
Abstract
Small-molecule discovery typically involves large-scale screening campaigns, spanning multiple compound collections. However, such activities can be cost- or time-prohibitive, especially when using complex assay systems, limiting the number of compounds tested. Further, low hit rates can make the process inefficient. Sparse coverage of chemical structure or biological activity space can lead to limited success in a primary screen and represents a missed opportunity by virtue of selecting the “wrong” compounds to test. Thus, the choice of screening collections becomes of paramount importance. In this perspective, we discuss the utility of generating “informer sets” for small-molecule discovery, and how this strategy can be leveraged to prioritize probe candidates. While many researchers may assume that informer sets are focused on particular targets (e.g., kinases) or processes (e.g., autophagy), efforts to assemble informer sets based on historical bioactivity or successful human exposure (e.g., repurposing collections) have shown promise as well. Another method for generating informer sets is based on chemical structure, particularly when the compounds have unknown activities and targets. We describe our efforts to screen an informer set representing a collection of 100,000 small molecules synthesized through diversity-oriented synthesis (DOS). This process enables researchers to identify activity early and more extensively screen only a few chemical scaffolds, rather than the entire collection. This elegant and economic outcome is a goal of the informer set approach. Here, we aim not only to shed light on this process, but also to promote the use of informer sets more widely in small-molecule discovery projects.
Background
Historically, high-throughput screening (HTS) has been the domain of pharmaceutical companies, which have used this activity to identify small-molecule leads for drug discovery. Over the last few decades, however, advances in technology and miniaturization, coupled with decreases in cost, have made HTS more accessible to academic centers. More recently, challenges in industry, including high costs, times to develop drugs, and drug development failure rates, have led to downsizing and to increased efforts to develop partnerships with smaller biotechnology companies and academic laboratories. 1 Thus, obtaining and using a suitable screening collection has become an important scientific as well as strategic consideration for an increasing number of institutions. 2
A number of initiatives to generate screening libraries have been reported in the literature; for example, EU-OPENSCREEN selected 200,000 compounds from a larger 1.4-million-compound collection. 3 Further, a joint European Compound Library comprises 321,000 compounds assembled from seven companies. 4 Each of these collections has been made available for use by academic laboratories. It is important to note that pharmaceutical companies not only have similar concerns regarding screening, but also must consider the novelty of compounds screened for intellectual property purposes. A study by scientists at Bayer found that, of their 4-million-compound collection, 54% of them were represented in PubChem in a generic form. 5 In order to counter this “novelty erosion,” a set of four target-class design teams aimed to add 500,000 compounds, with average properties including MW <400 and Fsp3 (fraction of sp3 content) ~0.4, where the existing collection had Fsp3 ~0.3. Similarly, at the Broad Institute, the Compound Management Team stores and handles a collection of ~800,000 compounds. This collection includes known bioactives, screening sets available from commercial vendors, and a collection of ~100,000 compounds derived from diversity-oriented synthesis (DOS). 6 Thus, the design of screening collections is an important component for any small-molecule discovery venture.
Over the last 10–15 years, the idea of screening subsets of larger compound collections has been increasingly appreciated. With small-molecule discovery becoming more common in an academic setting, concerns of cost, time, and resources required to conduct an HTS campaign have come to the forefront. 7 These needs have led to the development of innovative methods to design compound subsets, which we term “informer sets,” that capture the chemical or biological performance diversity of the larger collection. Indeed, the idea of a targeted library applied to physiologically relevant assays has been proposed to lead to higher-quality probes. 8 These subsets can be used for target identification, drug repurposing, predictive toxicology, or high-content screening, or as modifiers of other modalities like CRISPR. 9 An excellent compendium of such compound collections is available at https://www.probes-drugs.org/compoundsets.
Types of Informer Set
An early example of smaller subsets of compounds for screening is the 472-member ICCB Known Bioactives Library (https://www.enzolifesciences.com/BML-2840/screen-well-iccb-known-bioactives-library). This set was originally assembled at the Institute of Chemistry and Cell Biology (ICCB; https://iccb.med.harvard.edu/) and used in pilot screening before being obtained by vendors for commercial distribution. This step helped enable many academic laboratories to gain access to bioactive compounds, leading to new discoveries and the design and sale of additional compound sets.
