
Abstract
Mutations of DNA repair pathways contribute to tumorigenesis and provide a therapeutic target for synthetic lethal interactions in tumor cells. Given that tyrosyl-DNA phosphodiesterase 1 (Tdp1) repairs stalled topoisomerase-I DNA complexes, we hypothesized that inhibition of Tdp1 has synthetic lethal effects in some cancers. To test this, we screened tumor arrays for Tdp1 expression and observed that Tdp1 is expressed in many tumors, including more than 90% of human breast tumors. Subsequent chemical screening identified putative Tdp1 inhibitors. Treatment of control human mammary epithelial cells and the breast cancer cell line MCF-7 with compound CD00509 preferentially sensitized MCF-7 cells to camptothecin and decreased cell proliferation 25% more than camptothecin treatment alone. This suggests that CD00509 specifically targeted Tdp1 in vitro, and CD00509 increased the sensitivity of wild-type murine embryonic fibroblasts (MEFs) to camptothecin to a degree comparable to that of Tdp1−/− MEFs. In addition, consistent with poly ADP-ribose polymerase-1 (PARP-1) collaborating with Tdp1 in DNA repair, combined Tdp1 and PARP-1 inhibition was more detrimental to MCF-7 cells than either treatment alone, whereas the combination was not additively harmful to control mammary cells. We conclude that targeting Tdp1 in anticancer therapy preferentially enhances the sensitivity of some breast cancer cells to camptothecin and may be an effective adjuvant for breast cancer therapy.
Introduction
Deficits of DNA repair contribute to the survival and proliferation of tumor tissue and hence are implicated in cancer progression.1,2 Because the chemotherapeutic agents that target proliferative tumors are often harmful to normal cells and therefore carry significant adverse side effects, more specific targets that differentiate between tumorigenic and normal cells are needed.
The defects in DNA repair that many cancers have are an Achilles heel because these genetic deficits can have synthetic lethal interactions with inhibitors of other DNA repair processes. This is typified by the interaction between inhibitors of poly ADP-ribose polymerase-1 (PARP-1) and -2 (PARP-2) and BRCA1-deficient cancers. 3 Based on these observations and the facts that Tdp1 is highly expressed in some cancers and that it processes stalled DNA-topoisomerase I (TopoI) complexes, we and others hypothesized that Tdp1 function is essential for survival of those cancers and is therefore a potential anticancer target.4,5 Supporting this reasoning, reducing Tdp1 expression in some rhabdomyosarcoma cell lines enhanced the cytotoxicity of PARP-1 inhibitors and was independently cytotoxic. 5
Because Tdp1 repairs Topo I-DNA complexes such as those induced by the camptothecins (CPTs), inhibitors of Tdp1 may also enhance the sensitivity of cancer cells to the CPTs. Consistent with this thinking, increased Tdp1 expression counteracts the cytotoxicity of CPTs 6 and is frequently observed in cancers resistant to CPT therapy.4,5,7 Also, reduction of Tdp1 expression in rhabdomyosarcoma cell lines sensitizes them to CPT. 5
To identify an inhibitor that preferentially inhibits the growth of cancer cells but not noncancerous mammary epithelial cells, we devised an in vitro activity assay for Tdp1 and screened approximately 80,000 chemical compounds. Putative inhibitors identified by this screen were then evaluated in breast cancer cells for their ability to induce lethality, increase CPT sensitivity, and function additively with the PARP-1 inhibitor Rucaparib.
Materials and Methods
Expression and Purification of Tdp1 Proteins
N-terminally His-tagged Tdp1 protein was expressed as previously described. 8 Briefly, Tdp1 was expressed in Escherichia coli BL21 (DE3) cells using the pET expression system (Novagen), and after 2 h of induction with 1 mM isopropyl β-d-thiogalactoside, cells were harvested, and the dry pellets were stored at −80°C.
For Tdp1 purification, cell pellets were thawed on ice, resuspended in breaking buffer (100 mM KCl, 30 mM KPO4 pH 7.0, with protease inhibitors), and sonicated. After centrifugation at 20,000 × g (4°C) for 45 min, the supernatant was passed through a 22-gauge needle, filtered through a 0.22 µm filter, and loaded onto a Whatman Cellulose Phosphate P11 column. The column was washed with wash buffer (400 mM KCl, 30 mM KPO4 pH 7.0, with protease inhibitors) and eluted with wash buffer containing 800 mM KCl. The eluate was desalted using a PD-10 column (GE Healthcare) equilibriated with dialysis buffer [20 mM Tris, pH 7.9, 500 mM NaCl, 10 mM imidazole, and 0.2 mM phenylmethanesulfonyl fluoride (PMSF)] prior to passing through a NiSO4-charged HiTrap chelating column (GE Healthcare). The His-bind buffer kit (Novagen) supplemented with protease inhibitors was used to charge, bind, wash, and elute the column. The eluate was dialyzed against storage buffer (50 mM Tris-HCl, pH 7.5, 50 mM KCl, 1 mM EDTA, 2 mM DTT, and 50% glycerol) using a 10,000 MWCO Slide-A-Lyzer (Pierce). The protein was stored at −20°C.
Tdp1 in Vitro Activity Assay
The Tdp1 in vitro activity assay was designed as a linear quenched fluorescent substrate that negated interaction with DNA intercalators. A µFill microplate dispenser (Bio-Tek) was used to dispense 25 µL/well of a Tdp1 enzyme solution (0.02 µL purified Tdp1 (10 nM) in 10 mM Tris-HCl, pH 7.5, 50 mM KCl, 1 mM EDTA, 1 mM DTT, and 0.01% Brij-35) into wells of a black 384-well plate (Costar). Seventy (70) nL of each test compound (5 mM) was pinned into assay plates using an FP3-384 pin tool (VP Scientific) on a PlateMate™ Plus (Thermo Scientific) and incubated at room temperature for 30 min. During this time, the plates were read by a Varioskan® Flash multimode reader (Thermo) at Ex485/Em510 nm to identify false-positive compounds that had autofluorescence. The µFill dispenser was used to add 25 µL/well of linear oligonucleotide substrate [35 nM, (5′-/6-FAM/AGGATCTAAAAGACTT/3BHQ_1/-3′); Integrated DNA Technologies] in dH2O. The fluorophore 6-carboxyfluorescein (FAM) was coupled to the 5′ terminus, whereas the Black Hole Quencher (BHQ)_1 was coupled to the 3′ terminus. The whole plate was immediately read five times using a kinetic read on the Varioskan® (Ex485/Em510 nm). Tdp1 percentage inhibition was calculated for each compound by comparing the rate of increase in fluorescence throughout time for the compound-treated wells to that of DMSO control wells. All compounds considered for follow-up were retested using the primary screening assay, and if confirmed, an IC50 was determined using an eight-point concentration response curve. Concurrently, a counterscreen to identify false positives that quenched the fluorescence of the fluorophore was carried out using the substrate oligonucleotide without the quencher. Last, inhibitors were purchased and reconfirmed prior to the secondary cell-based assays.
