
Abstract
The authors describe a structure-based strategy to identify therapeutically beneficial off-target effects by screening a chemical library of Food and Drug Administration (FDA)–approved small-molecule drugs matching pharmacophores defined for specific target proteins. They applied this strategy to angiotensin-converting enzyme 2 (ACE2), an enzyme that generates vasodilatory peptides and promotes protection from hypertension-associated cardiovascular disease. The conformation-based structural selection method by molecular docking using DOCK allowed them to identify a series of FDA-approved drugs that enhance catalytic efficiency of ACE2 in vitro. These data demonstrate that libraries of approved drugs can be rapidly screened to identify potential side effects due to interactions with specific proteins other than the intended targets.
Keywords
Introduction
T
We previously used in silico docking to identify the compounds capable of enhancing catalytic activity of Angiotensin-Converting enzyme 2 (ACE2) to lower blood pressure and prevent cardiovascular disease.11,12 We used DOCK5.1 (University of California, San Francisco [UCSF]) to explore a structural feature in the hinge region of ACE2 (site 1, Fig. 1 ) that is implicated in conformational shuffling between the two isoforms of the enzyme. 13 A small molecule library of 139,735 compounds (molecular weight less than 500 Da) from National Cancer Institute (NCI) Developmental Therapeutics program plated set collection was docked into the selected site 1 and scored using grid-based scoring system. Compounds were ranked according to their combined energy scores for hydrogen bonding and van der Waals contact interactions. The highest scoring compounds were obtained and assayed in vitro for their abilities to enhance ACE2 catalytic activity. One of the compounds, [8-(2-dimethylaminoethylamino)-5-(hydroxymethyl)-9-oxoxanthen-2-yl]4-methyl-benzenesulfonate (from here on referred to as XNT) was shown to enhance the rate of catalysis by increasing the velocity of the enzyme approximately twofold (13). In vivo administration of XNT in the spontaneous hypertensive rat model induced a significant (71 ± 9 mm Hg) decrease in high blood pressure and resulted in improvements in cardiac function, including reversal of myocardial, perivascular, and renal fibrosis. 14
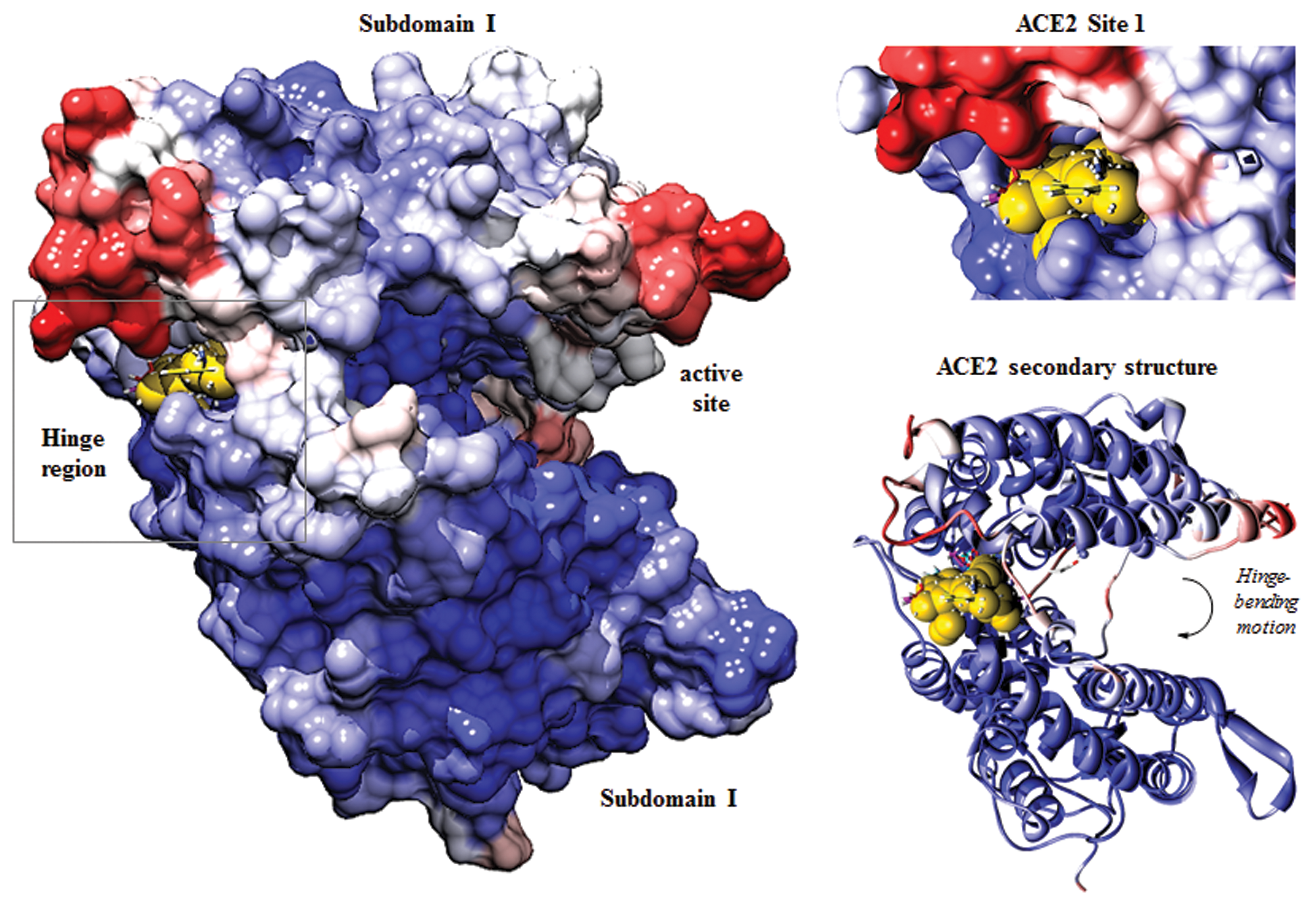
Angiotensin-converting enzyme 2 (ACE2) site 1 used as a structural basis for molecular docking of Food and Drug Administration–approved compounds. Site 1, located in the hinge region separating the two subdomains of the enzyme, is characterized by high solvent accessibility and flexibility. Molecular surface and secondary structure of ACE2 in the open conformation (PDB ID: 1R42) are colored by accessible surface area (ASA): red for the highest and blue the lowest ASA. Yellow spheres represent the site selected for in silico screening; 10 highest scoring small molecules are shown in their predicted binding positions and represented as sticks. Inset shows zoomed-in view of the site selected for molecular docking.