Informer Sets Focused on General Bioactivity
Many efforts to generate informer sets for screening rely on mining historical data, while also maximizing chemical diversity, to increase the likelihood of finding hits for follow-up evaluation. This category can be thought of as “generally bioactive.” For example, a group in Berlin used a maximum-common-substructure algorithm on the Derwent World Drug Index (WDI) to identify highly represented structures in the collection. 10 Importantly, the authors specified that a general-purpose library should be enriched with bioactive compounds, should have a high degree of chemical diversity, should be free of reactive or unstable compounds, and must be physically available. The resulting ~17,000-compound ChemBioNet library has been made available to the academic screening community in Europe. Similar data-driven approaches have been applied to historical screening data collected at the Broad Institute to develop assay performance profiles, which showed convergence of compounds with similar mechanisms of action (MoAs), which can also enable the prediction of new targets for compounds added to the collection. 11 Another effort was focused on 300,000 compounds, yielding a wide variety of active compounds. 12 In this case, a ranking of all compounds enables the selection of any size of informer set, based on the scale of screening that can be accommodated. In contrast to general activity, an approach focusing on performance diversity used analysis of Cell Painting13,14 and gene expression analysis to yield the ~2200 most diverse compounds (from an initial set of ~30,000 compounds), in terms of their general activity in cells. 15
Target-Focused Informer Sets
Target-focused informer sets appear to be the most common application for this screening approach. A notable early effort established “biospectra,” representing the activity of ~1500 compounds across a wide variety of in vitro targets. 16 The authors arrived at molecular property descriptors according to these biospectra, without requiring knowledge of the molecules’ targets. Later work, focused on ion channels, used existing activity data in ChEMBL on ~25,000 compounds to filter by potency, molecular weight, and undesirable chemical groups to arrive at a ~7000-compound set. 17 However, less than 5% of these compounds were commercially available, as many of the original set were part of medicinal chemistry campaigns. Targeted informer sets do not have to focus on protein classes alone; another report focused on compounds that bind RNA. 18 The authors took a structure-based approach to find compounds with favorable properties to bind RNA, and then validated the collection with a repeat RNA associated with myotonic dystrophy type 1. Kinases, of course, are an attractive set of proteins for this kind of informer set. A recent report developed and used an online tool (http://www.smallmoleculesuite.org) to apply cheminformatics and historical screening data to generate a new collection, LSP-OptimalKinase, consisting of 256 compounds. 19 The authors calculated that 1000 compounds are needed for a truly optimal kinase library. Another report focused on the Published Kinase Inhibitor Set (PKIS) to predict activity against unstudied kinases. 20
Most recently, targeted informer sets have focused on compounds that have a greater likelihood to inhibit protein–protein interactions (PPIs).21,22 FrPPIChem, for example, is a collection of 10,314 compounds developed by a French consortium. According to the authors, inhibitors of PPIs are, on average, heavier and more hydrophobic, contain more aromatic rings, and have different three-dimensional shapes than conventional drugs. 21 They identified a set of descriptors to select the compounds and validated the approach on the PPI between CD47 and SIRPα. Comparative analysis showed that, while the NCATS “Genesis” collection yielded a hit rate of 0.013% in the primary screen, the FrPPIChem collection had a hit rate of 15.7%, representing a 46-fold activity-rate enrichment. Finally, the Broad Institute has assembled or purchased a variety of target-focused informer sets for screening, focused on, for example, autophagy (1280 compounds), G-protein-coupled receptors (GPCRs; 5200 compounds), and ion channels (5000 compounds).
