
Abstract
ETV6 is an ETS family transcriptional repressor for which head-to-tail polymerization of its PNT domain facilitates cooperative binding to DNA by its ETS domain. Chromosomal translocations frequently fuse the ETV6 PNT domain to one of several protein tyrosine kinases. The resulting chimeric oncoproteins undergo ligand-independent self-association, autophosphorylation, and aberrant stimulation of downstream signaling pathways, leading to a variety of cancers. Currently, no small-molecule inhibitors of ETV6 PNT domain polymerization are known and no assays targeting PNT domain polymerization have been described. In this study, we developed complementary experimental and computational approaches for identifying such inhibitory compounds. One mammalian cellular approach utilized a mutant PNT domain heterodimer system covalently attached to split Gaussia luciferase fragments. In this protein–fragment complementation assay, inhibition of PNT domain heterodimerization reduces luminescence. A yeast assay took advantage of activation of the reporter HIS3 gene upon heterodimerization of mutant PNT domains fused to DNA-binding and transactivation domains. In this two-hybrid screen, inhibition of PNT domain heterodimerization prevents cell growth in medium lacking histidine. The Bristol University Docking Engine (BUDE) was used to identify virtual ligands from the ZINC8 library predicted to bind the PNT domain polymerization interfaces. More than 75 hits from these three assays were tested by nuclear magnetic resonance spectroscopy for binding to the purified ETV6 PNT domain. Although none were found to bind, the lessons learned from this study may facilitate future approaches for developing therapeutics that act against ETV6 oncoproteins by disrupting PNT domain polymerization.
Keywords
Introduction
The ETV6 gene, known also as TEL (
ETV6 is a modular protein composed of an N-terminal self-associating PNT (pointed, or SAM,
One well-characterized ETV6 translocation encodes the PNT domain fused to the PTK domain of neurotrophin tyrosine receptor kinase-3 (NTRK3). The resulting protein, named EN (ETV6-NTRK3), displays oncogenic properties including phenotypic transformation and soft agar colony formation of several experimental cell lines, as well as tumor formation in nude mice.6,17 The ETV6 PNT domain polymerization interfaces, called the midloop (ML) and end-helix (EH) surfaces, have hydrophobic cores surrounded by charged residues. 18 The mutation of one of two key hydrophobic residues to a charged residue (A93D on the ML surface, or V112E or V112R on the EH surface, according to the human ETV6 numbering) disrupts polymerization.14,18 The introduction of these mutations into EN-expressing cell lines prevents EN polymerization, PTK activation, and cellular transformation. 13 Similarly, mutation of the K99-D101 charge pair bridging the PNT domain interfaces weakens polymerization and inhibits the transformation activity of EN in NIH3T3 cells. 19 Co-expression of an isolated PNT domain also has a dominant negative effect, preventing cellular transformation. 13 Together, these studies indicate inhibition of PNT domain polymerization as a viable therapeutic strategy against cancers driven by ETV6 chromosomal translocations.
We hypothesized that small molecules that prevent the self-association, and hence oncogenic properties, of ETV6 chimeras containing the PNT domain might serve as a potential broad-spectrum therapy against many ETV6 PNT domain-containing oncoproteins and avoid toxicities associated with perturbing the normal activities of receptor PTKs. Furthermore, although approximately one-third of the 28 ETS transcription factor family members in humans possess a PNT domain, only ETV6 and perhaps closely related ETV7 polymerize.20 –22 Thus, a molecule that selectively inhibits ETV6 PNT domain polymerization may have few side effects.
Protein–protein interactions (PPIs) are challenging to disrupt, and hence we undertook complementary cellular and virtual screening strategies in the attempt to discover inhibitors of ETV6 PNT domain polymerization. Central to our approach is the use of PNT domains with monomerizing mutations in the EH or ML surfaces that can still associate with low nanomolar affinity through their remaining complementary wild-type interfaces.14,18 The soluble “heterodimer” serves as a model of the insoluble polymer, opening the door for in vitro and in vivo screens. In brief, both a mammalian cell-based assay utilizing a protein–fragment complementation approach with split Gaussia luciferase 23 ( Fig. 1A ) and a yeast two-hybrid assay 24 ( Fig. 1B ) were developed and used to screen chemical libraries for potential inhibitors. In parallel, large-scale virtual screening using the Bristol University Docking Engine (BUDE) was performed to identify theoretical compounds that might bind to the PNT domain polymerization interfaces.25,26 Subsequently, candidate compounds were tested for inhibitory effects in cellular assays and for binding to the isolated ETV6 PNT domain as monitored by nuclear magnetic resonance (NMR) spectroscopy. Although no inhibitory compounds were successfully identified, the development, validation, and implementation of these assays will be discussed herein.