Because the molecular docking selection strategy was useful in identifying compounds that enhance ACE2 catalytic activity and lower blood pressure in vivo, in this study we applied this selection strategy to a chemical library of 1217 Food and Drug Administration (FDA)–approved compounds with extensive information on bioavailability, toxicity, and safety (Distributed Structure-Searchable Toxicity Database Network, www.epa.gov/ncct/dsstox). Here we report identification of specific FDA-approved compounds that enhance the catalysis of ACE2. This rapid and economical molecular docking approach can be applied to other clinically relevant proteins to explore their structural features and identify off-target effects of the small molecule compounds.
Materials and Methods
Molecular docking
We used the crystal structure of the apo form of human ACE2 in the open conformation (PDB ID: 1R42) to provide the basis for molecular docking. 15 To prepare the site for docking, all water molecules have been removed. Protonation of ACE2 residues was done with SYBYL. 16 Intermolecular assisted model building and energy refinement (AMBER) energy scoring (van der Waals + coulombic), contact scoring, and bump filtering were done in DOCK5.1.0.17,18 Atomic coordinates for 1,217 FDA-approved compounds were obtained from the ZINC structural database (http://zinc.docking.org). Each of the molecules was positioned into the selected site (Site 1) in 1000 different orientations and scored with rank based on the predicted polar (H-bonding) and nonpolar (van der Waals) interactions. Compounds were selected to include protonation variants at medium pH (5.75–8.25). The scoring grid was set at 5 Å around the spheres selected for molecular docking. Molecular surface of the structure was explored using sets of spheres to describe potential binding pockets. The sites selected for molecular docking were defined using the SPHGEN program and filtered through the CLUSTER program. 18 The SPHGEN program generates sets of overlapping spheres to describe the shape of the selected site. The CLUSTER program groups the selected spheres to define the points that are used by DOCK to match (superimpose) potential ligand atoms with spheres. Intermolecular AMBER energy scoring (van der Waals + columbic), contact scoring, and bump filtering were implemented in DOCK v5.1.0. UCSF Chimera software package 20 was used to generate molecular graphic images. The UF High-Performance Computing Center linux cluster was used to run docking jobs by parallel processing. The 38 highest scoring compounds (based on the combined contact and electrostatic score) were obtained from NCI Developmental Therapeutics program for testing in ACE2 catalytic assays.