We screened a total of >80,000 compounds. These included the CCBN (Canadian Chemical Biology Network) collection consisting of 16,000 selected Maybridge compounds, 9989 selected Chembridge compounds, the ChemBridge DIVERset (50,000 compounds; ChemBridge, San Diego, CA), the KD2 library consisting of the LOPAC (Sigma) library, the Prestwick library, and selected compounds from BIOMOL and Microsource.
Small-Molecule Database and Protein Structures
The small-molecule databases and the crystal structure data of Tdp1 were respectively downloaded from ZINC 9 and PDB. 10 Three crystal structures of Tdp1 were used for ligand screening (PDB accession numbers: 1jy1, 1qzq, and 1nop).
Computational Screening and Molecular Docking
The molecular docking software ICM was licensed from Molsoft LLC for a Linux-based computer cluster of 400 central processing units. The binding pocket was identified by the PocketFinder program. The pocket was represented by a set of maps for van der Waals (carbon-based and hydrogen-based), electrostatic, hydrogen bonding, and hydrophobic interactions. Each compound (ligand) of the database was docked to the pockets one by one, and a score (ICM score) reflecting the quality of the docked complex was assigned to each compound. 11 The compound was fully flexible, and both the intramolecular ligand energy and the ligand–receptor interaction energy were optimized during the docking. The conformational sampling was based on the biased probability Monte Carlo procedure in the internal coordinate space. Due to the nature of Monte Carlo–based sampling, we repeated the database screening process five times. The results were combined, and the best score for each compound was retained.
Tumor Microarray
Three tumor microarrays of 17 common adult cancers were purchased from Imgenex, Inc. To assess Tdp1 protein expression, the microarrays were first deparaffinized by baking at 62°C and stained for Tdp1 expression using antihuman Tdp1 serum as previously described. 5 Tumors were considered Tdp1 positive if Tdp1 expression was observed in the cytoplasm, the nucleus, or both locations.
Cell Culture
MCF-7, MDA-MB-231, human dermal fibroblasts, and murine embryonic fibroblasts (MEFs) were cultured in Dulbecco’s Modified Eagle’s Medium (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Hyclone), 4 mL/L L-glutamine, and 1% antibiotic–antimycotic (Gibco). HCC-1937 cells were cultured in RPMI-1640 (Gibco) with 10% heat-inactivated FBS (Hyclone), 4 mL/L L-glutamine, and 1% antibiotic–antimycotic (Gibco). SUM-1315MO2 cells were cultured in Ham’s F-12 media (Gibco), supplemented with 10 mM HEPES (Gibco), 5 µg/mL insulin (Gibco),10 ng/mL EGF (Gibco), and 20% heat-inactivated FBS (Hyclone). Human mammary epithelial cells were cultured in Mammary Epithelial Cell Basal Medium (Lonza) and supplemented with MEGM SingleQuots (Lonza). All cells were grown at 37°C with 5% CO2 in a humidified environment.
MCF-7, MDA-MB-231, HCC-1937, and SUM-1315MO2 cells were a kind gift from Dr. Sam Aparicio’s laboratory at the BC Cancer Agency. Human mammary epithelial cells (CC-2551) were purchased from Lonza. Wild-type and Tdp1−/− MEFs were obtained as previously described. 12 Tdp1−/− mice were generated by insertion of a gene trap vector into intron 11 of TDP1. 12
Tdp1 Knockdown
Tdp1 was knocked down transiently by transfecting 8×104 cells with 100 nM of pooled small interfering RNAs (siRNAs; Dharmacon) in a 24-well plate using Lipofectamine 2000 (Invitrogen). Knockdown of Tdp1 expression was confirmed by immunoblotting and quantitative reverse transcription (qRT)-PCR. The sequences of the siRNAs and PCR primers are listed in
γH2AX Assay
MCF-7 cells were seeded in MEM + 10% FBS; plated at 20,000 MCF-7 cells per well of a 96-well, black, clear-bottom view plate; and incubated at 37°C, 5% CO2 overnight. The next day, cells were incubated for 1 h at 37°C, 5% CO2 with medium containing 4 µM CPT and the putative inhibitor at a final concentration of 30 µM. This medium was removed after 1 h and replaced with medium containing only the putative inhibitor at a final concentration of 30 µM. Following incubation at 37°C, 5% CO2 and removal of growth medium, the cells were fixed by adding 100 µL of 4% PFA to each well and incubating for 15 min at 37°C, 5% CO2. Subsequently, the wells were washed with 150 µL phosphate-buffered saline (PBS) and stored at 4°C. For immunofluorescence, cells in each well were permeabilized by incubation at room temperature for 30 min with 100 µL of 0.3% TritonX-100 in PBS, washed 3× with 150 µL PBS, and blocked with 100 µL of 3% bovine serum albumin (BSA) diluted in PBS for 20 min. The cells in each well were then incubated at room temperature for 1 h with 50 µL of 0.5 µg/mL anti-H2A.X antibody (Millipore JBW301) diluted in 3% BSA, washed 3× with 150 µL PBS, incubated at room temperature for 45 min with 4 µg/mL secondary antibody (Alexa Fluor 488) diluted in 3% BSA, washed 3× with 150 µl PBS, stained with 1 µg/mL Hoechst (10 mg/mL stock), and immediately washed 3× with 150 µL PBS. Fluorescence was quantified using the Cellomics Arrayscan.
Comet Assay
Comet assays were performed to assess DNA damage as previously described. 12 Briefly, MCF-7 cells were seeded at a density of 3×105 cells per well in a 6 well plate. After 24 h, the cells were treated for 2 h with 4 µM of CPT and/or 10 µM of test compound at 37°C, 5% CO2. Then, 1×104 cells were harvested, resuspended in 80 µL of 0.5% low-melting-point agarose, and pipetted onto microscope slides precoated with 1% normal-melting agarose. After addition of a second layer of 1% normal-melting-point agarose, the cells were lysed by immersing the slides in lysis solution (2.5 M NaCl, 100 mM EDTA, 10 mM Trizma base, 1% Triton X-100, and 10% DMSO) for 2 h. DNA was then denatured by incubation of the slides at 4°C in alkaline buffer (50 mM NaOH, 1 mM EDTA, 1% DMSO, and pH>13) for 25 min. Following electrophoresis at 25 V and 4°C for 25 min, the slides were neutralized in 0.4 M Tris-HCl and stained with SYBR-Green (Invitrogen, Carlsbad, CA). Images were acquired using a Zeiss Axiovert 200 microscope, a Zeiss AxiocamMR camera, and the Zeiss Axiovision imaging system. Comet tail analysis was done using the Comet Assay IV software by Perceptive Instruments (Haverhill, UK).
