
Abstract
The global impact of synthetic biology has been accelerating, because of the plummeting cost of DNA synthesis, advances in genetic engineering, growing understanding of genome organization, and explosion in data science. However, much of the discipline’s application in the pharmaceutical industry remains enigmatic. In this review, we highlight recent examples of the impact of synthetic biology on target validation, assay development, hit finding, lead optimization, and chemical synthesis, through to the development of cellular therapeutics. We also highlight the availability of tools and technologies driving the discipline. Synthetic biology is certainly impacting all stages of drug discovery and development, and the recognition of the discipline’s contribution can further enhance the opportunities for the drug discovery and development value chain.
Keywords
Introduction
Synthetic biology is the design and construction of new biological entities such as enzymes, circuits, modules, or systems, or the redesign of existing biological systems through reprogramming of genetic information for useful purposes. It has been developing since the 1960s, 1 but has expanded rapidly in the last 10 years, not least because of the plummeting cost of DNA synthesis, advances in genetic engineering, and a better understanding of genome organization and data science. Yet much of what surrounds this enigmatic and exciting field has been part of molecular biology for as long as we can remember. What distinguishes synthetic biology from traditional molecular and cellular biology is the focus on the design and construction of core components that can be modeled, understood, and optimized to meet specific performance criteria, and the assembly of these smaller parts and devices into larger integrated systems to solve specific problems. 2 So what makes this synthetic biology different, and what makes the time right for this field to promise so much to the pharmaceutical industry? In this review, we aim to capture some of the most tangible and promising areas for synthetic biology for drug discovery and development. We define how this blossoming field has already impacted the pharmaceutical industry and describe how it can further enhance the drug discovery value chain.
Speed, cost, and quality have long been the fundamental elements that control the productivity of the pharmaceutical industry. With a focus on improving the efficiency of drug discovery, all three are critical elements that have been incrementally improved by applying tools from synthetic biology. However, game-changing acceleration of drug discovery requires radically different approaches to the classical laboratory-based design-make-test cycles that underpin the iterative design of compounds. While the application of artificial intelligence and automated chemistry has strong support within the pharmaceutical industry,3,4 the potential application of synthetic biology beyond tool-based advances is relatively unappreciated and without specific acknowledgment. It is not just in the pharmaceutical industry that its impact has been underappreciated. A recent Economist article highlighted that the money made from organisms that had been genetically engineered underpinned 2% of the U.S. gross domestic product in 2017 ($388B) and is growing, from three sectors, pharmaceuticals ($137B), crops ($104B), and, less visible but even more lucrative, industrial biotechnology ($147B). 5 There is a rapid growth in the new synthetic biology industry, with start-up companies receiving ∼$6.1B investment since 2015. 6 The investment is accelerating, with Forbes reporting that synthetic biology start-ups received circa $3B in just the first 6 months of 2020. 7
There are numerous reviews and journal articles that reference the potential of synthetic biology to impact on drug discovery and produce new medicines.8,9 However, there are relatively few examples where they have been able to specifically describe how synthetic biology has already changed drug discovery. It is the authors’ opinion that many pharmaceutical scientists would not be able to identify a single synthetic biology influence on novel medicines, even though the directed evolution of therapeutic monoclonal antibodies has changed the face of modern drug therapy. In contrast to the minimal recognition within the pharmaceutical industry, perhaps two of the most exciting fields for synthetic biology, cell therapy and genetic reprogramming, have been combined within immuno-oncology to produce one of the most novel medical approaches, using chimeric antigen receptor T cells (CAR-T cells). 10
In this review, we align some of the exciting applications of synthetic biology to the drug discovery value chain ( Fig. 1 ), beginning with the toolkit of synthetic biology and the design-make-test-learn cycle, akin to the design-make-test-analyze cycle of medicinal chemistry. In each section, we also highlight the most disruptive innovative pieces of work, which, while a subjective choice, can at least serve as a next step for readers who wish to delve a little deeper into one or the other of the sections.
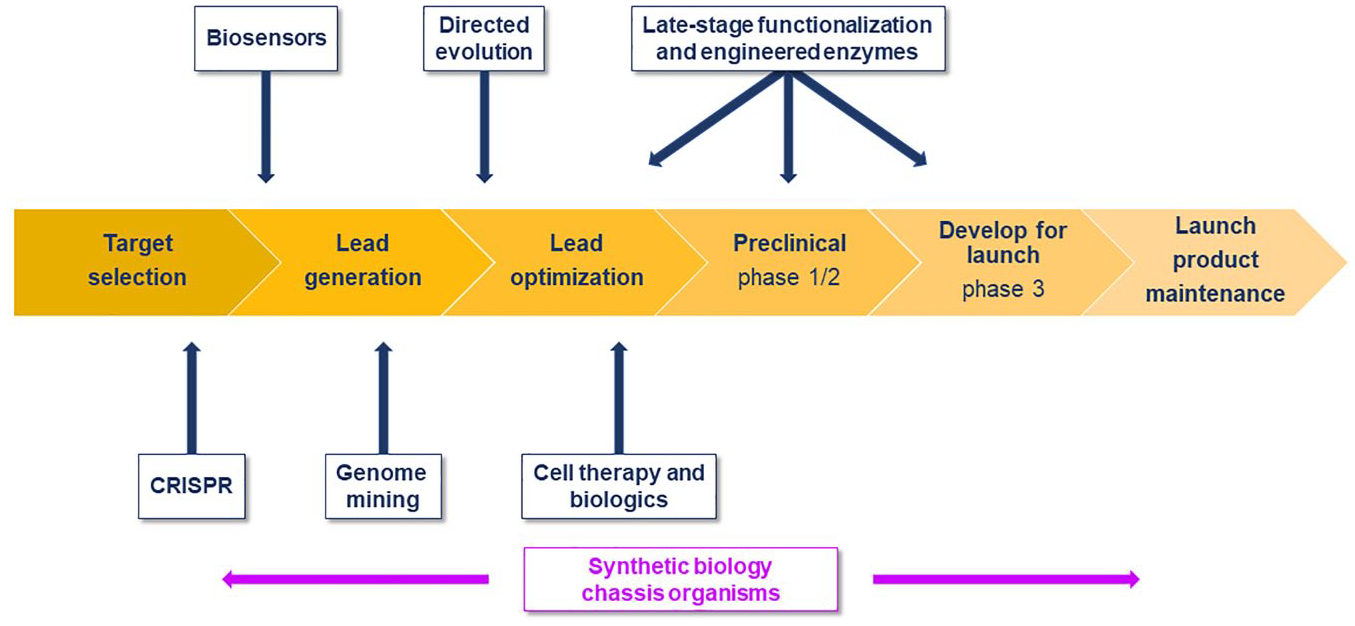
Synthetic biology contributions aligned to the drug discovery process.
The Design-Build-Test-Learn Cycle in Synthetic Biology
In recent years, we have seen major advances in the development of robust synthetic biology chassis organisms, the design of biological circuits, and high-throughput screening (HTS) technologies beginning to speed up modern drug discovery cycles. Substantial drops in DNA sequencing and synthesis costs have led synthetic biology to make major steps forward, for example, enabling the generation of DNA-encoded compound libraries, 11 genome mining for novel biosynthetic gene clusters (BGCs) for natural product discovery, 12 and advancing research toward the construction of programmable living therapeutics. 13
Synthetic biology chassis organisms like Escherichia coli and Saccharomyces cerevisiae play a key role in this field, as they are very well characterized, facilitate robust applications, and are cellularly reprogrammable. Modular design principles are important to easily exchange parts and to enable rapid generation and characterization of genetic devices and systems. In addition, an orthogonal mode of action is essential to separate biosynthetic functions from the chassis cell’s own intrinsic regulatory networks whose behavior is difficult to anticipate and might create background noise due to the environmental stimuli or cell cycle progression.
The concept of design-build-test-learn (DBTL) cycles is at the heart of synthetic biology applications. It enables efficient screening and optimization for desired functions of biosynthetic devices and systems of interest, ranging from proof-of-concept studies to advanced drug discovery screens and genetic circuit design in living therapeutics.