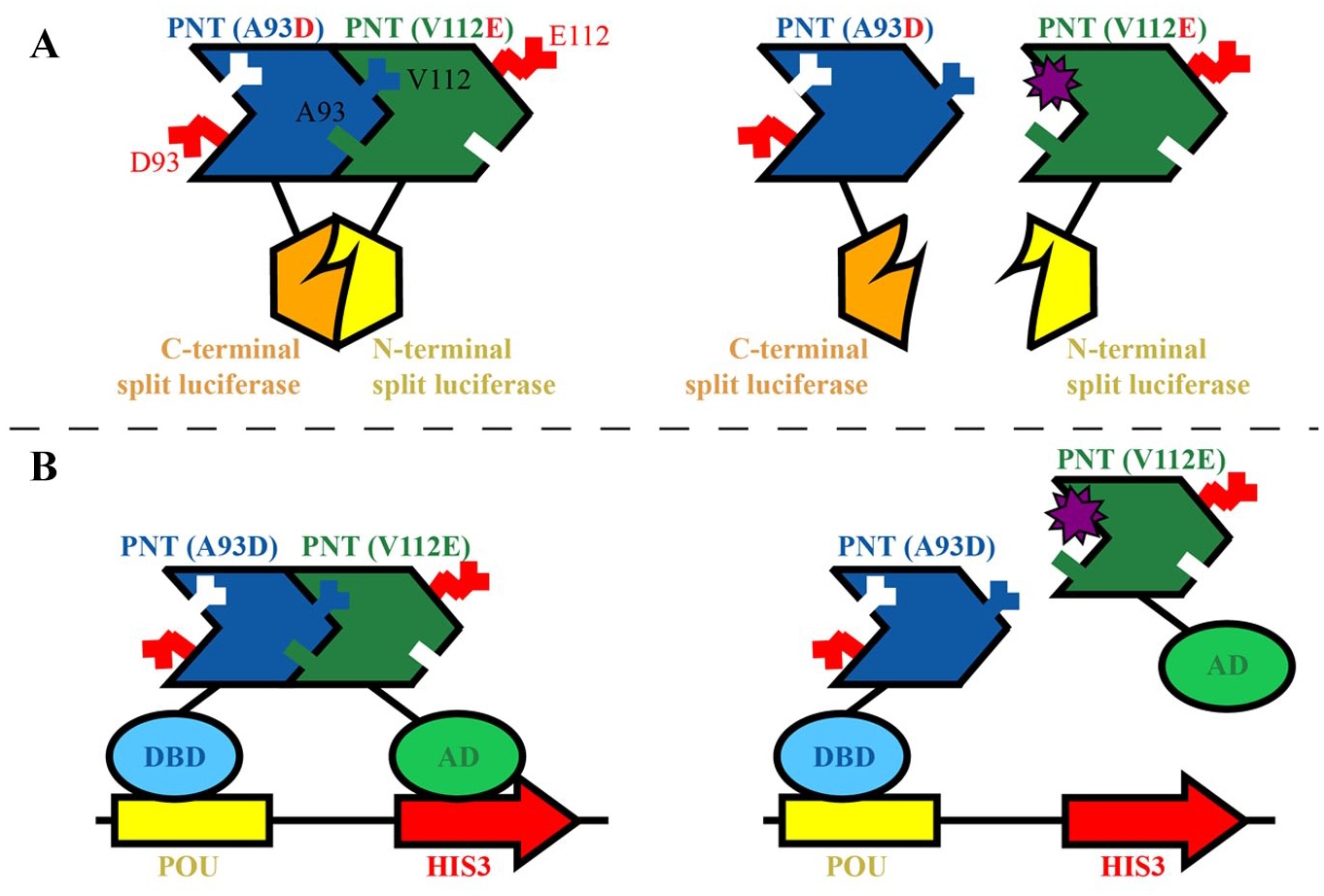
(
Materials and Methods
Chemicals for Screening
Screening compounds for the cellular assays consisted of 16,000 compounds from the Maybridge Hitfinder collection, 10,000 compounds from the ChemBridge DIVERset collection, 1120 compounds from Prestwick Chemicals, 1280 compounds from the Sigma LOPAC library, 2000 compounds from the Microsource Spectrum collection, 2697 compounds from the Selleck L1700 Bioactive Compound library, and 500 compounds from Biomol. The compounds were stored in 96-well plates at −25 °C as 5 mM stock solutions in DMSO. In addition, a small-molecule library targeting PPIs consisting of 1534 compounds was provided by the Perturbation of Protein-Protein Interactions (PoPPI) collaborative program (Leeds, UK). Candidate compounds from the BUDE screening assay were purchased from MolPort for testing both in cellular assays and by NMR spectroscopy.