MTT Assay
Cell proliferation and viability were measured by MTT assay, as previously described. 13 Briefly, 5×103 cells were plated in each well of a 96-well plate and cultured in phenol red-free media. MTT solution was prepared by dissolving 5 mg of MTT in 1 mL of 1× PBS. Following the described treatments, the culture media were supplemented with 10% MTT and incubated for 3 h. Subsequently, culture media were replaced with 100 µL of 100% DMSO, and cells were incubated for 30 min. All incubation steps were performed in the cell culture incubator (37°C with 5% CO2 in a humidified environment). Spectrophotometry was done at 565 nm using the Wallac VICTOR2 Multilabel Plate Reader (Beckman-Coulter).
Caspase-3/7 Activity Assay
Briefly, 5×103 cells were plated in each well of a 96-well plate and allowed to incubate in the cell culture incubator (37°C with 5% CO2 in a humidified environment). The cells were treated for 96 h with CD00509, Rucaparib, CPT, or a combination of these, as described. Caspase activity was measured using the Apo-ONE Homogeneous Caspase-3/7 kit according to the manufacturer’s protocol (Promega). Fluorescence of the cleaved caspase-3 and -7 substrate was measured at 520 nm using the Wallac VICTOR2 Multilabel Plate Reader (Beckman-Coulter).
TUNEL Assay
Briefly, 5×103 cells were seeded on a cover slip, placed in a six-well plate, and allowed to incubate in the cell culture incubator (37°C with 5% CO2 in a humidified environment). The cells were treated for 96 h with CD00509, Rucaparib, CPT, or a combination of these, as described. Nuclei with DNA breaks were visualized by colorimetric labeling of free 3′OH DNA termini using the Apoptag Plus Peroxidase In Situ Apoptosis Detection Kit according to the manufacturer’s protocol (EMD Millipore). The number of stained nuclei per 100 cells was recorded as indicative of apoptosis.
PARP-1 Inhibition
The PARP-1 inhibitor AG-014699 (Rucaparib) was obtained from Selleck Chemicals. 5×103 MCF-7 cells were seeded per well in 96-well microtiter plates. After 24 h, Rucaparib was added to the culture medium to a final concentration of 10 µM. Cell viability and proliferation were measured in triplicate every 24 h for 96 h by the MTT assay.
Statistical Analysis
Microsoft Excel was used to compute the group means and standard deviations for all treatment and control groups from cell viability data derived by the MTT and comet assays. GraphPad Prism software (La Jolla, CA) was used to analyze data from the primary screen, and γH2AX staining. Prism by GraphPad was used to perform two-way ANOVA to compare the differences in growth and viability under varying treatment conditions throughout time. Tukey’s test was used to assess statistical significance between individual groups. A p value of <0.05 was judged significant.
Results
Tdp1 Is Expressed in Many Cancers
To test if Tdp1 was highly expressed in cancers besides rhabdomyosarcoma and non–small cell lung cancer,4,5 we immunohistochemically assessed Tdp1 expression in 10 cores for each of 17 common adult cancers. We found that at least half of all the tumors assayed were Tdp1-positive, and that for 5 of the 17 cancers, ≥90% of the tumor samples were Tdp1-positive ( Fig. 1A ). The tissues of origin for these 5 cancers were thyroid, breast, liver, endometrium, and ovary.
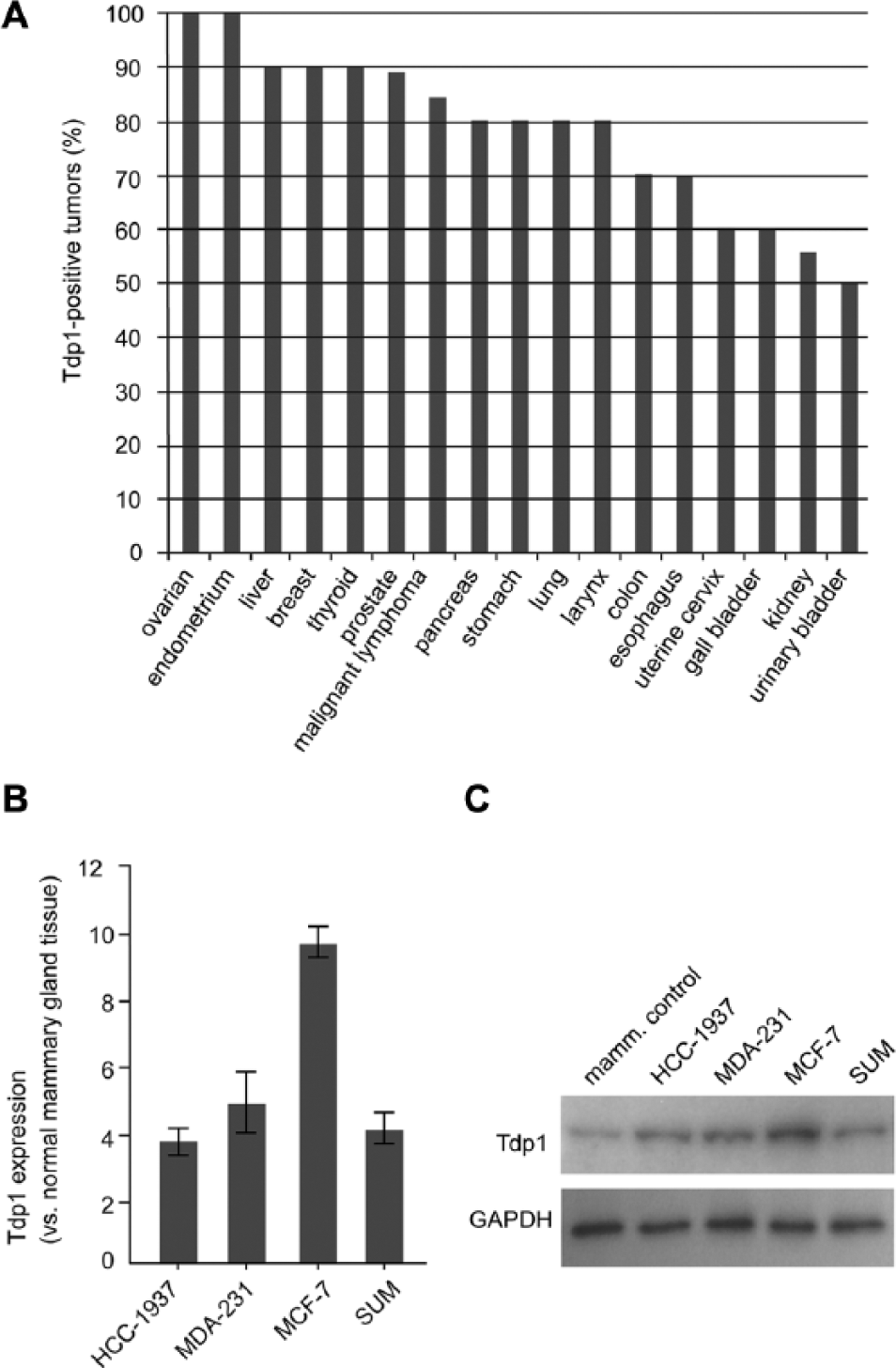
Tdp1 expression in cancer.