The design step relies on well-characterized biological parts and computer-aided approaches. These parts comprise means to specifically control gene expression or translation, including promoters, 14 terminators, 15 variation in codon usage, 16 ribosome binding sites (RBSs), 17 ribozymes, 18 and protein degradation tags. 19 We see an increasing number of synthetic promoters developed, allowing for orthogonal function and robust fine-tuning of gene expression. 20 Strategies employed include the combination of a core promoter region with binding sites for heterologous/hybrid transcription factors 21 or Cas9 coupled to transcriptional regulators.22,23 Also, inducible control of expression through external stimuli is often essential to have a direct control input to the system.24,25 The mentioned tools are used to fine-tune and characterize the behavior of genetic devices, including activators, repressors, reporters, and environment or drug/metabolite-responsive biosensors. Computational tools like AutoBioCAD, 26 Cello, and the Synthetic Biology Open Language (SBOL) 27 were developed to streamline this design step and facilitate laboratory automation and public reporting of data. 28
In the build phase, efficient DNA assembly tools, such as the Gibson-based assembly, 29 ligase cycling reaction, 30 BioBrick assembly (BASIC), 31 uracil-specific excision reagent (USER),32,33 and Golden Gate assembly,34,35 are used to ease and speed up the combinatorial assembly of parts and construction of combinatorial libraries.
The test phase relies on high-throughput analytics employing parallel cultivation platforms in microwell format 36 or microfluidic droplets 37 and the monitoring of reporter outputs like fluorescent proteins or liquid chromatography–mass spectrometry (LC-MS) analytics for compound detection. To further increase throughput, we might see methods like automated laser-assisted rapid evaporative ionization MS (LA-REIMS) used more in the future. For example, in a recent approach it was successfully used to perform rapid MS direct from agar plate yeast colonies without sample preparation or extraction. 38
In the learn step, various in silico modeling approaches are employed for simulation and optimization. This includes the usage of differential equations and machine learning algorithms as, for instance, recently shown for the rational tuning of G-protein-coupled receptor (GPCR) signaling, 39 engineering of synthetic gene networks in yeast, 40 or model-guided design of artificial yeast promoters. 41 Design of experiment (DoE) tools explore the multidimensional experimental space allowing for the modeling and optimization, for example lately employed to optimize the dynamic and operational range of a metabolite-responsive biosensor. 42
A main future driver within this cycle is the increasing application of automation and software development, integrating the different steps of the DBTL cycle in an industrial-like pipeline and enabling the characterization of thousands of parts and biosynthetic designs. 43 The concept was termed biofoundry (BioFAB) and has recently led to the establishment of a Global Biofoundry Alliance 44 coordinating activities in the field worldwide.
Synthetic Biology Chassis Organisms
Synthetic biology chassis organisms are key to rapid engineering and testing cycles to speed up drug discovery and optimize production. Detailed knowledge about the genome, function of proteins, and predictability and modeling of metabolism enable targeted genetic modifications to create, for example, platform strains for the production of certain molecule classes, construction of compound libraries, and implementation of biology circuits to screen for new drug leads. Synthetic biology tools as described above are well established, making the implemented systems easier to study and tune more efficiently. In the following, we summarize the role of the two main synthetic biology chassis and their role in the drug discovery pipeline.
The Prokaryotic Synthetic Biology Chassis: Escherichia coli
The gram-negative bacterium E. coli is the most studied in detail prokaryotic model organism. With its 4.6 Mbp genome, 4288 protein-coding genes, and 85% of the operons with proven function, 45 it is very well characterized and used in many applications within synthetic biology and natural product production. E. coli was successfully genetically engineered to produce complex natural products, including terpenoids, polyketides, phenylpropanoids, and alkaloids, as recently reviewed elsewhere. 46 To name a few, production was enabled for complex molecules like the antibiotic valinomycin,47,48 anticancer drug taxol precursor taxadiene, 49 and morphine precursor reticuline 50 ( Fig. 2 ).
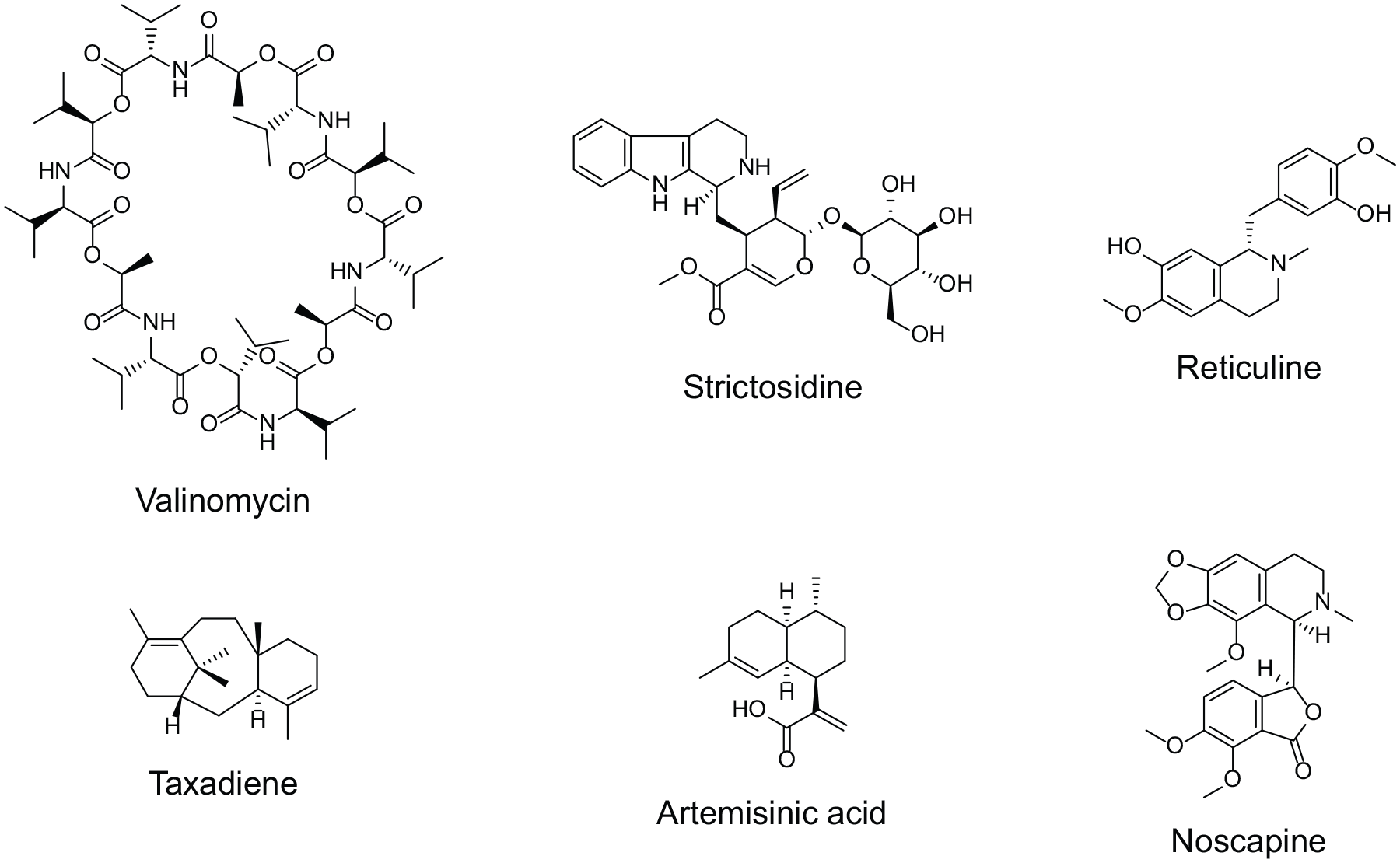
Structures of natural products synthesized in engineered E. coli or S. cerevisiae.
E. coli cells are an integral part of several high-throughput drug screening platforms. The industrially most relevant is the 2018 Nobel Prize-winning phage display technology used to identify and mature specific antigen binding domains for antibody engineering. 51 Libraries of antibody variable domains are expressed and presented on E. coli-produced bacteriophages that are used for panning rounds to identify superior antigen binders. In a different method, named phage-assisted continuous evolution (PACE), 52 the phage replication is dependent on the production of effective protein binders enabling the continuous in vivo evolution and selection of potent protein inhibitors. In another approach, E. coli was shown to be a suitable host to express cyclic peptide libraries and screen these for specific protein–protein interaction (PPI) inhibitors in vivo.53,54
Synthetic biology-engineered E. coli are being developed as cell therapies directly, as will be described in the Cell Therapy and Biologics section. Besides many examples in the preclinical stage, 55 we already see the first therapeutic bacteria that have entered phase I and II clinical trials for various indications, including hyperammonemia,56,57 phenylketonuria,58,59 and oral mucositis. 60 Despite holding great promise for many future applications, safety, regulatory, and public concerns have to be addressed before these new types of therapeutics become commercially available.