Vectors and Cloning for the Split Luciferase PCA
The split luciferase protein–fragment complementation assay (PCA) was based on the protocol of Remy and Michnick. 23 Sequences encoding ETV643–125 (residues 43–125 of human ETV6, encompassing the PNT domain; Genbank Gene ID 2120) with either an A93D or V112E substitution were cloned into either the modified mammalian expression vector pcDNA3.1/Zeo(+) or pcDNA3.1/Neo(+) at the 5′-end of sequences for humanized Gaussia luciferase (hGLuc) fragments ( Suppl. Table S1 ). The resulting constructs encoded either A93D- or V112E-PNT domain, a (GGGGS)2 flexible linker, and either hGLuc(1)1–93 or hGLuc(2)94–196. The latter are described herein as N-Luc or C-Luc, respectively. A control set of plasmids containing leucine zippers as the dimerization domains were also provided by Dr. Michnick. 23
Mammalian Cell Culture
Human embryonic kidney cells 293 (HEK293; ATCC, Manassas, VA) cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Waltham, MA) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO) and 1% antibiotic-antimycotic (Gibco). Unless otherwise noted, this medium was used in all experiments. Stably expressing transformants were treated additionally with either or both 400 µg/mL G418 (Gibco) and 50 µg/mL zeocin (Invitrogen, Waltham, MA). Cells were incubated at 37 °C with 5% CO2 and passaged upon reaching approximately 75%–85% confluency.
Transient Expression for Validation of the PCA
HEK293 cells were seeded in 96-well, clear-bottom, black microplates (cat. 6005182, Corning, Tewksbury, MA) at 15,000 cells/well and incubated at 37 °C. After 24 h, the medium was aspirated and 100 µL of fresh medium was added. For transfection of a single species of DNA, 20 ng/µL plasmid was added to OPTIMEM (Gibco), and for transfection of two species of DNA, 10 ng/µL each plasmid was added to OPTIMEM. Lipofectamine 2000 (Invitrogen) was diluted to 8% in OPTIMEM and added to the DNA at a 1:1 v/v ratio and incubated for 5 min at room temperature. After incubation, 10 µL of the total prepared DNA, OPTIMEM, and Lipofectamine 2000 was added to each well. Cells were incubated at 37 °C for either 24, 48, or 72 h. Prior to the luminescence reading, 50 µL of cell medium was removed and an equal volume of NanoFuel GLOW Assay (Nanolight Technology, Pinetop, AZ) for Gaussia luciferase was then added to each well. After incubation in the dark at ambient temperature for 5 min, the luminescence output was read for 1 s with a Varioskan LUX multimode microplate reader.
Establishment of a Stably Expressing PNT Domain PCA System
HEK293 cells were seeded in six-well microplates at 400,000 cells/well and grown overnight to approximately 80% confluence. The cell medium was changed and the cells were transfected with Lipofectamine 2000 utilizing 25 ng/µL DNA for single plasmid transfections or 12.5 ng/µL DNA for each plasmid in a double plasmid transfection. After 24 h, the medium was aspirated, fresh medium was added, and the cells were incubated again overnight. After 48 h, selection was introduced by incubating cells with medium supplemented with the corresponding antibiotic(s), replacing medium every 2–3 days, and splitting cells when 80% confluency was achieved. The resulting stable transformants were stored in liquid nitrogen.