ACE2 enzyme kinetic assays
Effects of 40 top-scoring compounds selected in virtual screening were tested in fluorescence-based kinetic assays using recombinant human enzyme and fluorogenic peptide substrate. Kinetic parameters for ACE2 in the presence of selected compounds were determined under steady-state conditions in the presence of saturating amounts of the substrate. Enzyme concentration was adjusted to ensure that <15% of the substrate was consumed at the lowest substrate concentration and product formation was linear with time. ACE2 assays for catalytic activity were carried out in a total volume of 100 µL, containing 75 mM Tris-HCl (pH 7.4), 0.1M NaCl, 0.5 µM ZnCl2, 10 nM ACE2, and 0.01% Triton-X. Small-molecule stocks were prepared by dissolving the compounds in DMSO to a concentration of 50 to 100 mM; a final concentration of 50 µM was used in all screening experiments. Compounds were preincubated in black 96-well plates with 10 nM human recombinant ACE2 (Enzo Life Sciences, Plymouth Meeting, PA) for 15 min at 37 °C. Reactions were initiated by addition of 25 to 250 µM fluorogenic peptide substrate Mca-APK(Dnp)-OH (AnaSpec, Fremont, CA) and monitored continuously for 30 min with Spectra Max Gemini M5 Fluorescence Reader from Molecular Devices (Sunnyvale, CA; λexcitation = 325 nm, λemission = 395nm). Initial velocities of ACE2 in the absence and in presence of 50-µM compounds were determined by measuring an increase in fluorescence upon hydrolysis of the substrate. Nonlinear regression analysis and fitting of the data into Michaelis–
Inner filter effect
Inherent small-molecule fluorescence may alter apparent initial velocities, kcat and Km, as a result of inner filter effect (IFE). IFE is a reduction in fluorescence signal due to absorption of the exciting light and/or emitted radiation in the presence of a second fluorophore. As a result, only a fraction of the photons reach the fluorophores and get picked up by the detection system. For this reason, we corrected observed fluorescence (Fobs) for IFE using the following equation:
High-performance liquid chromatography analysis of Ang II cleavage
ACE2 activity assays were carried out in a total volume of 100 µL, containing 100 mM Tris-HCl (pH 7.4), 0.1 M NaCl, and 0.5 µM ZnCl2; 10 nM ACE2 was preincubated for 5 min with 50 µM DMZ, and the reaction was initiated by addition of 50 µM substrate. Reactions were monitored for the first 15 min at λex = 328 λem = 392 and stopped after 2 h at 37 °C by adding 10 µL of 0.5 M EDTA, followed by heating to 100 °C for 2 min. Samples were centrifuged at 11,600 x g for 10 min, supernatants were collected and concentrated under vacuum. The resulting residue was resuspended in 50 µL of the mobile phase B and applied to a C18 reverse-phase high-performance liquid chromatography (HPLC) column (column: Bondclone 10 µm, 150 × 2.1 mm [Phenomenex, Torrance, CA]; HPLC system: Agilent 1100 series [Agilent Technologies, Santa Clara, CA]) with a UV detector set at 214 nm. A linear solvent gradient of 11% to 100% B over 15 min with 10 min at final conditions and 10 min reequilibration was used. Mobile phase A consisted of 0.08% (v/v) phosphoric acid, and mobile phase B consisted of 40% (v/v) acetonitrile in 0.08% (v/v) phosphoric acid. The peaks corresponding to full-length peptides and peptide hydrolysis products were compared with the standards, and peak areas were integrated to calculate the extent of hydrolysis. Peak identities were determined by matrix-assisted laser desorption/ionization–time of flight (MALDI-TOF) spectrometry using Applied Biosystems Voyager System 6031 (Applied Biosystems, Foster City, CA). Ang (1–7) resolved with a characteristic peak at m/z = 899.65, Ang II at m/z = 1046.52. 22
Statistical significance
Data are expressed as mean ± SD. Unpaired Student’s t test and one-way analysis of variance (ANOVA) were performed for statistical analysis. Differences were considered significant at p < 0.01 or p < 0.001, as indicated. All statistical analysis and curve fitting were performed with SigmaPlot 11.0 from Systat Software.
Results
Molecular docking of FDA approved drugs
In the previous study, we used molecular docking to target three structural features on the solvent-accessible surface of ACE2 and used in vitro assays to evaluate the effect of selected small molecules on enhancing catalytic activity. 13 Small molecules targeted at the ACE2 hinge region, (Site 1, Figure 1 ) produced the strongest effect on enzyme activity, increasing initial velocities by ~1.8- to 2.2-fold of control levels. In this study, we used the atomic coordinates and a scoring grid of Site 1 to dock a library of 1217 FDA-approved compounds. 23 The coordinates of the 1217 compounds were docked into the hinge Site 1 (DOCK5.1, UCSF), and ranked by grid-based energy scores (van der Waals + electrostatic). Figure 1 illustrates the binding pocket selected for high-throughput molecular docking and the positions of high-scoring small-molecule compounds. The functional effects of the top-scoring compounds were evaluated by measuring their effects on ACE2 maximal velocity and Km.
Effects of FDA-approved drugs on ACE2 catalytic activity
The highest scoring 38 compounds were tested under saturating substrate conditions for their ability to modulate ACE2 enzyme activity in vitro. Figure 2 demonstrates docking scores and initial velocities determined in the screening of 27 top-scoring compounds in the presence of 50 µM substrate. Ten compounds producing the highest initial velocities for ACE2 were selected for further evaluation. Their docking scores and kinetic parameters are reported in Table 1 ; chemical structures are shown in Table 2 (see also Table 3 ). The previously described compound XNT that increases ACE2 activity was used as a reference. Figure 3 demonstrates kinetic curves for ACE2 in the presence of selected compounds and 25 to 125 µM substrate.