Tdp1 Has Increased Expression in Breast Cancer Cell Lines
Observing the expression of Tdp1 in many breast tumors, we hypothesized that Tdp1 expression is increased in breast cancer tissue. To test this, we compared Tdp1 mRNA and protein levels for the patient-derived breast cancer cell lines HCC-1937, MDA-MB-231, SUM-1315MO2, and MCF-7 with noncancerous mammary tissue and cultured mammary epithelial cells ( Fig. 1B–C ). Indeed, Tdp1 expression was 4–10-fold higher in the breast cancer cell lines than in the controls.
Knockdown of Tdp1 Expression Reduces the Proliferation of MCF-7 Cells
Given this expression of Tdp1 in breast tumors and cell lines and the known interaction between Tdp1 and PARP-1, 14 we hypothesized that, like PARP-1 inhibition, the abrogation of Tdp1 processing of blocked 3′ DNA termini is lethal to breast cancer cells. Using siRNA to achieve a 65–85% knockdown of Tdp1 expression, we observed a 20% reduction in the proliferation of MCF-7 cells and no change in proliferation for HCC-1937, MDA-MB-231, and SUM-1315MO2 ( Fig. 2A–H ).
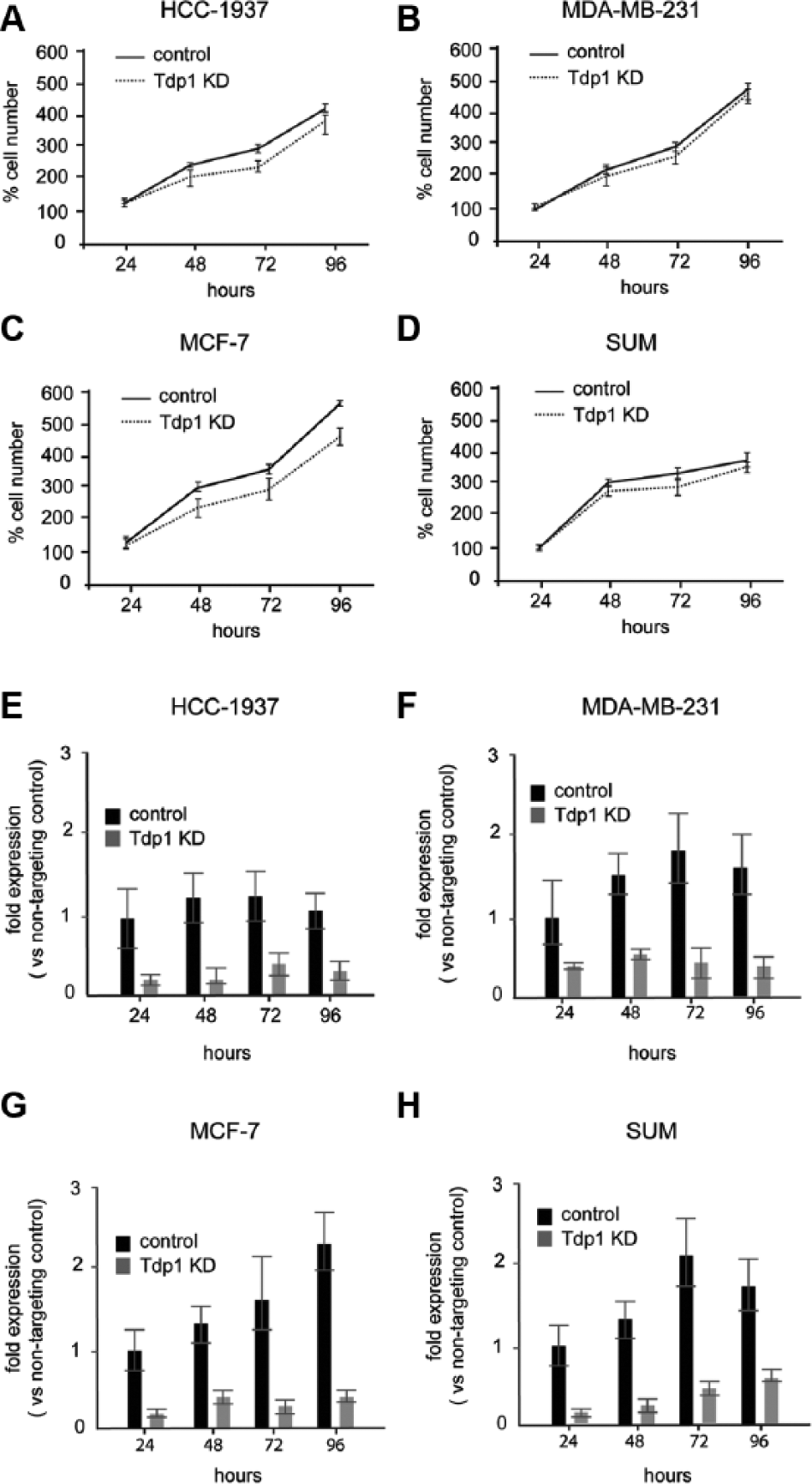
The effect of TDP1 knockdown on proliferation of breast cancer cell lines.
In Vitro Assay for Tdp1 Inhibitors
Given these findings and our prior observations with rhabdomyosarcoma, 5 chemical inhibition of Tdp1 may be an effective adjuvant for anticancer treatment. To screen for inhibition of Tdp1 activity, the hydrolysis of the phosphodiester bond between the O-4 atom of tyrosine and the 3′-phosphate of DNA,8,15 we constructed a single-stranded oligonucleotide substrate with a BHQ quencher instead of a tyrosine moiety attached to the 3′ end of the oligonucleotide via a phosphodiester bond and a 6-FAM fluorophore attached to the 5′ end ( Fig. 3A ). Basal fluorescence of this substrate is fully quenched. The recombinant Tdp1 enzyme had a Km of 23.8 and a Vmax of 0.0016 for cleavage of the phosphodiester bond between the quencher and the oligonucleotide.