High-Throughput PCA Screening
For screening, stable transformants of A93D-PNT/N-Luc(Neo) and V112E-PNT/C-Luc(Zeo) were plated at 40,000 cells/well in 96-well, clear-bottom, black microplates (Corning cat. 6005182) and incubated overnight at 37 °C. Compound plates were thawed and compounds were added to cells using a BioRobotics BioGrid Robot Microarrayer Model equipped with a 96-pin tool with either 0.7 or 0.4 mm diameter pins. After a 4 h incubation at 37 °C, a 1:1 ratio of NanoFuel GLOW Assay reagent was added to the cells and plates were incubated in the dark at ambient room temperature for 15 min. Luminescence output was then read with a Varioskan LUX multimode or BioTek Neo 2 microplate reader.
Secondary PCA
Compounds that yielded a lower luminescence in the initial PCA screens were retested at different concentrations against cells stably expressing the split luciferases fused to ETV6 PNT domains or leucine zippers. The latter served as a specificity control. Cells were seeded at 40,000 cells/well in 96-well plates and incubated at 37 °C overnight. The selected compounds were added to wells in duplicate at final concentrations of 1, 3, 10, and 30 µM. After 4 h incubation, cells were examined through a microscope for the presence of rounded, detached, or dead cells, or for compound precipitates (see Suppl. Fig. S1 for examples). Luminescence was read as previously described.
Yeast Two-Hybrid Assay Development
The two-hybrid assay consists of bait and prey plasmids and a yeast reporter strain, constructed for this study. The bait plasmids expressed, from a constitutive ADH1 promoter, the Oct1 POU DNA-binding domain alone (pIS341) or a fusion between the Oct1 POU DNA-binding domain and the A93D-PNT domain (pIS586). They are ARS-CEN (single-copy) plasmids with a TRP1 marker. The prey plasmids expressed the NLS-B42 activation domain (AD) alone (pIS580) or a fusion between the AD and V112E-PNT domain (pIS591). These genes are under the control of an inducible GAL1 promoter on 2-micron (multicopy) plasmids with a LEU2 marker.
Saccharomyces cerevisiae strain ISY361 was constructed from strain W303. It contains two integrated reporter genes. The HIS3 reporter with a minimal core promoter was integrated at an ade8 disruption with plasmid pIS452. 27 The lacZ reporter was integrated at a lys2 disruption with pIS341. 27 The expression of the two reporter genes is controlled by four upstream POU-binding sites. The genotype is MATα, ade2-1, his3-11,15, leu2-3,112, trp1-1, ura3-1, can1-100, lys2::POU ops-LACZ, ade8::POU ops-HIS3.
ISY361 was transformed with bait plasmid pIS586 and prey plasmid pIS591 to generate the strain ISY361+/+ expressing POU-A93D bait and AD-V112E prey. A strain containing bait plasmid pIS586 and prey plasmid pIS580 lacking the V112E domain was also generated to serve as a control and is referred to as ISY361+/–.
High-Throughput Yeast Two-Hybrid Screening
Yeast media were prepared with reagents obtained from Becton Dickinson (Franklin Lakes, NJ) and Sunrise Science Products (Knoxville, TN). Strains were grown overnight at 30 °C with agitation in Synthetic Complete (SC) medium lacking Leu and Trp and containing 2% glucose. Cells were harvested by centrifugation at 4700g for 5 min, pellets were rinsed twice with sterile distilled water, and cells were suspended at OD595 0.01 in SC medium lacking Leu, Trp, and His and containing 2% galactose instead of glucose. The suspension also contained 2 mM 3-amino-1,2,4-triazole, which is used to minimize the effect of the basal expression of HIS3.28,29 Cell suspension (100 µL) was distributed to wells of sterile clear, flat-bottom, polystyrene, 96-well microplates (cat. 3370, Corning Costar, Tewkbury, MA) using a dispensing eight-channel pipettor. Eight wells were reserved for blanks. Chemicals were added to each well using a Biorobotics Biogrid II robot equipped with either a 0.7 or 0.4 mm diameter 96-pin tool. The yeast plates were incubated at 30 °C in a humidified chamber without agitation for 48 h. The cells were suspended by gently vortexing for 1 min, and OD595 readings were obtained using an Opsys MR 96-well plate reader (Dynex Technologies, Chantilly, VA). OD595 readings of wells containing only medium were defined as 0% growth, and OD595 readings of wells containing yeast but no screening chemicals were defined as 100% growth.