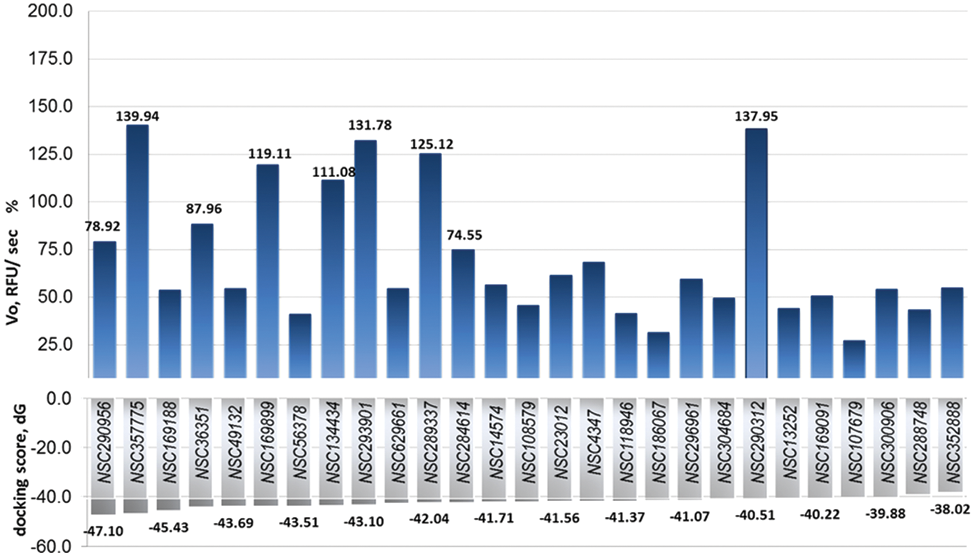
Docking scores and measured initial velocities for the highest scoring 27 compounds. Angiotensin-converting enzyme 2 (ACE2) experimental initial velocities were determined in the presence of 38 top-scoring Food and Drug Administration–approved compounds (50 µM) and compared to those in the absence of the drugs. Compounds are shown ranked by combined energy score (gray bars). Dark blue bars demonstrate change in initial velocity for the first 27 compounds,
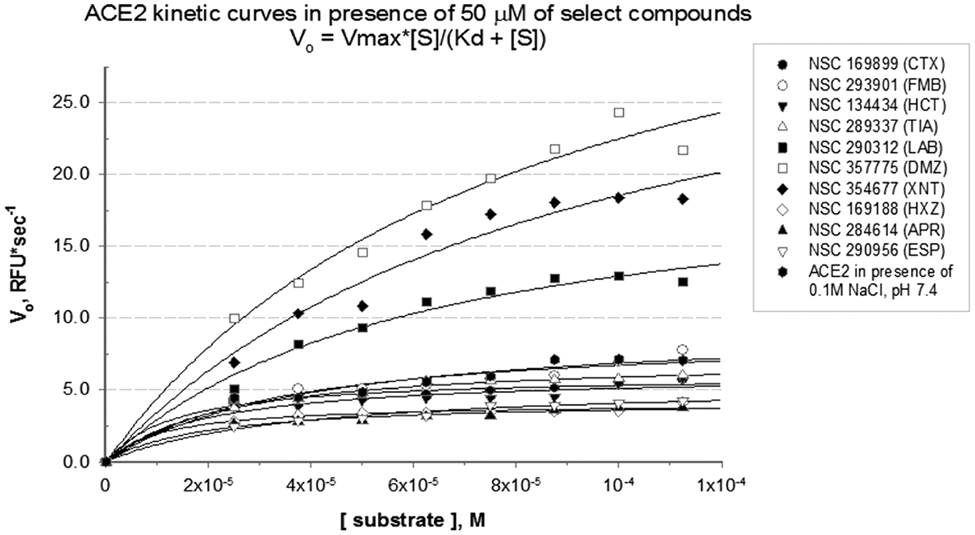
Food and Drug Administration–approved drugs enhance angiotensin-converting enzyme 2 (ACE2) catalytic activity. ACE2 kinetic parameters were determined for 10 compounds that demonstrated highest initial velocities. Kinetic constants for ACE2 in the presence of the 50-µM compounds and 25 to 125 µM substrate were determined by nonlinear regression fit of experimental data into Michaelis-Menten equation
Kinetic Parameters of ACE2 in the Presence of Select Compounds
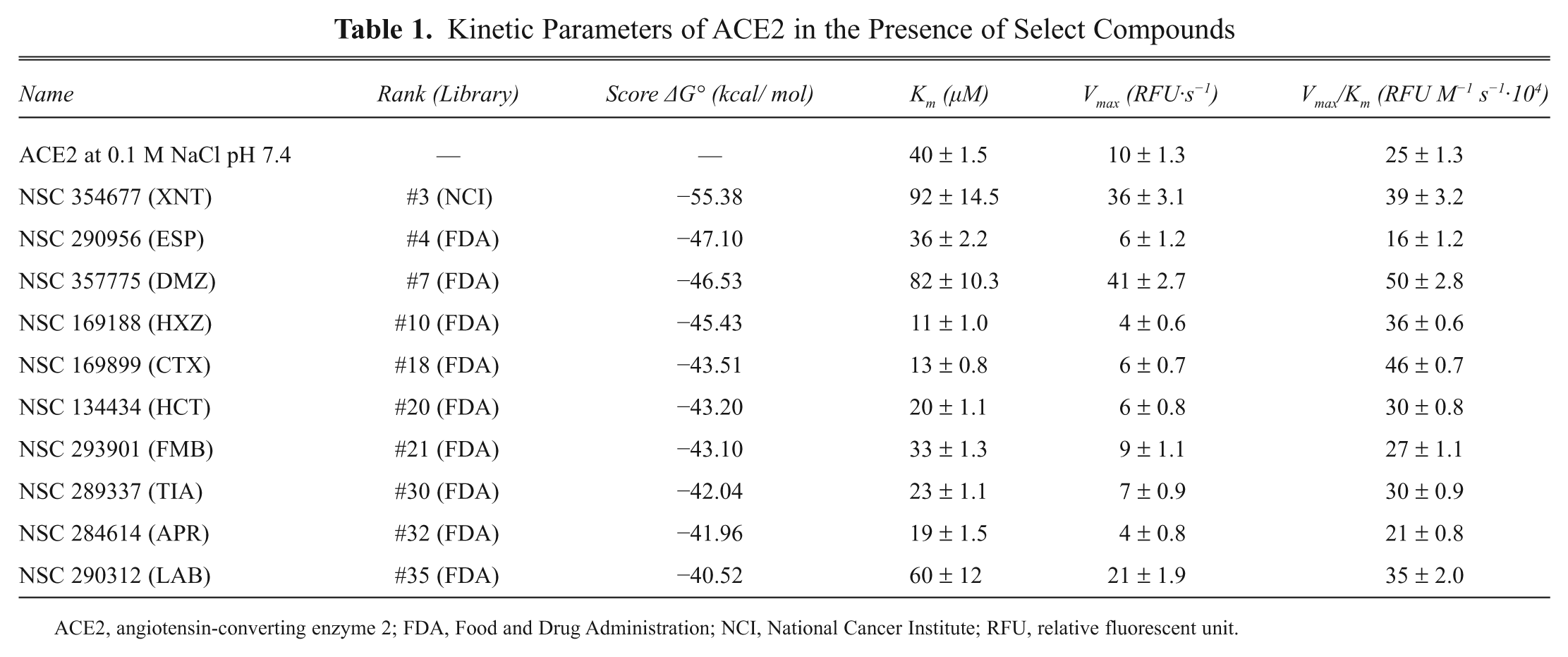