Although used in many studies and with a proven track record for drug screening approaches, E. coli has certain limitations when it comes to advanced chemical modifications of products like glycosylation of antibodies and monooxygenations of natural products. Also, looking at its potential for commercial drug production, E. coli-based bioprocesses have the disadvantage of being prone to phage contamination and come with increased purification costs due to the pyrogenic nature of its endotoxic membrane structures.
The Eukaryotic Synthetic Biology Chassis: Saccharomyces cerevisiae
Baker’s yeast S. cerevisiae is a very well-characterized eukaryotic model organism. It harbors 16 chromosomes, comprising a genome of 12 Mbp, with 6275 genes coding for 6049 proteins, where 90% have a characterized function. 61 S. cerevisiae is used for the commercial production of several drugs, including insulin, 62 vaccines, 63 and the antimalaria drug artemisinin. 64 Besides, S. cerevisiae is an established disease model organism to study, for example, aging, 65 neurodegenerative diseases, 65 drug toxicity, 66 and cancer. 67 For example, yeast single-gene deletion libraries, comprising more than 4000 strains, 68 were used to find synthetic lethal interactions using large-scale primary interaction screens, revealing insights into new combinatorial cancer therapies. 69
A prime synthetic biology undertaking is the ongoing Yeast 2.0 project aiming to resynthesize its entire genome. So far, seven chromosomes 70 have been synthesized, including specific modifications allowing for so-called genome scrambling (Synthetic Chromosome Recombination and Modification by LoxP-mediated Evolution [SCRaMbLE]). Through induction of a heterologous Cre recombinase, the genome can be cut and reassembled to easily create a large genotypic diversity. These can be screened for advanced phenotypes, for example, enabling better drug production, 71 as recently shown for heterologous production of penicillin. 72
In the field of drug screening, S. cerevisiae has successfully been used to find new antibody variants employing the yeast surface display technology. Developed in 1997 by Boder and Wittrup, 73 it has become an attractive eukaryotic alternative to the above-mentioned phage display. Yeast has the advantage to be able to efficiently secrete and display proteins and can be engineered to conduct posttranslational modifications like glycosylation similar to mammalian systems.74–77 In contrast to bacterial hosts like E. coli, S. cerevisiae has intracellular organelle systems, including extended membrane structures like the endoplasmic reticulum and Golgi apparatus. These are important to functionally express certain enzymes, like cytochrome P450 monooxygenases, which are essential for the biosynthesis of many natural products of high structural diversity. A prominent example is the production of the antimalaria compound artemisinic acid ( Fig. 2 ), where it was essential to have a functional, efficiently expressed P450 enzyme present to boost production by 25-fold to 25 g/L compared with the system used in E. coli.78,79
Recent impressive examples demonstrating the power of yeast as a synthetic biology chassis are the production of various plant-derived complex natural products.80,81 These include monoterpene indole alkaloids such as strictosidine, a precursor of the potent chemotherapeutic alkaloid vinblastine,82,83 and opioids such as noscapine84,85 and cannabinoids 86 ( Fig. 2 ). Advanced metabolic engineering approaches and implementation of up to 50 different genes from bacteria, mammals, and plants were necessary to establish production in these cases.
Besides the above-described classic synthetic biology chassis, other nonconventional chassis organisms can be more beneficial for the production of other compound classes. 87 This includes, for instance, Bacillus subtilis, 88 used due to its high secretion capacities for the production of enzymes and certain nutraceuticals; lactic acid bacteria for their application as living therapeutics; 89 the yeast Komagataella phaffii (formerly Pichia pastoris) for therapeutic protein production; 90 and Streptomyces, employed for its ability to produce high amounts of natural products, including polyketides, nonribosomal peptides (NRPs), and terpenes. 91
Choosing the right chassis organism is dependent on various parameters, including its final application, the compound class in focus, the importance and availability of synthetic biology tools, screening libraries, or even considerations toward the final industrial production of the compounds of interest.
Disruptive Science
The assembly methods30–35 have accelerated the assembly of parts to increase the speed of the DBTL cycle and, together with automation and software development, as described in Chao et al., 43 are set to be a main driver for future growth of synthetic biology.
Target Validation
One of the biggest reasons why drug discovery projects fail remains the identification and validation of targets. 92 Paradoxically, where targets are validated in the clinic following phenotypic or target-based approaches, second-generation medicines often build on this success. 93 Furthermore, where careful consideration is taken to validate targets and follow rigorous scientific principles, as exemplified by AstraZeneca 5R’s framework, success rates can be dramatically improved. 94 Hence, discovering novel targets and validating them is a vital part of establishing a successful pipeline and ensuring future medical advances. It therefore comes as no surprise that there has been significant interest and investment in tools that can help us achieve this.
The use of CRISPR-Cas995,96 tools fits perfectly into this arena. With the ability to exquisitely modify genetic sequences, this technology has allowed the generation of several new and important components of target validation that are poised to offer significant advantages in selecting the right targets.
At a very simple level, CRISPR-Cas9 has been used successfully in pharmaceutical research for several years to generate precise cell models replacing historic molecular biology approaches and introducing the ability to modulate gene function by ablation, downregulation, upregulation, or mutation, across cell types. Where previous technologies based on primer-driven mutagenesis, PCR, and subcloning or technologies such as siRNA screening could achieve many of these, they were laborious, were time-consuming, or could not be completed with the same level of confidence in the resulting cell population. However, in terms of target validation, CRISPR-Cas9 is poised to have a significant impact, at least in part due to parallel advances in complex cell systems. Although challenges remain in the cell types available and the cost of these techniques, the use of primary cells in these settings is growing, and the ability to sustain and differentiate stem cells presents an opportunity to modulate and explore disease in a way that previously would not have been possible. For example, recent reports include use of CRISPR-Cas9 in primary T cells to identify factors involved in HIV infection and pathogenesis, 97 while Martufi et al. 98 and Borestrom et al. 99 have used primary human fibroblasts and stem cell-derived kidney cells to probe genes involved in disease. This means that the cells that are now being used in validation experiments are closer to patient cells than ever before; hence, when interesting associations with disease are made, there is a higher confidence that these observations will translate into real effects in the clinic, if this target can be modulated with a drug.
Furthermore, CRISPR-Cas9 offers the ability to produce genome-wide screening sets that can ablate a gene function, which present very powerful tools that can be used to rapidly examine the role these genes play in different cell models. Pooled CRISPR-Cas9 screens whereby a population of cells are modified with a library of CRISPRs and then placed under a selection/growth condition, followed by next-generation sequencing (NGS) to determine which genes are enriched (resistant) or absent from a population (detrimental), have been used extensively over the last few years to implicate target validation.100–103 With arrayed CRISPR libraries, individual genes are targeted (usually with several different guide RNAs targeting the same gene) in isolated cell populations. Hence, it is easy to establish the direct effect or lack of effect each gene has within the array, providing higher confidence in the data output, but at a cost of requiring more complex platforms to run these at scale. Pooled screens also require fewer cells compared with array-based CRISPR and siRNA screening.
Shifrut et al. 104 nicely exemplify the combination of pooled CRISPR screening with primary human T cells, overcoming the issue of low lentiviral transduction rates for vectors carrying Cas9 in primary cells by introducing a new approach using single-guide RNA lentiviral infection with Cas9 protein electroporation (SLICE). Raising successful targeting to more than 80% of cells with a simplified protocol allowed Shifrut et al. to conduct large-scale genome-wide screening with increased confidence that the genes within the pool would be ablated in their experiment. Further, they were able to set defined conditions to identify genes that regulate T-cell proliferation in response to T-cell receptor (TCR) stimulation, in replicate screens and at differing doses of TCR stimulation, further increasing confidence in the targets identified. Finally, they combined their study with single-cell RNAseq to map pathways regulated by these genes and were able to repeat the screening in the presence of adenosine to test for targets that can allow the T cells to escape immunosuppression within adenosine-releasing cancers. These types of study have long been the promise of CRISPR as a synthetic biology tool, but this study is one that enables a paradigm shift in the ability to work with these types of complex cells with high confidence in the quality and completeness of the data generated.
To date, array-based screens have largely been used for smaller, more directed gene modulation,105–108 and at the time of writing, there were approximately five times the number of references for pooled screens compared with arrayed CRISPR in PubMed.
Yet, the greatest opportunity for these tools is to come. Without exception, large pharmaceutical companies are now investing in new genomic initiatives. Hailing a genomic era of discovery, patient NGS data and artificial intelligence are poised to identify many new targets across mainstream and rare diseases. 109 These data, when combined with powerful CRISPR arrays, will facilitate rapid confirmation of target association110,111 in cell, patient, and animal studies, and provide the foundations for new and improved medicines.