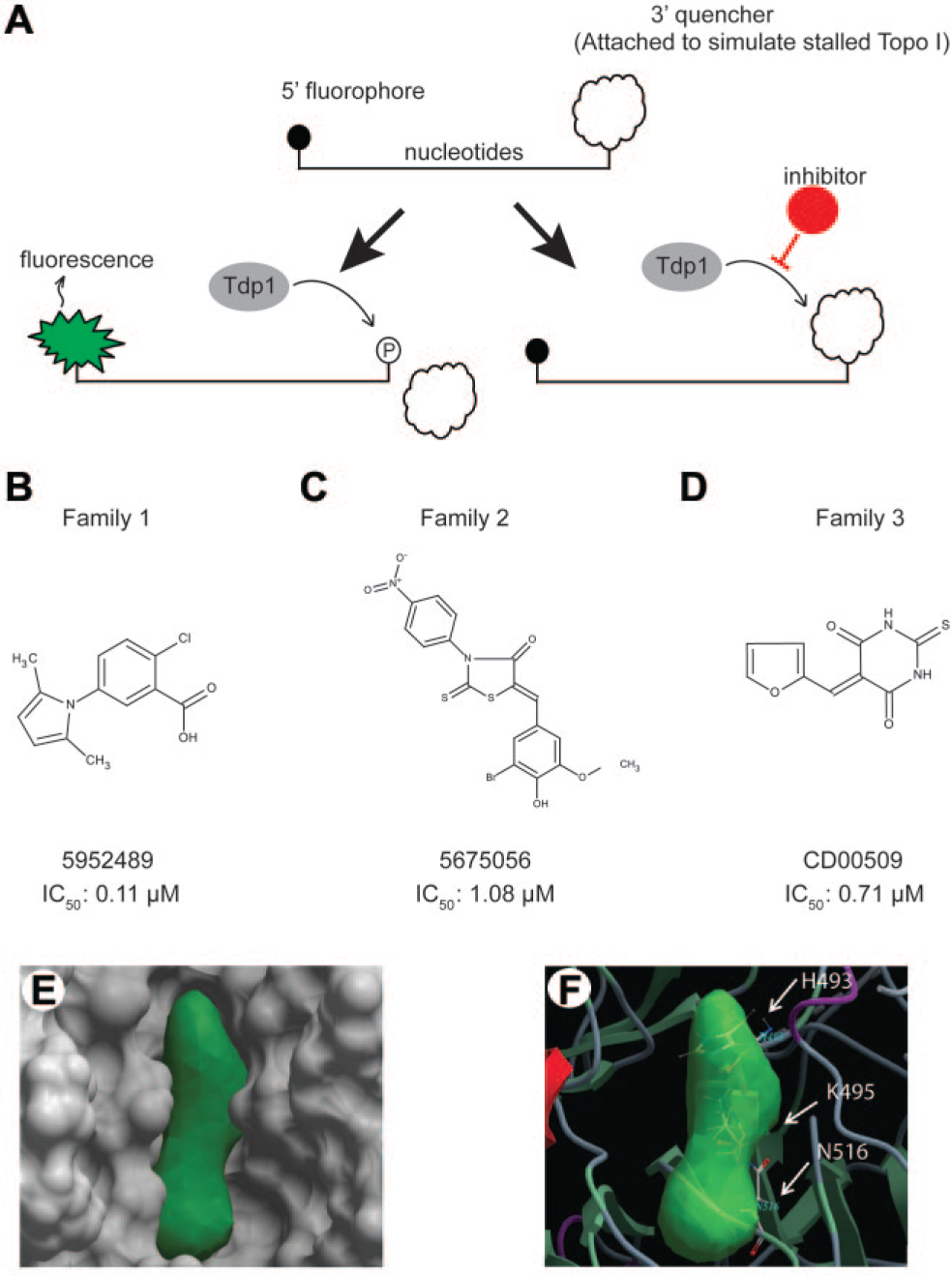
In vitro and in silico screens for inhibitors of Tdp1 activity.
The substrate and the purified Tdp1 were tested by conducting a pilot screen with the KD2 library of approximately 5000 compounds. To exclude false positives that intrinsically quenched the fluorophore, we screened all identified compounds using a single-stranded oligonucleotide with a fluorophore bound to the 5′ terminus but no quencher on the 3′ terminus. All compounds that quenched the fluorophore also had an autofluorescence more than fourfold that of DMSO control when premeasured at the same Ex485/Em510 nm. Therefore, only compounds that had less than fourfold autofluorescence compared to DMSO and that inhibited Tdp1 in the biochemical assay >70% were selected for retesting. Of the 31 compounds restested, 14 confirmed and inhibited Tdp1 >60%.
Identification and Characterization of Putative Tdp1 Inhibitors Using the in Vitro Assay
Having validated the in vitro assay methodology, we screened the CCBN collection (Canadian Chemical Biology Network) and the ChemBridge DIVERSet (ChemBridge) libraries for compounds that inhibited Tdp1 activity at low micromolar concentrations (
Top 17 Tdp1 Inhibitory Compounds Carried Forward to the Secondary Screen.
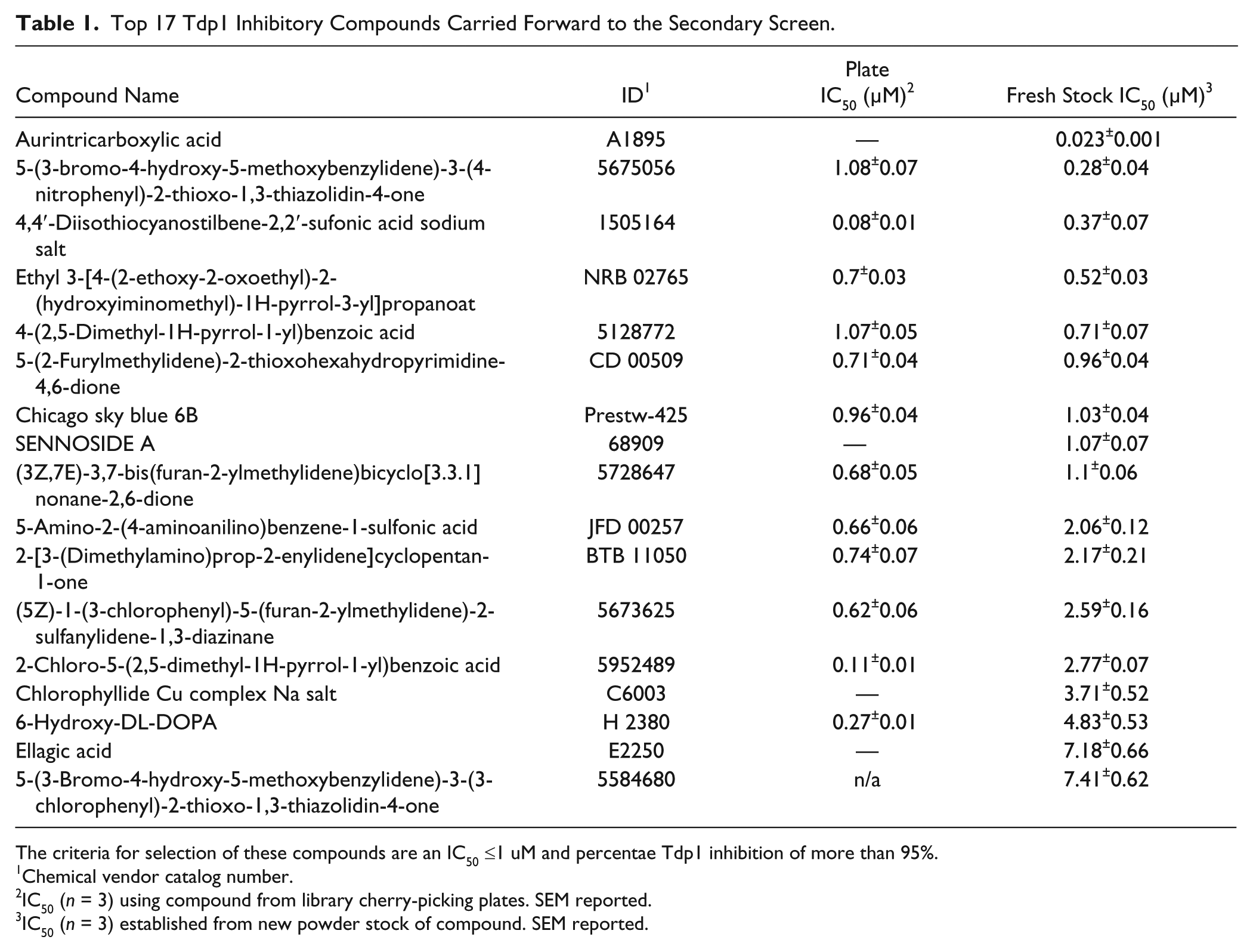
The criteria for selection of these compounds are an IC50 ≤1 uM and percentae Tdp1 inhibition of more than 95%.