ACE2, angiotensin-converting enzyme 2; FDA, Food and Drug Administration; NCI, National Cancer Institute; RFU, relative fluorescent unit.
Chemical Structures of Selected Food and Drug Administration–Approved Compounds
Effect of Diminazene (DMZ) on Enzyme Efficiency
ACE2, angiotensin-converting enzyme 2.
Two of the FDA-approved compounds (labetalol [LAB] and diminazene [DMZ]) were shown to increase maximal velocity at least twofold (21 ± 1.92 and 41 ± 2.69 RFU·s−1, respectively) compared to ACE2 alone (10 ± 1.30 RFU·s−1), whereas incubation with aprindine, minithixen, and hydroxyzine (APR, CTX, and HXZ) resulted in 2.1- to 3.5-fold reduction in Km. Despite the fact that many of the compounds demonstrated enhancing effects on either Vmax or Km, only 3 of the selected 10 compounds had a statistically significant effect on overall enzyme efficiency (Vmax/Km)—HXZ, CTX, and DMZ.
Interestingly, some of the known antihypertensive agents were identified in this screen. For example, labetalol (2-hydroxy-5-(1-hydroxy-2-((1-methyl-3-phenylpropyl) amino) ethyl) benzamide) is an antihypertensive drug marketed as Normodyne.