The compounds were tested at a final concentration of either 10 or 15 µM. Compounds showing growth inhibition were typically retested at two different concentrations in duplicate in medium lacking His and in medium containing 20 µg/mL L-His. Compounds showing more growth inhibition in medium lacking His than in medium containing His were retested in two or more replicates over a concentration range.
BUDE Virtual Screening
Virtual ligand screening was carried out on the University of Bristol’s high-performance computing system BlueCrystal with the docking program BUDE (version 1.2.9) 25 utilizing the University of California, San Francisco ZINC8 virtual ligand database. 30 Coordinates of monomer subunits were taken from the x-ray crystallographic structure of the polymeric ETV6T PNT domain (PDB: 1LKY) and used as representative of the wild-type interfaces in the A93D-PNT or V112R-PNT domain backgrounds. In brief, the protein structure, known as the receptor, was placed as a mol2 file with the origin at the wild-type A93 or V112 residue for the V112R- or A93D-PNT domain, respectively. Centered on the origin, the docking grid search volume was a 15 × 15 × 15 Å cube. Members of the ZINC8 library, consisting of greater than 8 million ligands (each having approximately 20 conformers per ligand generated), were tested for docking around the origin.
A second BUDE screen was carried out with residues of the intermolecular K99-D101 salt bridge set as the origins. In addition, an ensemble of 10 different structures, obtained from 10 ns steps of a 100 ns molecular dynamic (MD) simulation performed with GROMACS, 31 were used as the receptors. Docking was carried out using the top 200,000 compounds, and their conformers, that exhibited the lowest binding energies in the first BUDE screen, described above.
MD Simulations on Docked Candidates
Ligands that were targeted for the interfacial residues of the ETV6 PNT domain were ranked on their predicted free energy of binding. The top 500 compounds to each interface underwent a short 10 ns MD simulation of the ligand–protein complex to determine if they maintained a stable interaction. In brief, the MD simulations were performed with AMBER (version 16) 32 using the FF14SB-ildn force field, TIP3P water, and ligand parameters taken from the GAFF (General Amber Force Field). 33 The full 10 ns simulations were run with 2 fs integration step size while maintaining a temperature of 300 K and pressure of 1 bar. Resulting ligand root-mean-square deviation (RMSD) time courses were calculated for the trajectories relative to the initial, midpoint, and final poses using CPPTRAJ. 34 In addition, the trajectories were visualized using VMD (version 1.9.2) software. 35
BUDE Candidate Selection and Testing
Compounds identified by virtual docking underwent several iterations of selection. First, the top-ranked poses with the lowest calculated binding energies were manually inspected in Chimera (version 1.13.1). 36 For the ligands targeted to the interface, those that had duplicate conformers with low RMSDs during 10 ns MD simulations were preferentially selected over those with high RMSDs or that dissociated. The generated list of potential compounds was further refined by considering their commercial availability and selected to give a range of chemical diversity targeting the EH or ML surfaces of the ETV6 PNT domain. In total, 50 compounds were purchased to target the core interfacial residues. Of these, 16 targeted the ML surface around A93 in the V112E-PNT domain structure, 32 targeted the EH surface around V112 in the A93D-PNT domain structure, and 2 targeted both. In addition, 10 compounds were purchased to target the K99-D101 salt bridge (6 targeted D101 and 4 targeted K99).