Phenotype-Focused Informer Sets
Phenotypic screening does not always afford the luxury of knowing which cellular target will yield desired results. To address this challenge, several groups have developed informer sets focused on phenotypic screening. An effort from Novartis used high-throughput screening fingerprint (HTS-FP) 23 on 200 HTS available data sets. 24 Generally, the authors advise using collections <10,000 in size; including multiple compounds with the same target, to improve confidence in results; screening at multiple concentrations, to mitigate concerns over polypharmacology; and considering solubility and cell penetration, where possible. Barriers to this approach include errors in annotation and lack of uniformity in how HTS data or chemical structures are represented. To be fair, these challenges exist in any informer set assembly, but it is important to note that data harmonization is nonetheless a critical, but sometimes undervalued, activity. Exploring the mechanisms of cell death, Wolpaw and colleagues performed what they termed modulatory profiling of a collection of chemical modulators against 28 lethal compounds in fibroblast cell lines. 25 A more focused phenotypic informer set aimed to generate compounds active against malaria. 26 Using historical phenotypic data against the Plasmodium falciparum parasite from the Medicines for Malaria Venture, the authors collected 400 compounds and sent the compounds to 200 groups globally, resulting in data from 236 phenotypic screens. Some of the compounds were also active against bacteria, helminths, and the NCI-60 cancer cell line collection. At the Broad Institute, an alternative approach that we are taking is to use Cell Painting to identify “nuisance” compounds, which yield artifactual activity and can confound phenotypic screening results, due to such moieties as thiol-reactive groups (P.A.C. and B.K.W., unpublished results).
Some phenotype-based informer sets have been assembled with the intention of profiling a collection of cell lines or cell states. For example, the Cancer Therapeutics Response Portal (http://portals.broadinstitute.org/ctrp/) integrates genetic information, lineage, gene expression, and other cellular features available across a panel of cancer cell lines to identify small-molecule sensitivity patterns that reveal vulnerabilities in particular cancer types.27,28 In this case, 481 small-molecule probes and drugs were assembled, with a focus on selecting compounds that 1) individually, were highly target selective, and 2) collectively, impacted on a diverse set of nodes in cell circuitry. This set of compounds was tested over a 16-point concentration range against 860 cancer cell lines, with data and small molecule–gene expression correlations available on the portal.
Most recently, the development of an “MoA Box,” a chemogenetic collection of compounds designed to help understand MoA for phenotypes and target discovery, was reported. 29 The primary goal was to achieve broad representation across many human targets and modalities (>2100 mammalian targets covered). This internal project at Novartis used a novel web-based nomination form to crowdsource ideas from anyone at the company, which furnished ~30% of the collection. The other 70% was assembled in a more conventional fashion, by studying primary and patent literature and clinical phases reached and by optimizing the number of compounds per target (at least 5, no more than 10). Remarkably, the authors state that 83% of this collection has only one or two targets, so such a specific collection should be useful in the >300 screens to which it has already been applied.
Repurposing Informer Sets
A related approach to informer set construction is to develop “repurposing” libraries. These sets usually consist of approved drugs and related bioactives, to identify new indications for known drugs and discover new targets for phenotypic purposes. Groups at Calibr and Scripps reported recently on the ReFRAME collection (“Repurposing, Focused Rescue, and Accelerated Medchem”; https://reframedb.org), consisting of 12,000 compounds assembled by mining drug intelligence databases and patent literature. 30 The authors applied the collection to a screen for death of Cryptosporidium sp., identifying two hits, which were also effective in animal models at clinically relevant doses. Similarly, the more recent Broad Drug Repurposing Hub (https://clue.io/repurposing) contains approved drugs, compounds in phase 1–3 clinical trials, and preclinical tool compounds. 31 Approximately 90% of approved small-molecule drugs are covered in this set. Our recently replenished and expanded Repurposing Library consists of 6801 compounds plated as REPO 1 (Food and Drug Administration or globally approved), REPO 2 (clinical trial), and REPO 3 (preclinical). Associated data include extensive standardized curation information regarding reported target and pathway effects. These metadata will make this set highly effective as a reference collection of known bioactives as well as provide the potential for repurposing or targeted screening efforts.