Chemical vendor catalog number.
IC50 (n = 3) using compound from library cherry-picking plates. SEM reported.
IC50 (n = 3) established from new powder stock of compound. SEM reported.
In silico screen and compound selection
In parallel with the in vitro screen, we conducted a screen for compounds predicted to bind the Tdp1 catalytic site using molecular ligand docking. ICM (MolSoft) was used for screening of approximately 140,000 and 500,000 compounds from the U.S. National Cancer Institute (NCI) and the ChemBridge databases, respectively.
16
Each compound (ligand) was docked to the small-molecule binding pocket in a flexible-ligand rigid-receptor docking approach. Protein flexibility was addressed by using multiple receptor conformations from experimental structures. A docking score reflecting the “fit quality” of the ligand to the receptor was calculated and used for compound selection, in which low scores indicate a better fit. The best scoring compounds were also inspected visually and evaluated according to their chemical and druglike properties, as well as three-dimensional conformations of the docked ligand–receptor complex (
Fig. 3E–F
). Forty compounds were selected and obtained for in vitro validation (
CD00509 and 5675056 Increase γ-H2AX Foci in CPT-Treated MCF-7 Cells
Based on their sensitivity to Tdp1 knockdown (
Fig. 2C and 2G
), MCF-7 cells were selected for cell-based studies of the putative Tdp1 inhbitors. Because Tdp1 deficiency increases the number of blocked DNA 3′ termini arising from stalled Topo I and consequently the number of γ-H2AX foci following treatment with CPTl
17
, we tested if any of the 17 compounds identified above caused greater than baseline (
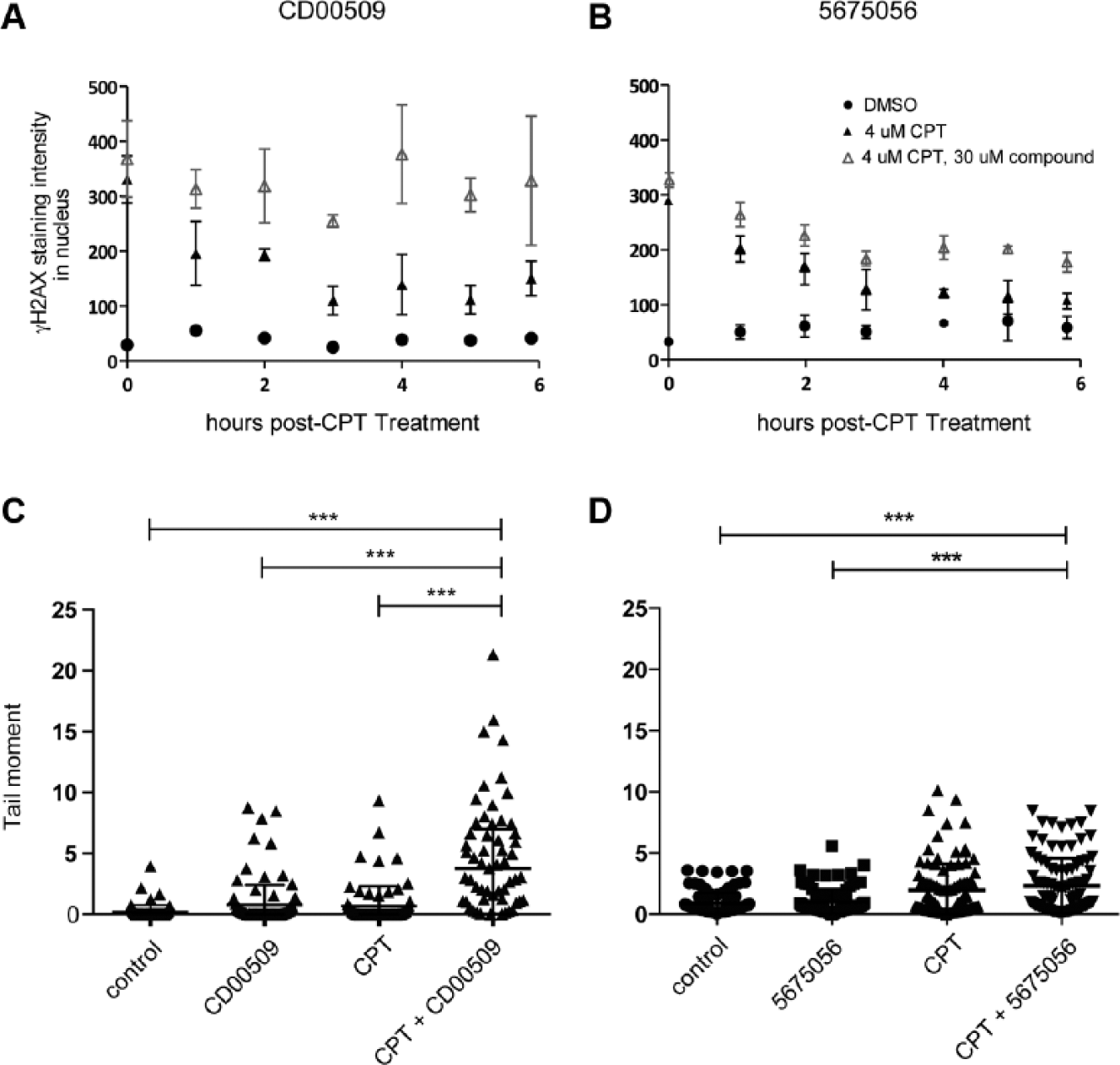
Tdp1 inhibitor CD00509, but not 5675056, is additive with camptothecins (CPT) in inducing DNA damage.