The ability to exquisitely control genes and connect them together to form functional circuits with desired outputs is not a new phenomenon. Classical molecular biology tools have been used historically to create impressive biological circuits in bacteria,112,113 yeast, 114 and mammalian 115 systems. The flexibility and fidelity of CRISPR-Cas9 means that these same things can be done in fractions of time and form more complex architectures. 116 Some may argue that CRISPR is not a product of synthetic biology, but rather a molecular biology tool. However, the high level of design, the use of standardized components, and the precise execution and engineering involved in the design of CRISPR tools certainly fit the definition provided in the introduction, and CRISPR is included by the European Union as a synbio tool.117,118 Through coupling a Cas9 with a designed RNA guide, CRISPR is a synthetic biology circuit all by itself. PRIME editors, consisting of a catalytically impaired Cas9 endonuclease fused to an engineered reverse transcriptase enzyme, and a prime editing guide RNA (pegRNA) offer a bespoke search-and-replace system for specific insertion, deletions and base-to-base swaps, with drug-inducible Cas9 allowing temporal control of gene editing for in vitro and in vivo applications at any desired point in the genome. 119
Disruptive Science
CRISPR-Cas9 95,96 is revolutionizing the way we do everything, from target validation to animal model development to the way we think about new therapeutics. It is not a surprise that the 2020 Nobel Prize for Chemistry was awarded to Emmanuelle Charpentier and Jennifer Doudna for the discovery of CRISPR-Cas9 genetic scissors.
Assay Development: Biosensors and Genetic Selections in Screening
Using synthetic biology to reprogram cells in an orthogonal and predictable manner offers many solutions to the drug discovery process. Previous efforts have already seen the creation of genetic circuits that can follow digital logic, are dynamic, or can even mimic electronic devices.120–122 Such circuits provide screenable or selectable outputs for diverse inputs, such as small molecules or metabolites, and can be incorporated into the drug discovery process as bioassays and biosensors for phenotypic screening. As more biological parts are characterized, the complexity of synthetic cellular systems will increase, and with that, so will their computational power. The potential translation of this power into the discovery of new and better drugs is promising, with examples and prospects outlined in this section.
Plasma membrane receptors, in particular GPCRs, play a key role in regulating the physiology of virtually every cell. 123 Given their fundamental importance, it is unsurprising that they are the largest family of proteins targeted by prescription drugs. 124 However, discovering hits against receptors remains challenging, as it is difficult to translate drug binding to a selectable or screenable output. Synthetic biology offers some solutions to this problem. Early, crude biosensor efforts took advantage of GPCR signaling cascades that increased cellular Ca2+ after drug binding, as this could be detected by the Ca2+-dependent fluorescence of recombinant aequorin. 125 Nevertheless, coupling receptors to a selectable signal transduction pathway, be it endogenous or synthetic, is a better and more direct way to assay receptor–ligand interaction. Barnea et al. were able to artificially rewire mammalian GPCRs, receptor tyrosine kinases, and steroid hormone receptor signaling by tethering transcription factors to receptors by a protease cleavable linker. 126 Ligand binding to the receptor recruits a signaling protein fused to a protease that frees the transcription factor, thus activating reporter genes. The authors were able to successfully identify a ligand for the orphan receptor GPR1. Other research has seen mammalian GPCRs transplanted into the yeast S. cerevisiae to create artificial signal transduction pathways that can then be used as a powerful tool to assess function or identify ligands, or as a pharmaceutical screening assay.127,128 Rewired receptors in yeast were used to make a bioassay for odorant screening, 129 and to identify peptide agonists against the chemokine receptor CXCR4 important in HIV infection. 130 Recently, Shaw et al. engineered a tunable refactored GPCR signaling pathway in yeast, making a significant advance in how future predictable artificial signal transduction pathways will be applied to drug discovery. 131
While the extracellular location of receptors makes them easily accessible as drug targets, many causes of disease are from aberrant PPIs that occur within the cell. 132 Synthetic biology approaches have been designed to select or screen for these interactions and monitor their disruption using chimeric proteins. For example, the protein–fragment complementation assay (PCA) functions where two proteins are each fused to a fragment of a third reporter protein; if interaction between the proteins of interest (POIs) occurs, the reporter protein is reconstituted and becomes active. 133 This approach has mapped the effects of small molecules on signal transduction pathways and both their on- and off-target effects,134,135 as well as identifying agonists of the glucocorticoid receptor. 136 An adaption of the PCA is the Förster resonance energy transfer (FRET) assay, which was used to screen small-molecule libraries for antiviral activity against poliovirus 137 or hepatitis C. 138 The bacterial reverse two-hybrid system (RTHS) is another option to assay PPI. Here, a POI is fused with the DNA binding domain of an obligate dimeric repressor. 139 When the POI fusions dimerize, a functional repressor is formed and binds operator sites engineered to prevent the expression of downstream reporter genes. Inhibitors of PPI can be screened by selecting for reporter gene activation. This strategy was used to find cyclic peptide inhibitors of HIF-1 heterodimerization, AICAR transformylase homodimerization, the HIV Gag-TSG101 interaction, and CtBP involved in cancer.140–143
Naturally evolved transcriptional repressors are often promiscuous allosteric regulators of bacterial physiology. 144 They are generally modular, with a DNA binding and a sensing domain; they are ordinarily used by bacteria to respond to other small molecules, secondary metabolites, the environment, and the cell cycle, but they are also functionally vital to sense antibiotics and regulate self-resistance. 145 Their modularity and ability to detect medicinally relevant molecules make them important biological tools that have been reprogrammed into synthetic transcription factors to regulate artificial eukaryotic genetic circuits. For example, a genetic switch for the antibiotic streptogramin was constructed in mammalian cells, 146 which led to the discovery of noncytotoxic antibiotics. 147 Furthermore, switches against tetracycline antibiotics 148 have been incorporated with a bacterial resistance mechanism TetX in yeast, thereby providing a genetic circuit to select not only for new tetracyclines but also for those that can evade antibiotic resistance. 149 A rewired Mycobacterium tuberculosis transcriptional repressor was used in human cells to screen for nontoxic drugs that increase the bacterium’s sensitivity to ethionamide. 150 Not only are bacterial repressors targets for drug discovery, but also their reappropriation to control gene expression allowed the construction of an inducible genetic circuit in mammalian cells for the HTS of cytotoxic drugs that preferentially targeted proliferation competent cells that mimic cancer. 151 The future potential to discover new small molecules by transcriptional repressor reprograming is immense, considering that more than 200,000 tetracycline family regulators have been identified, while the majority of their ligands have not been. 152 Furthermore, our ability to redesign synthetic transcription factors against new ligands exponentially increases this trove of possibilities for the use of genetic circuits in drug discovery. 153
Another interesting avenue for drug discovery is monitoring whether pharmaceuticals modulate the intracellular concentration of metabolites or small molecules. Following this premise, a naturally occurring riboswitch was adapted to control the fluoride-dependent expression of β-galactosidase in bacteria; this allowed the screen of a small-molecule library for fluoride toxicity agonists. 154 Considering synthetic aptamers that bind with high specificity and selectivity to a wide range of targets can be made and isolated, 155 the concept of sensing intracellular metabolites, or of controlling mammalian gene expression by any small inducer molecule, could be widely applicable.156,157
Disruptive Science
Barnea et al. 126 developed an assay where GPCRs can be reprogrammed to activate a reporter construct upon ligand binding. This technology provides a quantifiable measure of ligand interaction with a specific GPCR, useful in drug screens and the identification of ligands for orphan receptors.
Weber et al.’s 150 study creates an unnatural genetic circuit in mammalian cells using parts responsible for the persistence of tuberculosis. By combining parts from the host and the disease, this assay can screen drugs against tuberculosis for specificity, bioavailability, and cytotoxicity at the same time.