24
None of the compounds that significantly affect ACE2 enzyme efficiency were previously used for the treatment of hypertension or cardiovascular disease. Hydroxyzine is an antihistamine used in the treatment of allergies and hyperalgesia, minithixen is a dopamine receptor antagonist used as antipsychotic, and diminazene is an antiprotozoan chemotherapeutic used in veterinary medicine. More information on the clinical uses of selected compounds can be found in the
The magnitude of the effect of these compounds has particular significance when compared with clinical effects of ACE inhibitors used for the treatment of hypertension, which typically inhibit enzyme activity to levels below 10%.25,26 The FDA-approved drug diminazene (benzenecarboximidamide,4,4′-(1-triazene-1,3-diyl)bis-, dihydrochloride) identified in this study was shown to be the most effective in this group of compounds ( Table 1 ), and we selected it for further evaluation.
EC50 and Lineweaver-Burk analysis
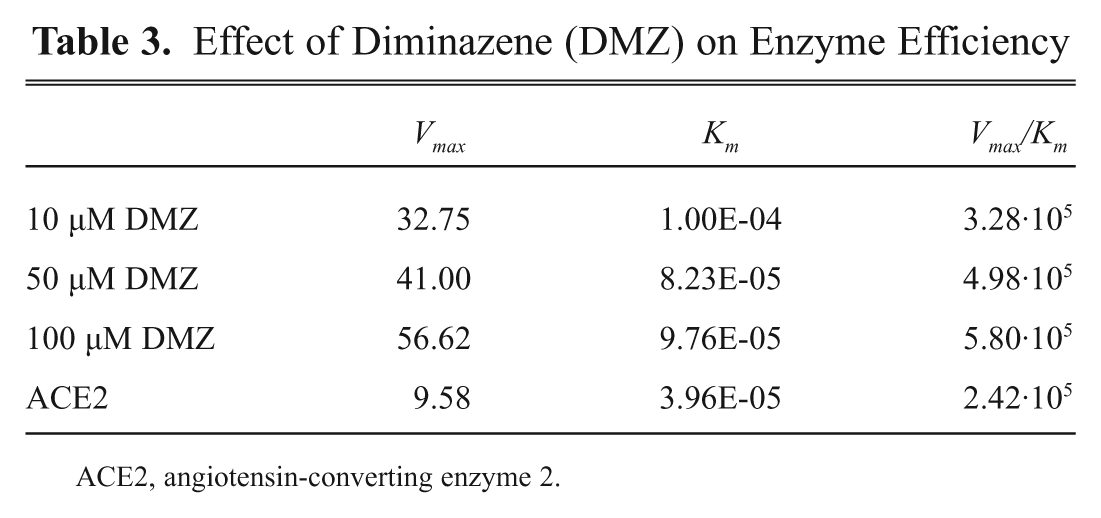
A Lineweaver-Burk (LB) plot was generated for five compounds that had the strongest effect on ACE2 (
Lineweaver-Burk double-reciprocal plot for angiotensin-converting enzyme 2 (ACE2) in the presence of 0 (○), 10 µM (■), 50 µM (▲), and 100 µM (◊) diminazene (DMZ). Despite the fact that even low concentrations of diminazene are sufficient to produce a boost in maximal velocity, overall enzyme efficiency responds more slowly to increasing concentrations of the compound and starts to approach 2.5-fold of the control levels only in presence of 100 µM drug.
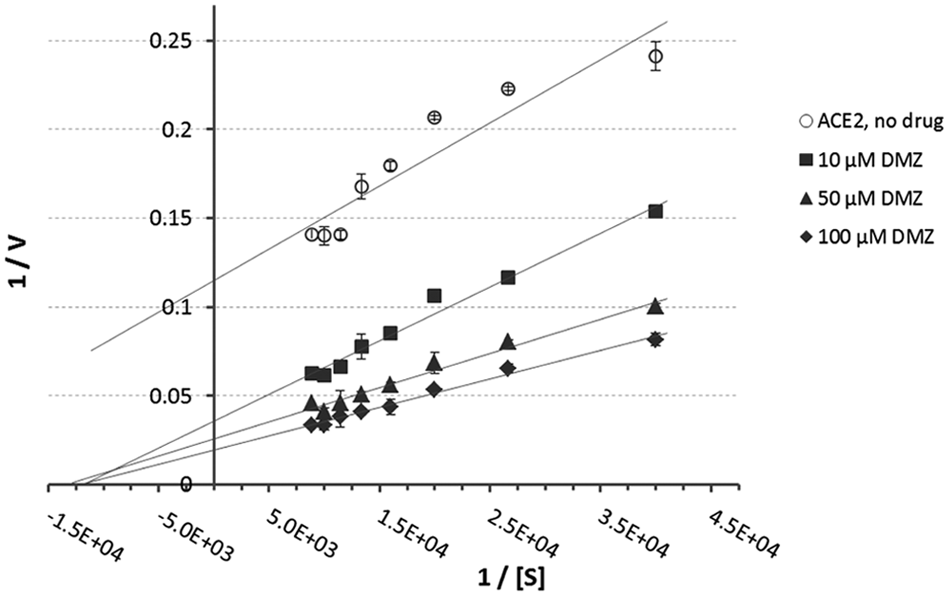
Titration of ACE2 with diminazene (0.01–1000 µM) results in a biphasic dose–response curve illustrated in Figure 5 . At low concentrations, the enzyme is activated with an EC50 of 8.04 µM, whereas at high concentrations, it is partially inhibited with an IC50 of 200.01 µM. Remarkably, even after the partial inhibition, ACE2 initial velocity remains significantly higher than in the absence of the drug. Previously discovered XNT and resorcinolnaphthalein (EC50 = 20 ± 0.8 and 20 ± 0.4 µM) 13 produced only about 70% of the maximal increase in initial velocity of diminazene.