To determine if treatment with CD00509 and 5675056 not only acutely increased sensitivity to CPT but also impeded repair of the Topo I-DNA complex after CPT treatment, we measured the persistence of γ-H2AX foci after removal of CPT. This showed a statistically significant increase in the number of γ-H2AX foci until 6 h after CPT treatment for CD00509 but not 5675056 ( Fig. 4A–B ). The prolonged persistence of γ-H2AX observed in CD00509-treated MCF-7 cells is consistent with cellular toxicity and DNA repair by less efficient mechanisms. 18
CD00509 Increases DNA Breaks Detectable by Alkaline Comet Assay of CPT-Treated MCF-7 Cells
To correlate γ-H2AX foci number with DNA strand breaks, we measured DNA strand breaks by the alkaline comet assay as previously described. 5 MCF-7 cells were treated with 10 µM of candidate Tdp1 inhibitors and 4 µM CPT for 2 h before analysis of DNA strand breakage. Treatment with CD00509, but not 5675056, sensitized MCF-7 cells to CPT, as shown by an increase in DNA breaks by alkaline comet assay ( Fig. 4C–D ).
CD00509 and 5675056 Do Not Decrease the Proliferation of Wild-Type Human Mammary Epithelial Cells
To determine if the Tdp1 inhibitors CD00509 and 5675056 were toxic to noncancerous cells, we measured the LD50 for human mammary epithelial cells. By MTT assay, neither of the compounds decreased the viability of mammary epithelial cells up to 10 µM Tdp1 inhibitor (
Combined Tdp1 and Toposiomerase-I Inhibition Reduces MCF-7 Cell Number More than Does Either Treatment Alone
Because CD00509 treatment increased the number of DNA strand breaks in CPT-treated MCF-7 breast cancer cells, we determined the effect of CD00509 and CPT on cellular proliferation. Treatment with 10 µM CD00509 alone reduced the number of MCF-7 cells by 16%, but it did not impede the proliferation of mammary epithelial cells ( Fig. 5A–D ). Treatment with 1 µM CPT alone reduced the number of MCF-7 cells by 32% and mammary epithelial cells by 24% after 96 h ( Fig. 5A–B ). Co-treatment with CD00509 and CPT reduced the proliferation of MCF-7 cells by 50% (18% more than did CPT treatment alone), whereas combination treatment in mammary epithelial cells was not additionally detrimental compared to CPT treatment alone ( Fig. 5A–B ).
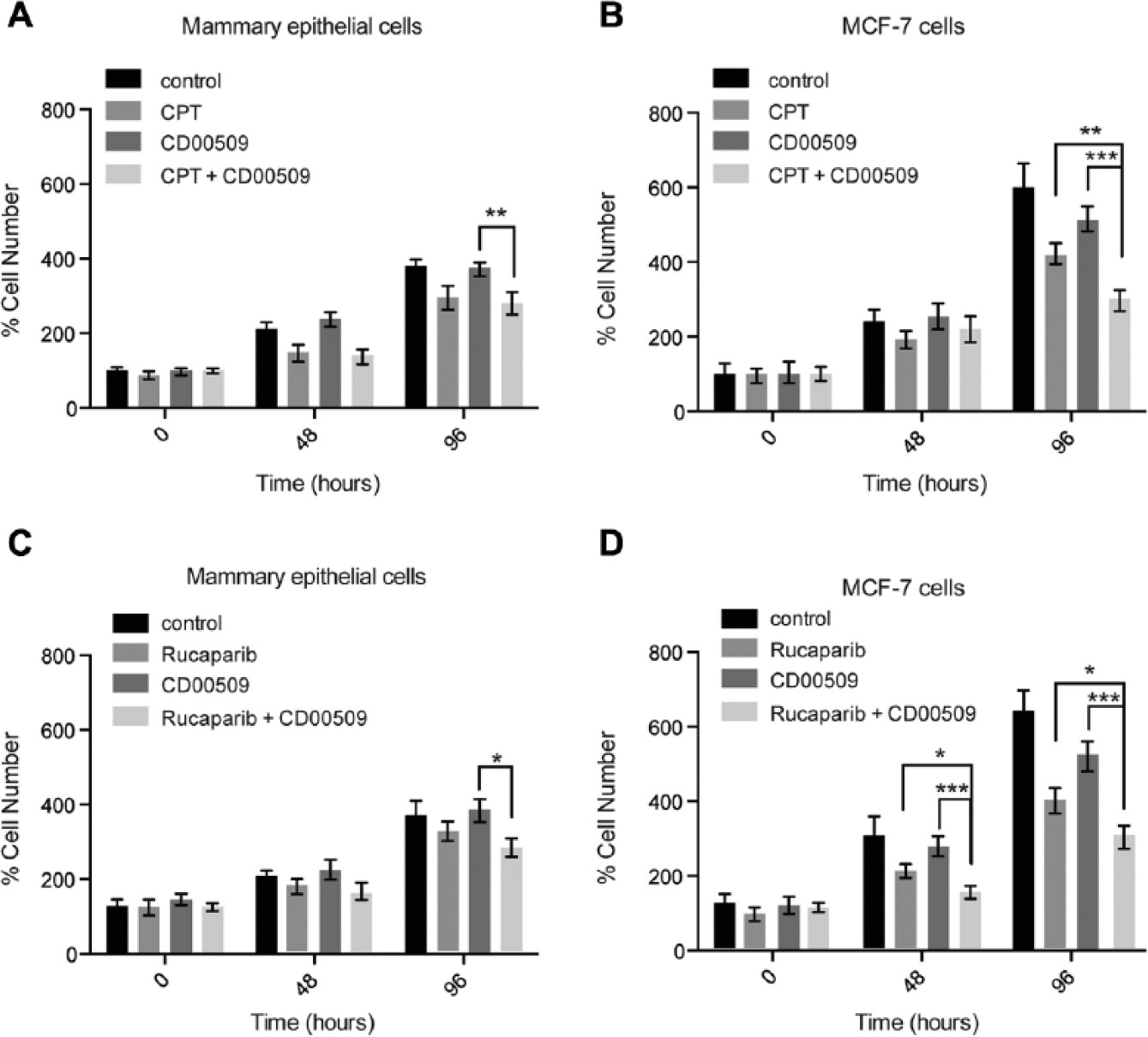
Co-treatment with CD00509 and CPT or CD00509 and Rucaparib is more lethal to MCF-7 cells than either treatment alone, as measured by MTT assay.