Hit Generation
While natural products have historically been a highly successful source of drugs,158,159 the modern drug discovery process has become focused upon the HTS of highly curated compound libraries in the search for leadlike 160 molecules as an easier source of equity for optimization. 161 Current screening practice encompasses the screening of thousands of fragments with x-ray-guided optimization to discover hit quality leads (micromolar activity), the HTS of 1–2 million compound libraries, and the recent development of DNA-encoded libraries (DELs) to screen libraries of billions of compounds. In such screens, the aforementioned synthetically engineered bioassays are implicit to the hit discovery process. In many ways, the ability to screen very large leadlike and druglike small-molecule libraries to find hits amenable to medicinal chemistry optimization has somewhat eclipsed natural products as drug modality. Historically, therapeutic natural products have been identified from biological samples collected from across the globe and HTS. Natural product drugs continue to be a valuable source of new drugs. 162 However, the screening of natural product extracts no longer drives the drug discovery engines of major pharmaceutical companies, even though many invested heavily in this area over the past 20 years and work continues in this area. 163 The modern fragment screening/HTS/DEL hit discovery paradigm is a very efficient process compared with the complexity of natural product chemical synthesis. Natural products can only compete if either they are already good enough to be the candidate drug or the candidate drug lies a short semisynthetic journey away. However, data science and synthetic biology offer new opportunities for natural products. Natural extract library screening can be replaced by the bioinformatic searching of BGCs to find putative natural products that could be synthesized. Now, DNA sequencing can find BGCs coding for pharmaceutical success in natural product drugs literally under our feet. Charlop-Powers et al. found gene clusters for 11 therapeutically important natural product families historically identified from across the globe, in the soils of Central Park, New York. 164 Less than 1% of the reads were aligned to known BGCs, which suggested a large reservoir of untapped biodiversity in the urban environment.
We are beginning to discover thousands of new BGCs identified through metagenome analysis. These clusters range from several kilobases to >100 kb in length and code for enzymes enabling complex small-molecule synthesis of large diversity. This covers various classes of secondary metabolites, including polyketides, NRPs, terpenes, and ribosomally synthesized and posttranslationally modified peptides (RiPPs). 165 Though the discovery of novel BGCs is progressing rapidly, only a fraction of these BGCs have been characterized and tested for their potential to produce novel drug leads.
Most of the BGCs are derived from nonculturable microorganisms, which makes the heterologous expression in established hosts necessary. As mentioned above, well-characterized synthetic biology chassis organisms have been established, which have a plethora of synthetic biology tools available to optimize and establish production. Gene synthesis enables the expression of codon-optimized versions of the BGC adapted to each host strain, combining these with well-characterized genetic parts to control transcription and translation. This refactoring not only has the advantage to remove regulatory elements, enabling controlled production, but also allows for the generation of combinatorial libraries. For example, it is possible to substitute enzymes with homologous variants to create derivatives or completely random enzyme clusters that generate novel chemical diversity.166,167 Another important point is the precise control of gene expression of the BGC members. For many BGCs, it was shown that gene expression is necessary to be fine-tuned to achieve optimal production.168–170 Using genetic BioBricks as described above, many gene cluster variants can be generated reusing a set of regulatory parts. Libraries can be screened for setups with optimized expression levels and increased titers. In addition, we see more platform strains becoming available, providing sufficient precursor and co-factor supply, easing the discovery and production for specific product classes of interest.171–173
Synthetic genetic circuits can be used to establish switches to specifically turn on BGC-derived pathway genes, allowing the de-coupling of growth from production. 174 For example, feedforward regulation has been described for many biosynthetic pathways of natural products, 175 allowing for dynamic regulation and the adaption to precursor availability and activating product export. Positive and negative feedback loops can be used to specifically allocate cellular resources to secondary metabolism. 176
The effort required to express the diversity of natural products encoded in these cryptic BGCs and test them in HTS to identify function in an unguided fashion would be enormous. However, a quirk of genome organization may offer clues to the purpose of the cryptic natural products that can be mined bioinformatically.
Along with the enzyme-coding genes, many BGCs contain other genes not involved in product synthesis, including transcription factors and genes that could be self-protective. These self-protective genes encode drug efflux transporters, detoxifying enzymes, and sometimes even resistant versions of the protein targeted by the BGC. 177 Many BGCs contain more than one of these self-protecting genes. These resistance genes offer a clue as to the function of the encoded natural product.
A search of the genomes of 86 Salinispora bacteria for protein-coding genes for lipid transport and metabolism associated with BGCs identified a 22 kb polyketide synthase–nonribosomal peptide synthetase (PKS-NRPS) hybrid gene cluster that, when heterologously expressed, produced thiolactomycin analogs previously shown to be fatty acid synthase II inhibitors. 178 An examination of an uncharacterized Aspergillus nidulans BGC identified a putative gene (inpE) with no obvious role in natural product synthesis. 179 InpE has homology to the b6 subunit of the proteasome, suggesting that the product of the inp BGC might be a proteasome inhibitor. Through use of a serial promoter exchange approach to sequentially replace six promoters in genes of the cluster with a regulatable promoter, the cluster was successfully expressed and the product of the BGC was identified as fellutamide B ( Fig. 3 ). By deleting inpE in A. nidulans and activating the expression of fellutamide, Hsu-Hua et al.179 were able to demonstrate that inpE is required for resistance of the internally produced fellutamide B.
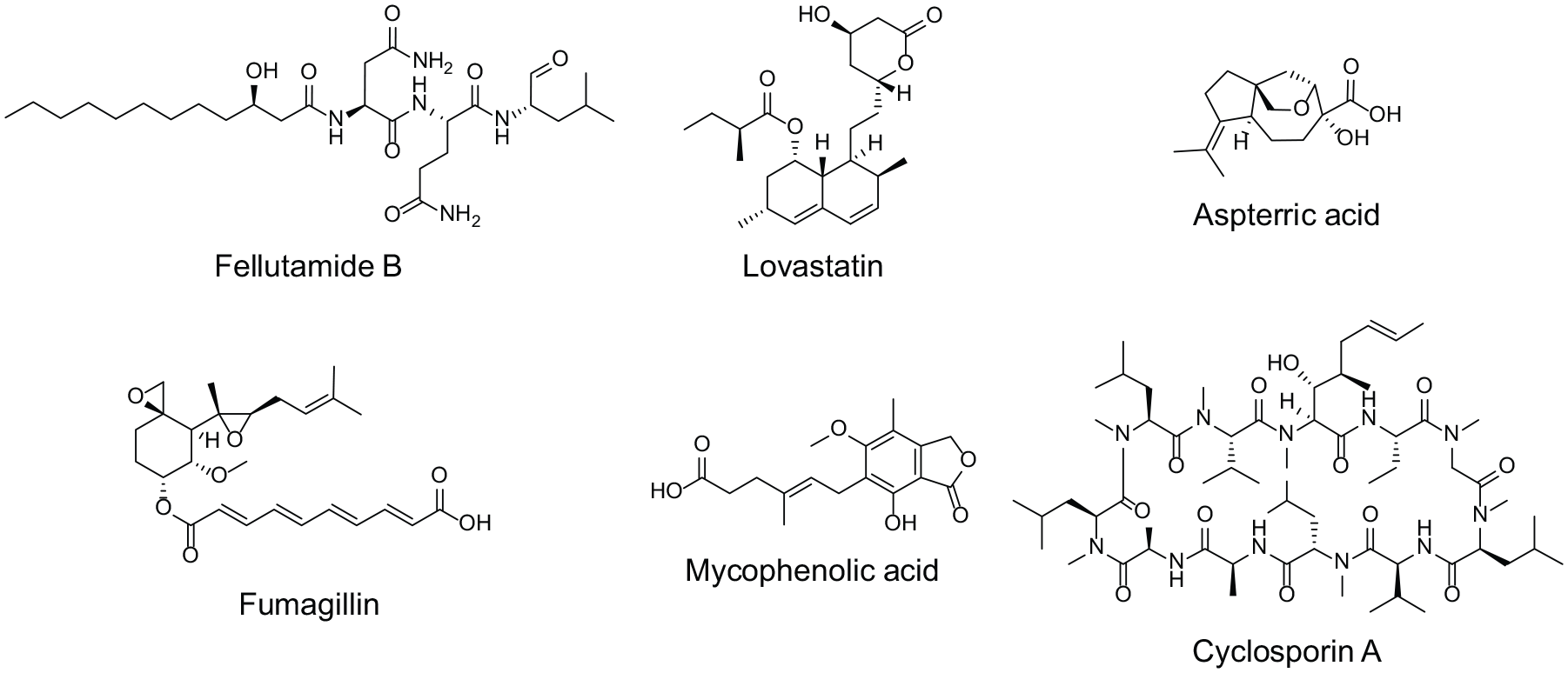
Examples of natural products with in-cluster resistance genes. As suggested by Keller, 177 the identification of other genes in BGCs offers a tactic to reveal the targets of natural products encoded by BGCs, and focus on useful clusters to characterize.