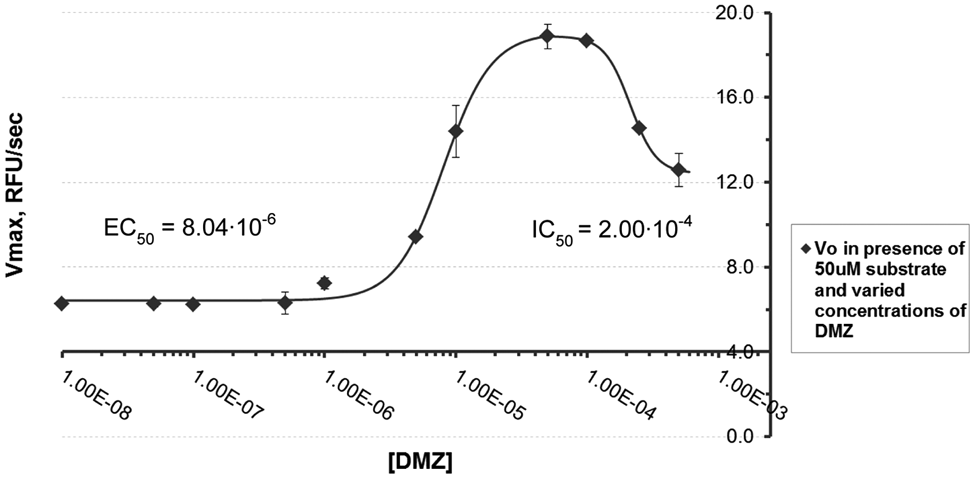
Angiotensin-converting enzyme 2 (ACE2) dose–response curve for activation with diminazene (DMZ). In total, 10 nM ACE2 was incubated with 10 nM to 1 mM diminazene for 5 min at 37 °C. The reaction was initiated by addition of fluorogenic peptide substrate Mca-APK-(Dnp)-OH and monitored for 15 min. Diminazene enhances ACE2 activity in a dose-dependent manner with an EC50 of 8.04 µM (concentration to achieve 50% increase in initial velocity). Titration results in a biphasic dose–response curve: at low concentrations of DMZ, the enzyme is activated, whereas at high concentrations, it is partially inhibited with an IC50 of 200 µM.
HPLC analysis of Ang II cleavage
It has been determined that vasoconstrictor peptide angiotensin II (Ang II) is a preferred natural substrate for ACE2. 27 Angiotensin 1–7 (Ang 1–7), generated by enzymatic cleavage of Ang II, is one of the main effectors responsible for beneficial effects of ACE2 on the cardiovascular system. 28 Classical photo- and fluororimetric methods could not provide accurate detection and quantification of Ang 1–7 formation, and therefore we used a liquid chromatography/mass spectrometry (LC-MS)–based assay system to directly analyze ACE2-catalyzed hydrolysis of Ang II and validate our kinetic assay data.
A 2-h incubation of 50 µM Ang II with 10 nM ACE2 in the presence of 50 µM DMZ resulted in a 1.5-fold increase in Ang (1–7) formation compared to that in the absence of the drug. MALDI-TOF spectrometry was used to determine peak identities and confirmed the generation of Ang (1–7) (899 m/z), which provides direct evidence for Ang II cleavage by ACE2. Figure 6 shows HPLC chromatograms for ACE2 in the presence and absence of DMZ.