To ascertain whether CD00509 and CPT co-treatment reduced cell number by impairing cellular proliferation or apoptosis, we checked for activation of caspases-3 and -7 by a colorimetric assay and for DNA fragmentation by TUNEL assay. Both measures suggest that the combined treatment preferentially increased apoptosis in MCF-7 cells compared to control mammary epithelial cells (
CD00509-Treated Tdp1+/+ MEFs Are Comparably Sensitive to CPT as Tdp1−/− MEFs
After establishing that CD00509 was not cytotoxic to Tdp1+/+ and Tdp1−/− MEFs and that Tdp1−/− MEFs were more sensitive to CPT treatment than Tdp1+/+ MEFs (
Inhibition of MCF-7 Proliferation Is More Severe with Combined CD00509 and PARP-1 Inhibition than Either Treatment Alone
Because PARP-1 poly-ADP ribosylation of DNA-binding proteins at DNA breaks facilitates the recruitment of DNA repair proteins,
19
and cancers frequently have compromised DNA repair processes
20
that poorly tolerate further impairment, we investigated the effect of the PARP-1 inhibitor Rucaparib (AG014699) on MCF-7 cells. Rucaparib treatment decreased MCF-7 cell number by 40% (
Fig. 5D
), and the combination of Rucaparib with CD00509 decreased MCF-7 cell number by 56% (15% more than Rucaparib treatment alone) (
Fig. 5D
). Neither Rucaparib nor CD00509 treatments alone significantly impaired the proliferation of mammary epithelial cells (
Fig. 5C
), although the combination of Rucaparib and CD00509 decreased the number of mammary epithelial cells by 25% (
Fig. 5C
). Combined CD00509 and Rucaparib treatment also caused an increase in caspase-3/7 activation and TUNEL staining in MCF-7 cells but not mammary epithelial cells, indicating that the decrease in cell number is likely due to apoptosis (
Discussion
We find that Tdp1 is highly expressed in many cancers and that its subcellular localization is variably nuclear and cytoplasmic, suggesting that further analysis of its subcellular localization might provide a basis for cancer stratification. In addition, we find that Tdp1 inhibition may be an effective adjuvant in anticancer therapy when combined with the use of other DNA repair inhibitors such as Topo I poisons. Through the screening of chemical libraries using in silico and in vitro efforts, we identified CD00509 as a potential Tdp1 inhibitor. Furthermore, consistent with such inhibition, we found that treatment with CD00509 increased MCF-7 sensitivity to CPT and additively suppressed cell proliferation when combined with the PARP-1 inhibitor Rucaparib.
The suppression of MCF-7 cell proliferation by CD00509 and Rucaparib occurs in the absence of identified BRCA1 or BRCA2 mutations.21,22 This suggests that Rucaparib and CD00509 have potential synthetic lethal interactions with other deleterious mutations carried by the MCF-7 cell line. For example, the mutation in PIK3CA (NP_006209.2:p.E545K) causes MCF-7 cells to proliferate more rapidly than other breast cancer cell lines ( Fig. 2A–D ); 23 therefore, one might hypothesize that because Tdp1 and PARP-1 help maintain DNA replication forks,24,25 inhibition of these enzymes by CD00509 and Rucaparib causes replication fork collapse and cytotoxicity.
Induction of synthetic lethality to selectively kill cancer cells has been discussed for at least 20 years. 26 According to data annotated by the Cancer Cell Line Encyclopedia, Tdp1 is highly expressed in more than 30 cancers. 21 Because Tdp1 is a member of the phospholipase D (PLD) family, known inhibitors of the PLD family such as vanadate, paromomycin, lividomycin, and several teracyclines have been tested on Tdp1, but they are only weak inhibitors. 27 A recent paper by Walker et al. describes the utility of fluorescence-based oligonucleotide assays in the determination of Tdp1 activity in whole-cell extracts 28 and validates our independently developed sensitive and high-throughput approach to Tdp1 inhibitor screening. Removing the extra Gyrasol assay sensor step used by Walker et al., our design of the quencher and fluorophore on the small substrate permitted real-time kinetic monitoring of Tdp1 activity and decreased the background fluorescence to a signal-to-noise ratio >10.
The most effective Tdp1 inhibitors identified to date are two phosphotyrosine mimetics, methyl-3.4-dephostatin and the steroid NSC88915.29,30 Examination of the structure–activity relationship between these and CD00509 reveals no close structural similarities. CD00509 is an alkylidene barbiturate, a class of compounds notable for reactivity interference. 31 The specificity of CD00509’s action in increasing the sensitivity of Tdp1+/+ MEFs to CPT, however, suggests that this compound may be specifically targeting the Tdp1 enzyme and thus an exception to the PAINS (Pan Assay Interference Compounds) filter, which identifies structural features that appear as frequent hitters (promiscuous compounds) in many biochemical high-throughput screens. Nonetheless, further studies are required to determine the specificity of CD00509 and its cytotoxicity when combined with CPT.
More than 20 distinct diseases have been associated with defective DNA repair. 32 This highlights the importance of DNA repair in disease, including cancer, which has a high turnover and heightened sensitivity to DNA repair inhibition. The relative contribution of closely related and potentially redundant DNA repair pathways needs better delineation, however, to pave the way for effective targeted drug development.
In the context of such targeted drug development, there is a need for combination therapies that improve anticancer activity in cancers resistant to conventional therapies such as Topo I poisons. To the best of our knowledge, no such Tdp1 inhibitors are being developed in the preclinical or clinical stage, although a co-inhibitor of Tdp1 and Topo I has been described. 33 Herein, we confirm the potential efficacy of Tdp1 inhibition and the potential utility of combination therapies with PARP-1 inhibitors.
In summary, we provide further evidence that Tdp1 is a valid anticancer target and report a novel high-throughput method for identifying Tdp1 inhibitors using a single-stranded oligonucleotide reporter capable of identifying compounds with significant specificity and efficacy for inhibiting Tdp1. We identified CD00509, a potent and specific Tdp1 inhibitor, and show that it intrinsically restricts the growth of some patient-derived breast cancer cells and that, with or without PARP inhibition, it further sensitizes patient-derived breast cancer cells to Topo I poisons.
Footnotes
Acknowledgements
We thank Dr. Derek van Pel for his criticism, suggestions, and review of this manuscript. We also wish to acknowledge the generous support of the Centre for Drug Research and Development. C. F. Boerkoel is a scholar of the Michael Smith Foundation for Health Research and a clinical investigator of the Child & Family Research Institute.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by grants from Puget Sound Partners for Global Health, Seattle, WA, USA (to H Interthal); the Michael Smith Foundation for Health Research [CI-SCH-O1899(07-1) to C. F. Boerkoel]; the Child & Family Research Institute (to C. F. Boerkoel); the Child & Family Research Institute Pediatric Oncology Research Program (to C. F. Boerkoel); the Canadian Institutes of Health Research (PPP-248178); the Centre for Drug Research and Development; and GenomeBC.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.