The fungal metabolite lovastatin blocks cholesterol synthesis by inhibiting 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, an enzyme required for the synthesis of fungal ergosterol. To prevent toxicity in the host strain, the Aspergillus terreus lovastatin gene cluster encodes a HMG-CoA reductase proposed to be resistant to lovastatin, 180 a feature common in related statin BGCs. 181
Fumagillin inhibits methionine aminopeptidase 2, required for the removal of N-terminal methionine residues from nascent proteins. The BGC for fumagillin in Aspergillus fumigatus contains an additional “in-cluster” methionine aminopeptidase 2 gene, maintained in analogous BGCs in other fungi. 182
Dihydroxyacid dehydratase (DHAD) is a key enzyme in the production of branched chain amino acids (BCAAs) in plants but not present in animals, making it an excellent target for new herbicide discovery. Although no natural products were known to target DHAD, Yan et al.
183
hypothesized that a fungal natural product might exist, as BCAA biosynthesis is required by plants. So, they went hunting for fungal BGCs containing an additional copy of the DHAD homolog. They identified four-well conserved genes across multiple fungal genomes, encoding for a sesquiterpene cyclase homolog (astA), two cytochrome P450 genes (astB and astC), and a homolog of DHAD (astD). Heterologous expression of astA, astB, and astC in S. cerevisiae produced the tricyclic aspterric acid, a previously identified natural product with a potent activity against Arabidopsis thaliana, through an unknown mode of action. The group showed that aspterric acid was a 0.5 µM inhibitor of A. thaliana DHAD, but did not inhibit AstD DHAD even up to its solubility limit of 8 mM. Other identified BGCs containing resistant copies of the target gene include echinocandin, which targets β-1,3-
Disruptive Science
Smanski et al. 174 provide a good review of the area of resistance genes in filamentous fungi and highlights how identification in BGCs with collocated resistance genes can reveal the targets of natural products encoded by BGCs, and other references184–187 illustrate how this information can be used as a drug discovery tactic.
Lead Optimization
Directed Evolution and Synthetic Biology
Hit identification and lead generation are followed by lead optimization, a process in which an initial lead compound is subjected to iterative rounds of (chemical) modifications and characterization to give insight into its structure–activity relationship and metabolic stability. These iterative rounds of modifications are a trait that lead optimization shares with directed evolution. 187 In directed evolution, a genetically encoded molecule (nucleic acids or proteins/peptides) is subjected to iterative rounds of modification/mutagenesis, followed by the selection/screening of a user-defined goal, while ensuring the inheritance of the advantageous characteristic of the evolved molecule (genotype–phenotype linkage). For example, linear peptides targeting G-protein subunit α 188 and cyclic peptides inhibiting HIV protease 189 have evolved by directed evolution.
To further increase the chemical space accessible for lead optimization, one can take a look at natural products and their biosynthesis for inspiration. NRPs are synthesized by NRPSs, and the peptides often contain noncoded amino acids that might be further modified by, for example, N-methylation. However, directed evolution of NRPSs presents additional challenges for lead optimization. The correlation between the enzyme primary structure and the structure of the produced peptide is often elusive, as the sequence of the NRP is not genetically encoded. Attempts to diversify natural products by domain swapping or mutation of NRPSs often result in greatly reduced enzymatic activity accompanied by low product titers. Heterologous expression of NRPSs further limits diversity due to the absence of noncognate substrates in the heterologous host. Despite these shortcomings, some successful cases have been reported. Directed evolution was used to restore the functionality of a heterologously expressed AdmK-CytC1 hybrid. 190 This hybrid was also capable of producing new derivatives of andrimid, a peptide antibiotic that inhibits prokaryotic acetyl-CoA carboxylase. Additional derivative compounds of andrimid have been obtained by directed evolution of AdmK and expression in the native host Pantoea agglomerans. 191 Recent advances in the design and engineering of NRPs might simplify the diversification of natural peptide products and the generation of novel NRPs in the future. 192
Unlike NRPs, RiPPs are genetically encoded peptides that are synthesized on ribosomes and further modified by RiPP biosynthetic enzymes. The precursor peptides contain a conserved leader peptide and a variable core peptide, and the latter is posttranslationally modified and proteolytically processed to the mature form. The discovery of novel posttranslational modifications, previously thought unique to NRPs, such as N-methylation, 193 largely increases the chemical space accessible by RiPPs. Lanthipeptides are a class of macrocyclic RiPP that contain characteristic lanthionine moieties. Phage display and yeast display techniques have been adapted to allow for screening of in vitro-generated libraries of lanthipeptides, 194 enabling directed evolution of this modality in the future. The accessible chemical space can be increased further by using synthetic biology to create nonnatural hybrid RiPPs that contain moieties from different classes of RiPP. 195
The advantage of genetically encoded molecules is the ease of library generation by means of in vitro mutagenesis, for example, by error-prone PCR (epPCR). However, in vitro-generated libraries that can be screened in vivo often have size limitations due to the transformation efficiency of the host organism, limiting the solution space that can be probed in practice. A simple octapeptide composed of amino acids encoded by standard genetic code already has 2.56 × 1010 possible combinations, and the DEL would have to be even larger than this to account for codon degeneracy. Additionally, the screening of in vitro libraries requires the isolation and identification of beneficial mutations after each iterative round, which is laborious and time-consuming. Continuous directed evolution avoids these limitations by performing all steps of directed evolution—library generation/mutagenesis, selection/screening, and inheritance—in vivo, allowing for a continuous process.
Recent years have seen a surge in method development allowing for continuous directed evolution in a variety of host organisms, such as PACE, 52 in vivo continuous evolution (ICE), 196 CRISPR-AID, 197 orthogonal replication (OrthoRep), 198 and EvolvR. 199 Both classes of the previously described peptides, NRPs and RiPPs, are promising lead compounds for drug development. 200
Disruptive Science
With the growing number of continuous directed evolution techniques and the improving toolbox provided by synthetic biology, these techniques, as described in several references,52,196–200 have the potential to revolutionize the drug development process, from hit discovery all the way to lead optimization.
Late-Stage C–H Functionalization of Drug Leads Using Engineered Enzymes
During early drug discovery, bioactive molecules can be structurally modified by selective additions, deletions, and/or replacement of specific atom(s). This process, referred to as late-stage functionalization (LSF) or molecular editing, is generally faster and more cost-effective than de novo synthesis in generating libraries of drug lead analogs. 201,202 Even a subtle structural change can have dramatic effects on the properties of a drug, as exemplified by the addition of a single methyl group drastically increasing kinase inhibitor selectivity203,204 or the important role played by a halogen-bonding interaction. 205 Thus, LSF has great potential to accelerate the discovery of drug lead derivatives exhibiting optimized activity, safety, and/or drug metabolism/pharmacokinetic (DMPK) profile. To rapidly build large libraries of molecules derived from a single druglike scaffold, unactivated C–H bonds have high value as points of diversification (i.e., C–H bonds are replaced with carbon–heteroatom or carbon–carbon bonds). Over the last two decades, new methods in transition metal, photoredox, and metallophotoredox catalysis and other chemistry fields have proven to be powerful tools for LSF in drug discovery laboratories. 202 However, predicting and controlling regioselectivity still remains a challenge in structurally complex molecules. 206 Therefore, additional methodologies that achieve a high degree of C–H selectivity are highly desirable to further expand into novel chemical space in a fast, cheap, and sustainable manner. In the last few years, many impressive advances have been made in the field of synthetic biology, which have great potential for filling the space left by synthetic chemistry methodologies.
To create nonnatural biological systems for new applications, synthetic biology borrows and combines tools from different disciplines, including molecular biology, protein engineering, metabolic engineering, and bioinformatics. The directed evolution of enzymes has contributed enormously to propelling synthetic biology advances forward. The relevance of this methodology is illustrated by the 2018 Nobel Prize in Chemistry awarded to Prof. Frances Arnold for her outstanding contributions to directed evolution.207,208 In addition to the directed evolution of enzymes, rational and semirational design have also proven to be a very powerful strategy when either x-ray crystal structural data or high-quality homology models, combined with mechanistic data, are available. The generation of huge mutagenesis libraries is now both time- and cost-effective using well-established methods, for example, epPCR, DNA shuffling, single-site saturation mutagenesis, and combinatorial active-site saturation test (CAST). 209 Furthermore, high-throughput enzyme expression using heterologous hosts is efficient in many cases. One bottleneck in directed evolution can be HTS using agar plates or microtiter plates for large (>1012) mutant libraries. Nevertheless, the optimization of ultra-HTS technologies relying on flow cytometry and chip-based microfluidic screening for their use in directed evolution campaigns will likely open new doors in the near future. 210 Importantly, machine learning is attracting increasing attention in prediction and decision-making in the field of enzyme engineering. 211 As a result of all these efforts, engineered robust and selective enzymes currently have enormous potential to expedite diverse projects in the pharmaceutical industry, including drug discovery.
