir3.1/3.4 and Kir3.1/3.2 channels, as well as Kir2.1 channels.41,50,51 As noted previously, this assay has been applied to identify compounds displaying neuronal Kir3.x subunit selectivity. Kir4.1 inhibitors and Kir6.2 channel activators have also been recently discovered and characterized using Tl+ assays. HEK293 cells expressing either Kir4.1 or Kir6.2/SUR1 channels were screened in 386-well format using the Tl+-sensitive dye Thallos-AM.85,86 VU0134992 (IC50 = 1 µM), the most potent Kir4.1 inhibitor identified in the screen, was 30-fold more selective for Kir4.1 channels over Kir1.1, Kir2.1, and Kir2.2 channels, but showed equal activity toward GIRK and Kir4.2 channels. 85 Through the use of whole-cell patch clamping, glutamate and isoleucine residues lining the pore of the Kir4.1 channel were found to be critical for the activity of VU0134992. This same group of investigators identified VU0071063 (IC50 = 7 µM) as an activator of Kir/SUR1 channels. 86 Importantly, VU0071063 was more potent than the KCO diazoxide in activating the Kir6.2/SUR1 channel and inhibited glucose-dependent insulin release when tested in mouse pancreatic islet cells at a concentration of 10 µM.
As described above for MPSD assays, “no-wash” Tl+ assay kits are also available that allow a homogenous assay format. One major limitation to the Tl+ assay is that some cells contain endogenous Tl+ transport pathways that can interfere with the Kir channel efflux under study. In some cells, this “background” Tl+ fluorescence signal can be of significant size and can cause a higher rate of false-positive hits. Furthermore, HTS using MPSD and ion flux fluorescent assays often requires high-end instruments such as FLIPR (Molecular Devices) or FDSS (Hamamatsu, Hertfordshire, UK) workstations that integrate liquid handling with fluorescent imaging. In addition to the cost and experimental limitations, biosafety and disposal issues must be taken into account with the use of toxic heavy metals such as Tl+.
Electrophysiology: APC Systems
While fluorescent MPSDs and ion flux analysis have been valuable for HTS assays of Kir channels, electrophysiological procedures that allow for the direct measurement of Kir currents provide a unique advantage over these technologies. The traditional, whole-cell patch clamp technique has been considered the “gold standard” for investigating the molecular pharmacology of K+ channels and has been applied for studies of both recombinant and native Kir channels ( Fig. 3C ). However, this procedure is labor-intensive, requires technically skilled personnel to carry out the experiments, and is extremely low in compound screening throughput. Numerous APC instruments including the QPatch, Patchliner, IonFlux, PatchExpress, IonaWorks, and SynchroPatch have been introduced over the past 25 years.87,88 By enabling whole-cell recording to be made from multiple cells in parallel, APC systems have gained popularity for lead optimization and secondary screening of K+ channels. Most APC systems utilize a planar array-based format containing a micron-sized aperture (or apertures) in the center of silicon, borosilicate glass, or polydimethylsiloxane “chips” ( Fig. 3C ). Cells are added in suspension to a multiwell recording plate and negative pressure is applied to attract cells to the apertures. Further application of negative pressure causes the patch of membrane immediately beneath the aperture to rupture, thus establishing the whole-cell configuration. Alternatively, electrical access to the cell can be obtained using a pore-forming antibiotic such as nystatin or amphotericin B. Most APC systems now incorporate microfluidic networks, temperature control, and cell population recording features into the device. This later feature allows K+ current averaging to compensate for cell-to-cell variations in current amplitudes and kinetics.
To date, only a limited number of APC experiments have been carried out with recombinant Kir channels. As described above, an APC system was used in a secondary screen of the Kir2.1 channel blocker ML133. 41 In this report, HEK293 cells stably expressing Kir2.1 channels were dispensed into the 384-well recording plate of an IonWorks Quattro (Molecular Devices). After perforation of the cell membrane using an internal solution containing amphotericin B, IK1 was measured either during voltage steps to various potentials or by using ramps from −100 to +100 mV. Kúsz et al. 89 employed a Patchliner APC (Nanion Technologies, Munich, Germany) to examine the blocking activity of diterpenoid alkaloids, isolated from the plant Euphorbia dulcis, on Kir3.1/3.4 channels stably expressed in HEK293 cells. Some of the diterpenoids displayed IC50 in the 1–12 µM range for blocking the GIRK currents while having no inhibitory effect on hERG channels. Finally, as part of the Comprehensive In Vitro Proarrhythmia Assay, CHO cells expressing Kir2.1 channels were analyzed with a QPatch 48-well APC (Sophion Bioscience, Copenhagen, Denmark). 90 Kir2.1 currents measured using the QPatch displayed the expected shift in the reversal potential in the presence of different external K+ concentrations and were blocked by Cs+ and Ba2+. Small-molecule blockers of the Kir2.1 channel, including PA-6, chloroquine, and ML133, were also validated in the QPatch experiments.
Given the interest in applying HTS to native cardiac cells, APC instrumentation has been used to measure hERG (IKr) and other Kv currents from human induced pluripotent stem cell (hiPSC)-derived cardiomyocytes (hiPSC-CMs).91,92 Unfortunately, available hiPSC-CMs express low levels of Kir channels, thus limiting Kir channel experimentation with these cells.93,94 There are currently no reports of APC technology being successfully applied to study IK1 in primary cardiac cells. While many APC systems now provide high-quality voltage clamp data that approach traditional whole-cell patch clamp currents, some instruments have sacrificed data quality in exchange for higher screening throughput.87,88 In addition, as in the case with top-line fluorescent plate readers, the high initial startup price and consumable costs involved in using APC systems often limit their application to large academic and pharmaceutical research facilities.