Testing of Compound Binding by NMR Spectroscopy
Candidate compounds were tested in vitro for binding to the 15N-labeled PNT domain via 15N-HSQC monitored titrations recorded at 25 °C with Bruker Avance 500 or 600 MHz spectrometers. Isotopically labeled ETV643–125 with either an A93D or V112E substitution was expressed in Escherichia coli and purified as described previously. 19 Purchased compounds were dissolved to 50 mM stock solutions in DMSO. Purified protein samples were at a final concentration of 150 µM and volume of 450 µL in a standard buffer (20 mM MOPS, 50 mM NaCl, and 0.5 mM EDTA at pH 7.0 for the A93D-PNT domain and pH 8.0 for the V112E-PNT domain) with D2O (5% v/v) added for signal locking. All compounds were tested at a minimum 2:1 molar ratio and a maximum 20:1 molar ratio compound to protein. Control titrations with DMSO were also recorded.
Results
Development of a PCA to Screen for Inhibitors of PNT Domain Association
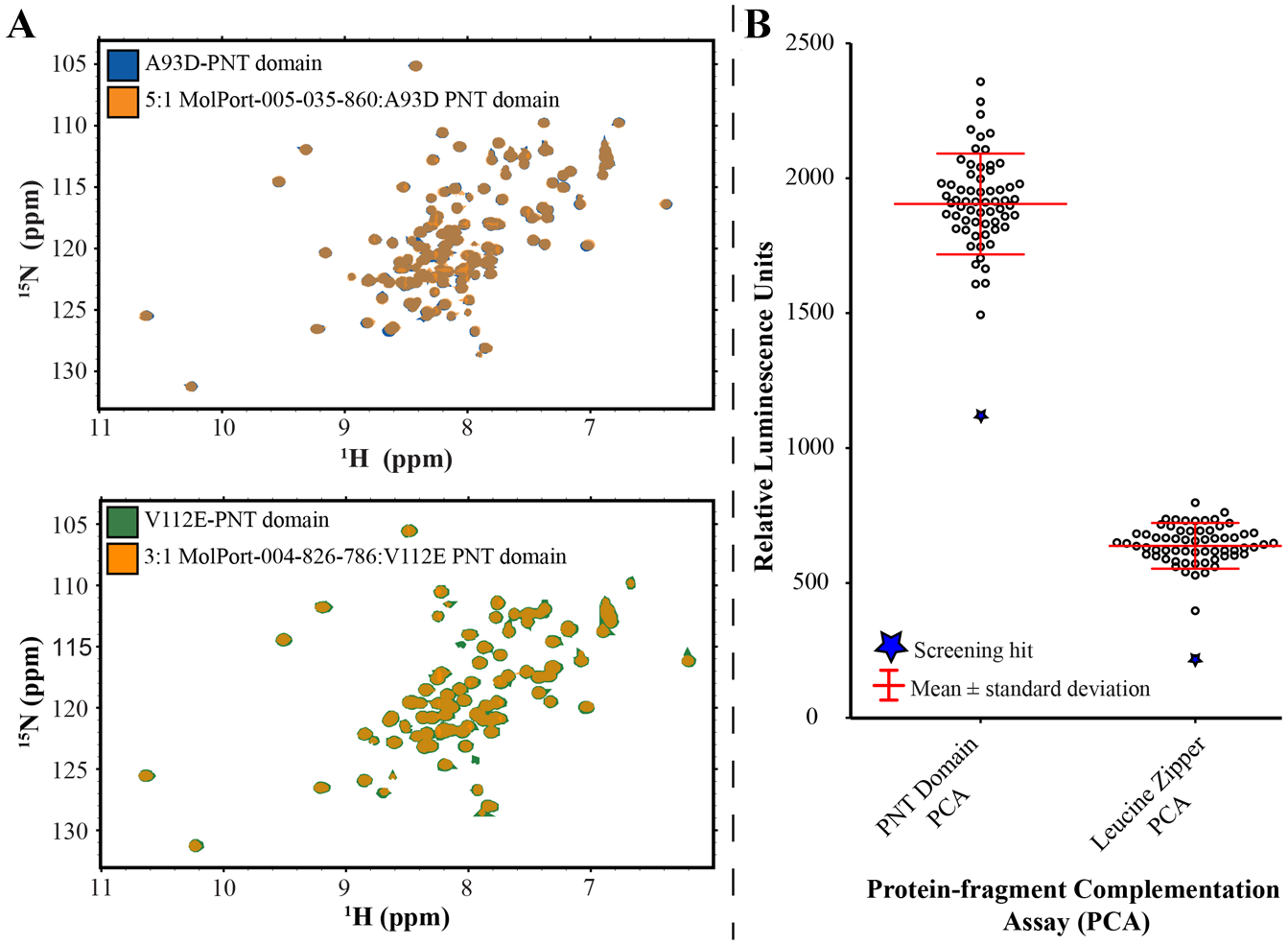
Initially, we established and characterized a PCA for monitoring heterodimerization of the A93D- and V112E-PNT domains based on the split Gaussia luciferase methodology. 23 Various fusion proteins containing the mutant ETV6 PNT domains and either N-Luc or C-Luc fragments were transiently expressed in HEK293 cells ( Suppl. Table S2 ). Cell culture medium alone or cells exposed to the reagents needed for the transient transfections showed luminescence readings of 19 ± 2.4 (mean ± SD) on a Varioskan LUX multimode microplate reader. When expressed alone, V112E-PNT/C-Luc, A93D-PNT/C-Luc, and A93D-PNT/N-Luc also produced relatively low luminescence readings of ~600, either 48 or 72 h posttransfection. Co-expressing A93D- and V112E-PNT domains that can heterodimerize, but fused to the same luciferase fragment, also resulted in a low luminescence reading of 70 ± 59. Introduction of the complementary N-Luc and