Currently, synthetic biology’s toolbox contains fascinating biocatalysts that exhibit improved stability, activity, substrate scope, and/or selectivity engineered to fulfill industrial demands. These enzymes can provide access to drug lead analogs that would be challenging, inefficient, or unsafe to prepare by nonenzymatic chemical reactions.
Figure 4
shows relevant examples of heme-, flavin-, S-adenosyl-
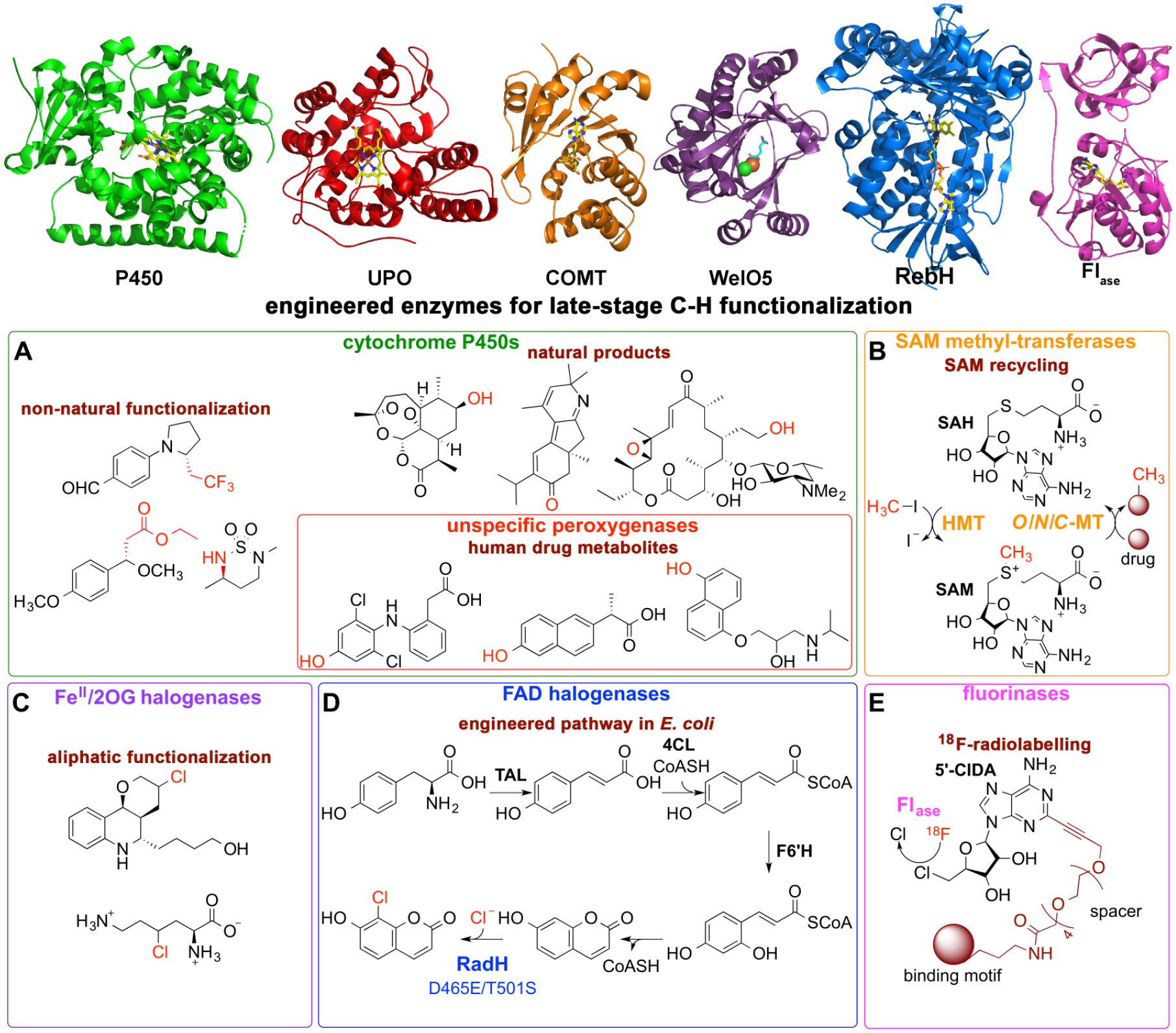
Enzymes harboring great potential for late-stage C–H functionalization of drug leads. Enzyme, reaction, and pathway engineering can provide industrially relevant compounds as the examples shown here. (
In addition, LSF approaches can rapidly synthesize human drug metabolites. Drug development processes should include toxicity assessments of human drug metabolites that are generated at a level higher than 10% of the parent drug exposure, following the metabolites in safety testing (MIST) guidance reported by the U.S. Food and Drug Administration (FDA). 228 P450 enzymes are the key players in phase I drug metabolism in human liver, turning over the majority of commercially available drugs.229,230 The pharmaceutical industry therefore needs efficient methods to synthesize products resulting from the action of human P450s on new drug candidates. Microbial P450s are often preferred for the synthesis of putative human drug metabolites, mainly due to the expense of purified recombinant human P450 or hepatic microsomes. 231 Various studies show that mutagenesis can be used to tune the substrate scope, selectivity, and/or activity of microbial P450 enzymes, which often results in improved mimics of the desired human P450 isoform. For example, enzyme engineering was used to obtain P450 variants that produce major human metabolites of omeprazole, 232 diclofenac, 233 naproxen, and propranolol ( Fig. 4A ). 234 Engineered unspecific peroxygenases (UPOs) are an attractive alternative to P450 for synthesizing human drug metabolites.235,236 Oxyfunctionalization chemistry catalyzed by UPO is comparable to that performed by P450s. 237 However, UPO reactions only require hydrogen peroxide as a co-substrate, in contrast to P450 catalysis, which requires a redox partner(s)/domain, NAD(P)H, and dioxygen. Remarkably, despite the complex catalysis of wild-type P450s, engineered P450 enzymes have been used to catalyze even nonnatural chemistries ( Fig. 4A ), 238 for example, cyclopropanations, 239 which is of current interest in pharmaceutical development.
The presence of halogen atoms (F, Cl, Br, I) can improve the potency and pharmacokinetic profile of many drugs.
240
Indeed, around 30% of pharmaceuticals on the market today contain a halogen. Flavin-dependent halogenases are attractive catalysts for the replacement of C–H bonds with C–halogen bonds in drug leads due to their high stereo- and regioselectivity.
241
Unfortunately, wild-type flavin-dependent halogenases generally suffer from narrow substrate scope, low catalytic efficiency, and poor stability under industrial conditions. Enzyme, reaction, and pathway engineers, using synthetic biology tools, have made significant progress in increasing the potential of various halogenases for their implementation in lead optimization. For example, variants of the flavin-dependent halogenase RebH exhibiting a significantly higher thermostability than the wild-type enzyme were discovered by only three rounds of directed evolution.
242
In a subsequent study, the substrate scope of one of these thermostabilized variants was expanded using random mutagenesis and a substrate walking approach.
243
Halogenases have been successfully incorporated into biosynthetic pathways to catalyze late-stage halogenations of druglike scaffolds in an appropriate host. For example, a double mutant of the flavin-dependent halogenase RadH was successfully used in a metabolic engineering effort, directed toward the synthesis of a chlorinated coumarin from glucose in E. coli (
Fig. 4D
).
244
While flavin-dependent halogenases act on electron-rich substrates, FeII/2OG-dependent halogenases catalyze the halogenation of unactivated aliphatic sp
3
carbon centers (
Fig. 4C
). So far, only a few carrier protein-independent FeII/2OG halogenases have been discovered (e.g., WelO5, AmbO5, and BesD), but engineered variants are already available.245–247 They seem to exhibit great potential for C–H functionalization in the pharmaceutical industry. Another interesting halogenase type is an adenosyl-fluoride synthase, often referred to as fluorinase, which catalyzes the conversion of fluoride ion and SAM to 5′-fluoro-5′-deoxyadenosine and
Many of the biocatalysts described here and in previous works255,256 look ready to be incorporated into screening kits for use in drug discovery laboratories. The challenges to be overcome before they are more widely used in industry have been identified for each enzyme class, and these are being tackled by numerous research groups. The dream, which is now closer to being realized, is to routinely perform and analyze thousands of enzyme-catalyzed C–H functionalization reactions at the microgram scale in a few days. Importantly, the rapid establishment of structure–activity relationships using these technologies will be translated into improved drug discovery timelines and ultimately speedier delivery of novel medicines to patients.