Chemistry-Focused Informer Sets
Of course, chemical structure itself is also a sensible approach to selecting compounds for an informer set. Several papers have noted the importance of the chemical scaffold tested and its overall impact on screening campaigns.32,33 A recent evaluation of a natural product-like flavonoid library, which contained a diversity of scaffolds, stereochemistries, and appendages, revealed that, at least by Cell Painting, the chemical scaffold was the dominant feature in predicting performance diversity. 32
Many reports in the literature have taken a cheminformatics approach, for example, to remove compounds with undesirable properties.34,35 However, chemical diversity is also an important consideration. O’Hagan and Bell used a hybrid hierarchical k-means clustering strategy to identify subsets of compounds based on chemical structure. 36 Their effort aimed to reduce a 200,000-compound library to more manageable subsets for testing (e.g., 96, 384, 1152, and 1920 compounds). As a result, the average molecular similarity of the subset was much lower than that of the total collection. A notable component of this effort was that their work was virtual and so did not result in a physical collection. However, even if there were such a physical collection, it can be appreciated that sometimes it is not practical to replate all of these compounds to create an informer set. Instead, a plate-based approach, where diversity is taken into account on a per-plate basis, may be more tenable. The latter approach is the one we took with the compound collection at the Broad Institute.
Over the past two decades, chemists at the Broad Institute developed a collection of 100,000 small molecules derived from DOS.6,37 Advances in combinatorial chemistry in the 1990s enabled the synthesis of large compound collections for screening, while advances in DOS chemistry have greatly expanded the chemical space accessible to biological testing.6,34,38 However, as we have noted, it is not always practical to screen all 100,000 compounds of the DOS collection. Challenges include the cost of the reagents, access to the proper instrumentation, and the time and effort required to complete such a screen. Here, we found that an attractive solution was to sample each scaffold (which, again, corresponds to a library emerging from a single synthetic strategy) by testing one or two plates per library. This strategy led to a collection of ~10,000 compounds, or approximately thirty-one 384-well plates, which is much more feasible for screening, and which we call the DOS Informer Set. The results of this pilot screen can be used to identify scaffolds for which greater activity is observed, followed by in-depth screening of only those scaffolds (
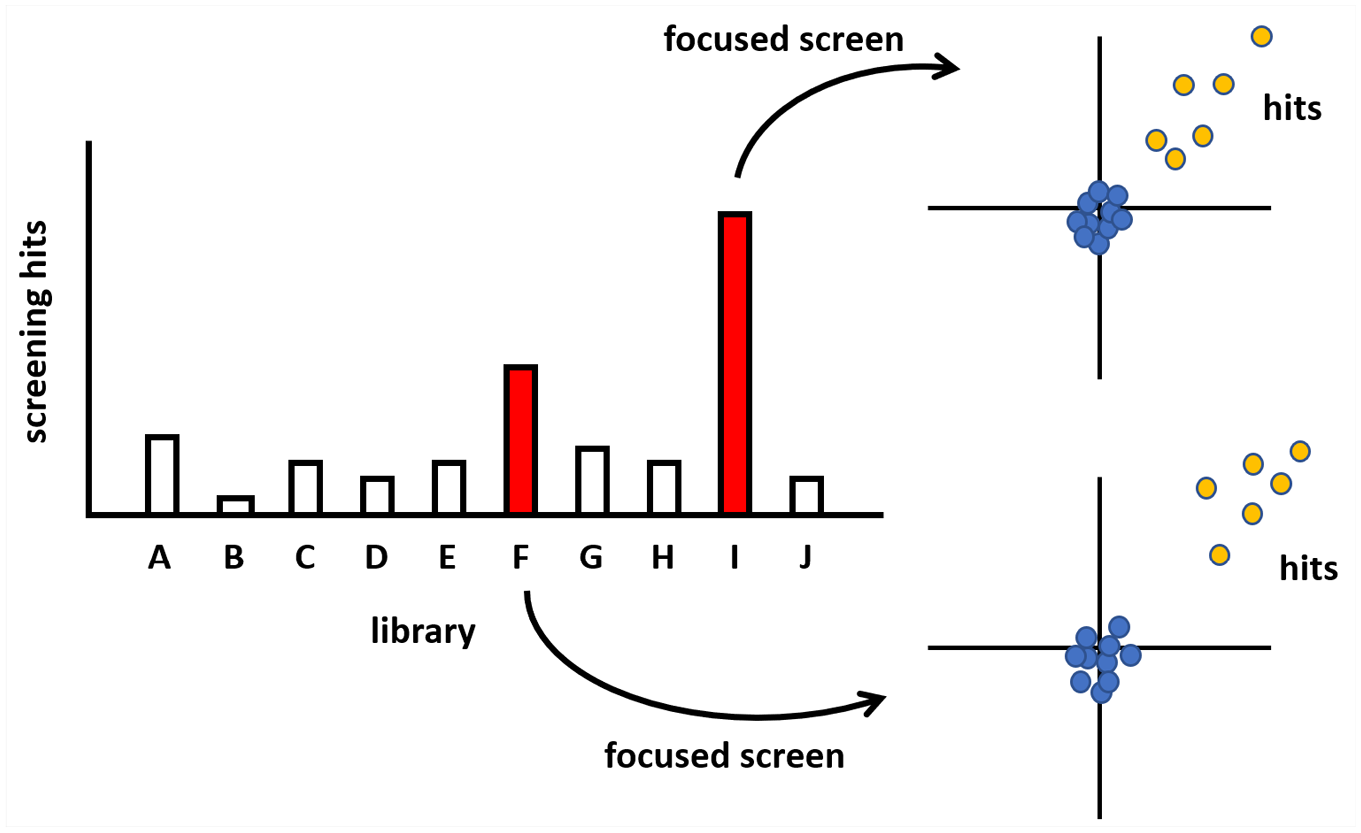
Illustration of the screening process using the DOS Informer Set. Each library, representing distinct chemical scaffolds, yields different levels of activity in a particular high-throughput screen. By screening a small subset of each library, this pattern can be discerned, followed by in-depth screening of active libraries. This strategy leads to far fewer compounds needing to be screened: on the order of 10,000–20,000 compounds, for example, as opposed to the full 100,000-compound collection.