C-Luc luciferase fragments, each linked to the A93D-PNT domain, yielded a higher luminescence of 2500 ± 170. However, this was still low when compared with combinations of A93D-PNT/C-Luc and V112E-PNT/N-Luc or A93D-PNT/N-Luc and V112E-PNT/C-Luc, which showed readings 48 h posttransfection of 320,000 ± 2500 and 91,000 ± 17,000, respectively. The luminescence readings of these combinations diminished 72 h posttransfection. The higher luminescence readings of the A93D-PNT/C-Luc and V112E-PNT/N-Luc combination compared with the reciprocal combination may indicate that one orientation of the luciferase fragments is more favorable to reconstitution of the active enzyme than the other. Based on these initial studies, the constructs encoding A93D-PNT/C-Luc and V112E-PNT/N-Luc were expressed stably in HEK293 cells to facilitate high-throughput screening.
There are no known inhibitors of ETV6 PNT domain polymerization to use as controls. However, the isolated PNT domain has a dominant-negative effect on cells expressing EN, reverting morphology back to wild-type. 19 Thus, we transiently transfected the A93D-PNT domain into the stably expressing HEK293 cells to test for an expected reduction in the luminescence reading due to competition with the A93D-PNT/N-Luc for the V112E-PNT/C-Luc-binding interface ( Fig. 2A ). At 24 h posttransfection, the A93D-PNT domain caused a significant decrease in luminescence with this cell line, but not with control cells expressing luciferase fragments fused with leucine zippers. At 48 h posttransfection, both systems were affected, but the decrease was most pronounced with the cells for the PNT domain PCA. To a much lesser extent, a transiently transfected empty pcDNA3.1 vector also reduced luminescence for the two systems 48 h posttransfection. Collectively, these controls define the expected sensitivity to inhibition of PNT domain heterodimerization.