Disruptive Science
O’Hagan and Deng 248 provide an innovative SAM recycling system involving only one regenerating enzyme, which enormously facilitates the implementation of SAM-dependent methyltransferases in biocatalysis. The authors discovered that S-adenosyl homocysteine, a by-product in the methyltransferase reactions, can be converted by a halide methyltransferase into SAM using methyl iodide as the methyl source.
Butler et al. 232 recently reviewed natural enzymes that have acquired the ability to catalyze nonnatural chemical reactions after modifying in vitro their amino acid composition, co-factor, available reagents, environment, or combinations thereof. This work covers a rapidly growing research area of considerable interest to organic synthesis.
Sun et al. 250 provide a comprehensive review of the current role of biocatalysis in drug development, as well as the challenges to overcome in this field and the most promising areas under exploration. Interestingly, both academic and industrial viewpoints are provided.
Cell Therapy and Biologics
The panacea of synthetic biology of the future may be the ability to control how patient cells behave in vivo. The ability to remove, modify, and replace these cells is already a reality in oncology treatments where the body’s own immune system can be reprogrammed to hunt and destroy cancerous cells, through CAR-T-cell therapy. 257 Synthetic biology can further enhance the special and temporal aspect of CAR-T cells. “Kill” switches, peptide-specific switchable CAR-T cells (sCARs) can improve reversible tumor-specific activation. In an inducible CAR (iCAR) system, two incomplete CAR molecules that can heterodimerize in the presence of a small molecule can function as a switch to activate the T-cell response. 258 CAR-T cells can also be engineered with AND-gate logic, where activation of one CAR receptor activates the expression of a second CAR receptor for a second antigen, further increasing the specificity of targeting.
Yet, there are other ways in which cells can be utilized to deliver medicines more efficiently than systemic treatments. The generation of sensor actuator circuits presents a real possibility that cells can be generated to replace or repair defective cellular processes. There are a series of animal studies that suggest that cellular medicines are on the verge of being able to translate to much needed improvements in therapy. For example, Shao et al. demonstrated that it is possible in mice studies to generate sentinel cells that can reduce feeding and manage diabetic-glucose peaks in response to light signals, 259 or electrogenetic engineered electrosensitive B cells that can release insulin in vivo upon wireless electrical stimulation. 260 Additionally, Chowdhury et al. have engineered an E. coli strain to specifically lyse within the tumor microenvironment. 261 On lysis, they released an encoded nanobody antagonist of CD47 (CD47nb), an antiphagocytic receptor that is commonly overexpressed in several human cancer types. They showed delivery of CD47 nanobody by tumor-colonizing bacteria increased activation of tumor-infiltrating T cells, stimulated rapid tumor regression, and prevented metastasis, leading to long-term survival in a syngeneic tumor model in mice.
The use of bacterial systems is certainly not a new concept, with anecdotal evidence of bacterial infections curing cancers throughout civilized history and numerous clinical studies from the early 20th century that showed partial efficacy, albeit with some significant side effects. 262 Bacteria are unsurprisingly good at negotiating the complex biology of mammalian systems; with an evolutionary need to avoid and hide from immune systems, they are able to specifically hunt out the necrotic centers of solid tumors and hide deep within them. However, these approaches were soon surpassed and largely forgotten by the developments made in small-molecule drug research that have built the modern pharmaceutical industry. However, with the developments made in synthetic biology, it now seems that bacteria have a similar potential, with researchers now examining how human peptides can be used to arm bacteria with “weapons of mass destruction” that are desirable, with the potential to specifically target and destroy tumor masses. 263 Of course, not all bacteria are pathogenic, and there are other opportunities with the microbiomes of our digestive tract and skin systems that are very important to the healthy balance of human life. E. coli and other bacterial hosts have been engineered to diagnose diseases and produce and deliver therapeutics in situ. So-called living therapeutics were successfully engineered to target diseases like inflammatory bowel disease, 264 diabetes mellitus, 265 and cancer. 266 Advanced engineering of the bacterial hosts is necessary, including sensing inputs, controlling gene expression, building memory, producing and delivering active compounds, and genetic switches for biocontainment. 267 For example, in a recent study, bacterial biosensors that trigger a differential response to the healthy or diseased mammalian gut were identified, enabling the future design of specific diagnostic and therapeutic biosynthetic circuits. 268
It is an exciting prospect that these bacteria can be given medically beneficial properties to target chronic skin and metabolism disorders directly, while there also remains a possibility to use these as routes of administration for small molecules targeting other medical conditions.
Legislation on cell therapies is changing in the United States at least, and we are seeing a number of late-stage clinical studies that should pave the way for the future expansion of this field.
Disruptive Science
Yeo et al. 251 describe the first clinical efficacy of CAR-T cells infused in three patients with advanced chronic lymphocytic leukemia that targeted CD19 and contained a co-stimulatory domain from CD137 and the TCR ζ chain. It heralded a new era of cancer therapy. This therapy was later developed in the clinic and taken to market by Novartis, and in 2017 Tisagenlecleucel became the first FDA-approved medicine that included a gene therapy step.
Merging Workflows
Projects merging machine learning, medicinal chemistry, and synthetic biology into a common workflow have the potential to push the boundaries of modern drug discovery. We see more examples of applying machine learning approaches in synthetic biology to make genetic modules and designs more predictable. This includes, for instance, the prediction of promoter designs 269 and automated tools recommending engineering strategies to improve the microbial chassis performance. 270 We have seen the first synthetic biology approaches creating compound libraries to explore new chemistries, and the combination of these with intracellular selection regimes for hit discovery. 271 Also, deep learning methods are increasingly applied in modern drug discovery. 272 Machine learning approaches are further developed to better predict structure–activity relationships, for example, by applying recurrent neural networks using molecular descriptors as inputs. 273 However, projects combining all three disciplines of synthetic biology, medical chemistry, and machine learning are just beginning to emerge. A recent example is from the field of protein drugs, where deep learning was used to optimize therapeutic antibodies through exploring the high-dimensional protein sequence space. 274 Screening and deep sequencing of relatively small libraries (104) were used to train deep neural networks that accurately predicted antigen binding based on antibody sequence and allowed efficient exploration of a large in silico library of ~108 variants.
Summary and Outlook
Synthetic biology has impacts throughout the drug discovery value chain and into the development phases and production of pharmaceuticals. The plummeting cost of synthetic DNA, the increasing detailed understanding of the genome, its organization, gene regulation, and the availability of chassis organisms underpin the successes so far and the future potential.
The CRISPR field is exploding with profound impacts on target identification and validation. The development of assays, which underpin the modern drug discovery process, owes much to synthetic biology through engineering of biological circuits.
Genome mining enabled by combination of the availability of genomic sequences, data science, and synthetic biology may catalyze a resurgence in natural product drug discovery. This may be particularly so for “difficult-to-drug” targets such as PPIs and phosphatases, where the modern hit discovery engines of HTS, DEL, and fragment-based lead generation may fail, but where nature has found a solution. Combinatorial biosynthesis, where components of BGCs are permuted, may allow further biosynthetic diversification without resorting to difficult chemical synthesis. Artificial intelligence and machine learning may be able to augment the refactoring of BGCs to increase the predictability of forming further novel molecules.
Directed evolution coupling genetically encoded libraries, mutation, and selection pressure has revolutionized the development of therapeutic antibodies. Coupling to biological circuits allows in vivo directed evolution for drug discovery and the evolution of new proteins with novel function, such as enzymes to catalyze new transformations for chemical diversification in drug discovery, and “green chemistry” for bulk drug production. While engineered enzymes are already used in individual steps in bulk drug production, whole pathways can be engineered for biosynthetic production. While the successes have been achieved with stepwise directed evolution, continuous directed evolution can enable many more generations to be explored, allowing a deeper search through structure–activity space.
Synthetic biology is enabling new advances in cell therapy, already in oncology with CAR-T, and providing exciting opportunities in cell reprogramming, precise genome editing to correct genetic defects, and reengineering for cell and tissue regeneration. But further, cells can themselves be engineered to sense their environment and respond to treat acute and chronic diseases.
Finally, the merging of workflows between modern technologies such as artificial intelligence and machine learning with synthetic biology and chemistry is emerging to further push the boundaries of drug discovery.
Synthetic biology certainly is impacting all stages of drug discovery and development, and the recognition of the discipline’s contribution can further enhance the opportunities for impact on the drug discovery and development value chain.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: M. G, E. R, and L. H. S are funded through the AstraZeneca Postdoctoral Programme.