Finally, there is value in using screening data itself to drive the focused synthesis of compounds with an enrichment of biological activity. Starting with a small informer set, the Schreiber laboratory and collaborators used Cell Painting to annotate a collection of 10 triads of isomeric compounds generated through photochemical and thermal rearrangements. 39 The multiplexed data collected on these compounds provided a proof of concept for selecting scaffolds on which to focus using cell-based data and reinforced that chemical descriptors can be insufficient to predict biological performance.
Case Studies Involving Screening the DOS Informer Set
IDE Inhibitors
An example of the utility of a chemistry-focused informer set comes from the search for inhibitors of insulin-degrading enzyme (IDE). This zinc metalloprotein induces endogenous degradation of insulin to modulate the duration of its activity in the bloodstream, and IDE has been identified in genome-wide associate studies for type 2 diabetes (T2D). A long-standing therapeutic hypothesis holds that IDE inhibition may increase insulin activity, compensating for insulin resistance in T2D-affected individuals. However, no viable probes had been discovered for IDE. In 2014, researchers from Harvard performed in vitro selections on a DNA-encoded library of ~14,000 macrocycles and discovered 6bK, a 20-membered macrocycle with an IC50 of 50 nM. 40 6bK lowered blood glucose levels in a mouse model of diet-induced obesity (DIO). Through structural analyses, the authors determined that 6bK bound an exo-site adjacent to, but not overlapping with, the active site of the IDE protease. As a result, 6bK was very selective among proteases, but also inhibited the degradation of glucagon, which can bind to the IDE active site as well.
To identify inhibitors selective for insulin degradation, we developed an exo-site-focused screen, using a fluorescence polarization assay to measure displacement of a fluorescence analog of 6bK. 41 Screening the 10,000-compound DOS Informer Set revealed that a number of compounds from azetidine-focused libraries not only were weakly active, but also contained a biaryl group similar to the active benzophenone group on 6bK. We then screened nearly 9000 compounds containing an azetidine scaffold 42 from the full 100,000-compound DOS collection, several of which resulted in IDE inhibition in orthogonal assays. The most potent hit, BRD8283, inhibited protease activity with an IC50 of 100 nM. Remarkably, BRD8283 showed strong selectivity for inhibiting insulin degradation, with extremely low effect on glucagon degradation, and >500-fold selectivity for IDE over other metalloproteases. Medicinal chemistry to optimize the potency and substrate selectivity of BRD8283 resulted in related analog 63, which had an IC50 of 0.5 nM against IDE, strong insulin selectivity, and >10,000-fold selectivity for IDE. X-ray crystal structures showed that IDE bound to 63 blocked the binding of insulin but enabled an IDE–63-glucagon ternary complex that preserved glucagon cleavage.