Other Screening Technologies
Yeast cell growth measurements and the liposome flux assay (LFA) represent two additional K+ channel screening technologies. The K+-dependent growth of yeast, such as Saccharomyces cerevisiae, represents a unique system for the discovery of new Kir channel modulators. 95 Electrochemical influx of K+ into budding yeasts occurs through two primary K+ channels: TRK1 and TRK2. Yeast lacking these channels (trk1Δ/trk2Δ yeast) requires a high-potassium (100 mM)-containing media for growth. However, the expression of exogenous K+ channels, including Kir channels, enables growth of the trk1Δ/trk2Δ yeast in a low-potassium (2 mM) media. Zaks-Makhina et al. 95 developed an HTS assay by expressing the Kir2.1 channel in the trk1Δ/trk2Δ yeast and monitoring yeast growth in low-potassium media. Treatment with the Kir channel blocker Cs+ abolished growth in the trk1Δ/trk2Δ yeast expressing the Kir2.1 channel, thus validating the assay. A library of 10,000 small molecules was then screened for their ability to inhibit yeast growth in low-potassium media. One compound, 48F10, identified in the yeast Kir channel screen, was found to block whole-cell Kir2.1 currents expressed in HEK293 cells with an EC50 of 60 µM. In a follow-up study, trk1Δ/trk2Δ yeast growth was monitored to screen a library of natural product compounds. 96 Through use of this assay, gambogic acid was identified as a relatively potent (EC50 = 100 nM) and selective blocker of the Kir2.1 channel when applied chronically to the trk1Δ/trk2Δ yeast. Unfortunately, gambogic acid was a much less potent blocker of the channel when it was applied acutely (EC50 = 10 µM). One drawback to Kir channel HTS in yeast is that the transmembrane potential in yeast is much more negative than cultured mammalian cells. This large potential could alter the binding of ionized blockers within the pore of the Kir channel. In addition, the yeast cell wall presents a barrier that may impede access of compounds to the channel.
The LFA represents another HTS system with applications for Kir drug discovery. 97 In this procedure, K+ channels are purified and reconstituted into lipid vesicles in the presence of KCl. A concentration gradient for K+ efflux from the proteoliposomes is then established by adding the vesicles into a NaCl solution. K+ efflux is initiated by the addition of a proton ionophore, such as carbonyl cyanide m-chlorophenylhydrazone (CCCP), which allows the influx of protons to balance the efflux of K+. The influx of protons into the vesicles is then quantified by monitoring the proton-dependent quenching of a fluorescent dye. Thus, K+ flux through the channels can be directly correlated with the rate of fluorescent dye quenching. Through use of the LFA, a library of 100,000 compounds was screened to identify both inhibitors and activators of the GIRK2 (Kir3.2) channel. 97 One inhibitor, RU-GIRK-1, was shown to inhibit K+ efflux with an IC50 of 350 nM when the GIRK channels were activated following application of the Giβγ subunit and PIP2 to the vesicles. In comparison, the antiparasitic agent ivermectin was identified as an activator of the GIRK channels using the assay in the absence of Giβγ-mediated gating. In summary, LFA provides an efficient and low-cost method for K+ channel screening. However, drug potencies obtained using LFA will need to be rigorously evaluated with potencies measured using ion flux and APC systems before LFA becomes a widely accepted Kir channel HTS method.
Conclusions
This review has provided an overview of Kir channel pharmacology and a description of channel screening technologies. Kir channels represent potential drug targets for the management of AF, hypertension, anxiety, chronic pain, and other disorders. Therefore, it is essential to identify new Kir channel blockers and activators that can be tested in preclinical models of these diseases. The combination of HTS ion flux fluorescence and moderate-throughput APC instrumentation constitutes the current state of the art for discovering new Kir channel modulators. However, this approach relies on measurements obtained in nonexcitable HEK293 and CHO cells overexpressing Kir channel subunits that do not replicate the phenotypic properties of primary neuronal, cardiac, and renal epithelial cells. To address this limitation, hiPSC-derived neurons and hiPSC-CMs will be increasingly utilized in the future for APC analysis of K+ channels. Although the density of Kir2.1 channels has been reported to be low in hiPSC-CMs, a recent study suggests that Kir expression in some hiPSC-CM cell lines may approach those levels found in primary adult cardiomyocytes. 98 In addition, transfection of hiPSC-CMs with the KCNJ2 gene produces Ba2+-sensitive IK1, stabilizes the resting membrane potential, and reduces excitability of the cells. 94 Another limitation of Kir channel HTS technologies has been the need for high-end fluorescent plate readers and APC instruments. However, the introduction of less expensive devices, including fluorescent plate readers (manufactured by Tecan, Männedorf, Switzerland; BMG LabTech, Cary, NC; and BioTek Instruments, Winooski, VT) and APC systems (Fluxion Biosciences, Alameda, CA), is bringing the cost of equipment within range of academic and small business core facilities. This should be accompanied by a reduction in the costs of equipment maintenance and consumable supplies. These improvements along with the introduction of new screening technologies will be pivotal for the discovery of potent and selective Kir channel modulators.
Footnotes
Acknowledgements
The author thanks Dr. Shirley H. Bryant for his mentorship during the author’s early career and his pioneering work into the role of ion channels in myotonia congenita.
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by U.S. Public Health Service award NS-071530 and National Science Foundation award CBET-1606882 to the author.