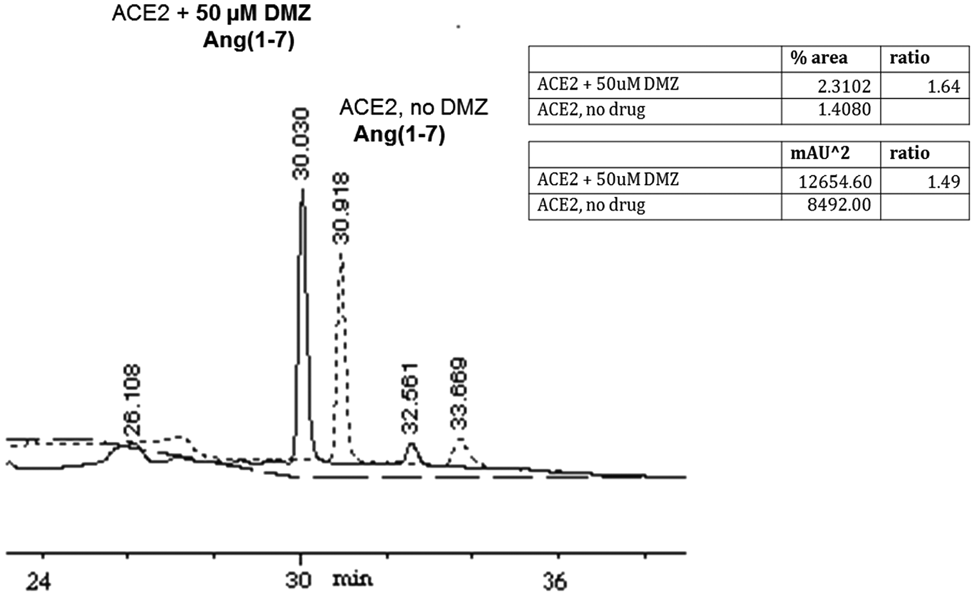
Diminazene (DMZ) enhances cleavage of natural substrate angiotensin II (Ang II). High-performance liquid chromatography (HPLC) analysis of angiotensin II cleavage was performed by incubating 10 nM angiotensin-converting enzyme 2 (ACE2) with 50 µM Ang II in the presence of 50 µM DMZ and in the absence of the drug. Peptide products were separated by HPLC, and their identities were assigned by matrix-assisted laser desorption/ionization (MALDI). Mass spectrometry confirmed the peak for Ang (1–7) at 899 m/z. Two-hour incubation with DMZ resulted in a 1.5-fold increase in Ang (1–7) formation compared to that in the absence of the drug (as determined by integrating area under the curve [AUC]).
Discussion
We designed a study that focuses on a newly discovered enzyme that is a key regulator of hypertension because high blood pressure is a frequently observed and documented off-target effect. We targeted a specific structural pocket in the hinge-bending region of ACE2 (using the coordinates of the crystal structure of ACE2 in the open conformation) and found three FDA-approved compounds that significantly modulated ACE2 activity: hydroxyzine (HXZ), minithixen (CTX), and diminazene (DMZ). Hydroxyzine and minithixen act by increasing specificity for the synthetic substrate, which is reflected by reduction in Km (KmHXZ = 11 ± 1.0 µM, KmCTX = 14 ± 0.8 µM vs KmACE2 alone = 40 ± 1.5 µM). In contrast, diminazene and labetalol produce a large increase in Vmax, which comes at a cost of loss in substrate specificity (KmDMZ = 82 ± 10.3 µM; Table 1 ). These data are consistent with the hypothesis that ACE2 conformational changes associated with substrate binding and/or product release may be rate limiting. This becomes particularly important in proteolytic cleavage of longer substrates, where fast turnover might be more difficult to achieve.
The effectiveness of FDA-approved diminazene as an ACE2 activator was confirmed by analysis of cleavage of the octapeptide angiotensin II, which is considered the main effector of the renin-angiotensin system (RAS) and the most physiologically significant natural substrate for ACE2. ACE2 EC50 values for diminazene fall in the low micromolar range (8.0 µM) and demonstrate stronger response to this FDA-approved compound than to XNT, a non-FDA-approved drug-like small molecule shown to reduce high blood pressure in the in vivo model systems.13,14
Repurposing or repositioning is a strategy that takes advantage of the information available on drugs that have been approved for clinical use to enable their use in achieving therapeutic goals not intended originally. Because new relevant drug targets are constantly emerging, strategies to validate their targeting in humans are urgently needed.
This study has implications on the specificity of some known drugs. For example, Normodyne, a beta-blocker used to treat hypertension, enhances ACE2 activity in vitro. Effects observed in vivo may be due to the interaction between this drug and a combination of molecular targets, possibly including ACE2. Drugs that are useful in treating psychosis, such as minithixen, enhance ACE2 activity, suggesting that low blood pressure may be considered a possible side effect of this treatment due to the promiscuity of these agents. However, because minithixen is considered safe in humans, experiments may be designed to test its utility in treating hypertension. Similarly, the antibacterial and antifungal drugs diminazene and hydroxyzine, respectively, enhance ACE2 activity, suggesting that these compounds, or related analogs, may be useful in controlling blood pressure. These findings suggest that identification of off-target proteins that FDA-approved drugs modulate can be achieved by molecular docking. This strategy is both an informative way to illuminate possible undesired side effects and a rapid way to screen approved compounds for new clinical purposes.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.