This work reveals the economic and operational efficiency of initially screening an informer set, rather than directly embarking on a full-collection screen. By screening <20,000 compounds (~10,000 in the DOS Informer Set plus ~9000 in azetidine-based libraries), we were able to efficiently sample the chemical space represented by the DOS collection and found a set of related structures that enabled medicinal chemistry and structure–activity analysis (SAR) analysis.
Cas9 Inhibitors
The biomedical advancements enabled by CRISPR/Cas9-based genome editing have been tremendous. However, the need for precise control of Cas9 activity, both at multiple concentrations and over time, has led to the search for anti-CRISPR molecules, primarily focused on protein-based reagents. To identify small-molecule inhibitors of Cas9 from Streptococcus pyogenes (SpCas9), the Choudhary laboratory at the Broad Institute developed a high-throughput discovery platform consisting of a suite of assays aimed at facile detection of Cas9 activity in vitro and in cells. 43 For primary screening, we used a fluorescence polarization assay to measure the displacement of SpCas9:gRNA from a fluorescently labeled PAM-containing DNA oligonucleotide. Rather than screen the entire DOS collection, we tested a similar ~10,000-compound DOS Informer Set and found that members of the Pictet–Spengler, spirocyclic azetidine, and Povarov libraries had hit rates >1%. The spirocyclic azetidine compounds were ruled out due to nonspecificity, leaving the Pictet–Spengler and Povarov libraries for evaluation.
Screening these individual libraries (2000–3000 compounds for each library) revealed that the Pictet–Spengler compounds had significant autofluorescence and cytotoxicity; however, the Povarov scaffold compounds yielded hits that were more suitable for follow-up studies. In particular, BRD7087 showed dose-dependent inhibition of SpCas9 in several secondary assays, including cell-based activities. We synthesized or obtained 641 analogs of BRD7087, resulting in the identification of BRD0539, which showed stronger inhibition of SpCas9 and target engagement by CETSA. 44 Again, by screening only ~15,000 compounds, we were able to identify cell-active inhibitors of SpCas9 that served as starting points for medicinal chemistry and SAR analyses, resulting in the identification of even more potent inhibitors.
Outlook
We believe that the use of informer sets in screening has a bright future and should be a strategy that researchers increasingly use, especially at the beginning of a discovery project. The increase in efficiency and cost-effectiveness afforded by judicious screening of library subsets enables sufficient sampling of the larger collection’s chemical space, to home in on scaffolds with greater activity toward the desired target or phenotype. Although not meant as a comprehensive screening activity, there is the possibility of missing hits that would have been found in a large-scale screen, so consideration of this possibility should be made during a screening project. Overall, it is important to note a few critical lessons from these activities. First, we have found that the clustering of screening hits in particular sublibraries is not limited to target-based screens. For example, we also identified scaffolds for deeper testing in screens for pancreatic beta-cell apoptosis and proliferation. 45 Second, we identified different libraries in each of the screens performed. If the results had been due to artifacts or to nonspecific activity, we might have observed that some scaffolds result in high hit rates, regardless of the modality. However, the fact that, in each case, we found different scaffolds for testing suggests that there may be specificity in the molecular interactions for each of the proteins and phenotypes tested. Indeed, the SAR in the above two vignettes bore this out: there was a molecular explanation for the outcomes that we observed. Together, these results provide further confidence that an informer set-based approach to screening can yield effective directions for small-molecule discovery.
Footnotes
Acknowledgements
We thank Jayme Dahlin for helpful comments.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: P.A.C. is an advisor for Pfizer, Inc. and nference, Inc. J.A.B. is an employee and shareholder of Vertex Pharmaceuticals. S.L.S. is a shareholder and serves on the board of directors of Jnana Therapeutics; is a shareholder of Forma Therapeutics and Decibel Therapeutics; is a shareholder and advises Kojin Therapeutics, Kisbee Therapeutics, and Eikonizo Therapeutics; serves on the SABs of Eisai Co., Ono Pharma Foundation, Exo Therapeutics, and F-Prime Capital Partners; serves on the board of advisers of the Genomics Institute of the Novartis Research Foundation; and is a Novartis Faculty Scholar.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.