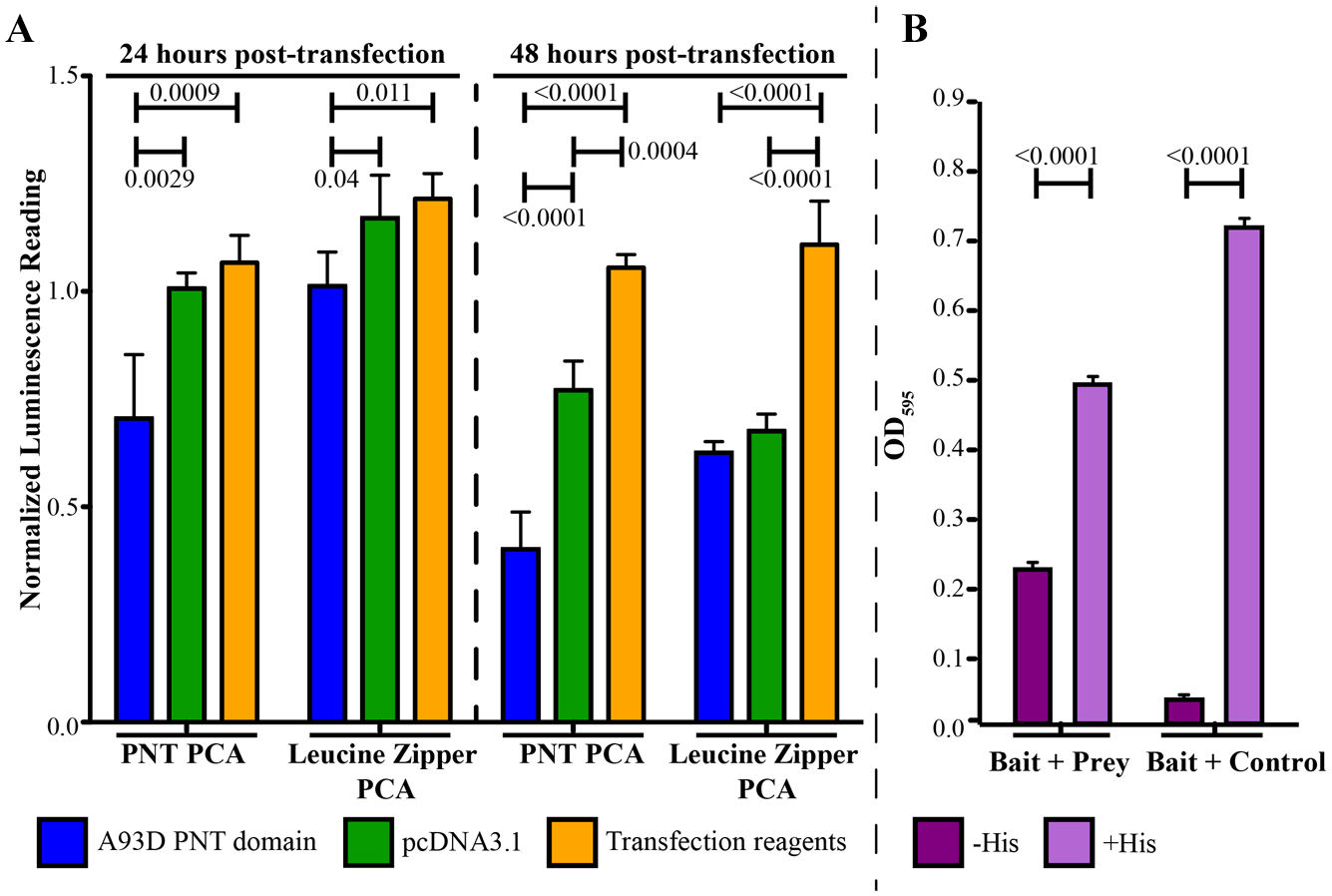
(
High-Throughput Screening Using the PNT Domain PCA
In total, ~18,000 compounds were screened with the PNT domain PCA assay. Plates were analyzed individually due to the limited stability of the luminescence signal over time. Thresholds for hit identification are generally expressed in terms of standard deviations from the mean and can be set empirically based on the number of compounds from the library that can be reasonably screened in secondary assays. 37 Thus, for this assay, compounds of interest were identified as having a luminescence reading that was more than 2 standard deviations away from the average luminescence reading of a plate ( Fig. 3A ), leading to an overall ~1% hit rate. Statistical analysis of 146 plates screened, which encompasses all plates that contained controls, comparing the positive control (the PNT domain PCA) and negative control (media), gave an overall Z′ factor for the screening assay of 0.33. 37 However, due to the variation of the luminescence signal over time, calculating the Z′ factor per batch (i.e., per set of plates run at one time) resulted in a higher Z′ factor of 0.71 ± 0.12 (n = 17 batches). Furthermore, on a plate-by-plate analysis, the Z′ factor increased to 0.86 ± 0.07.
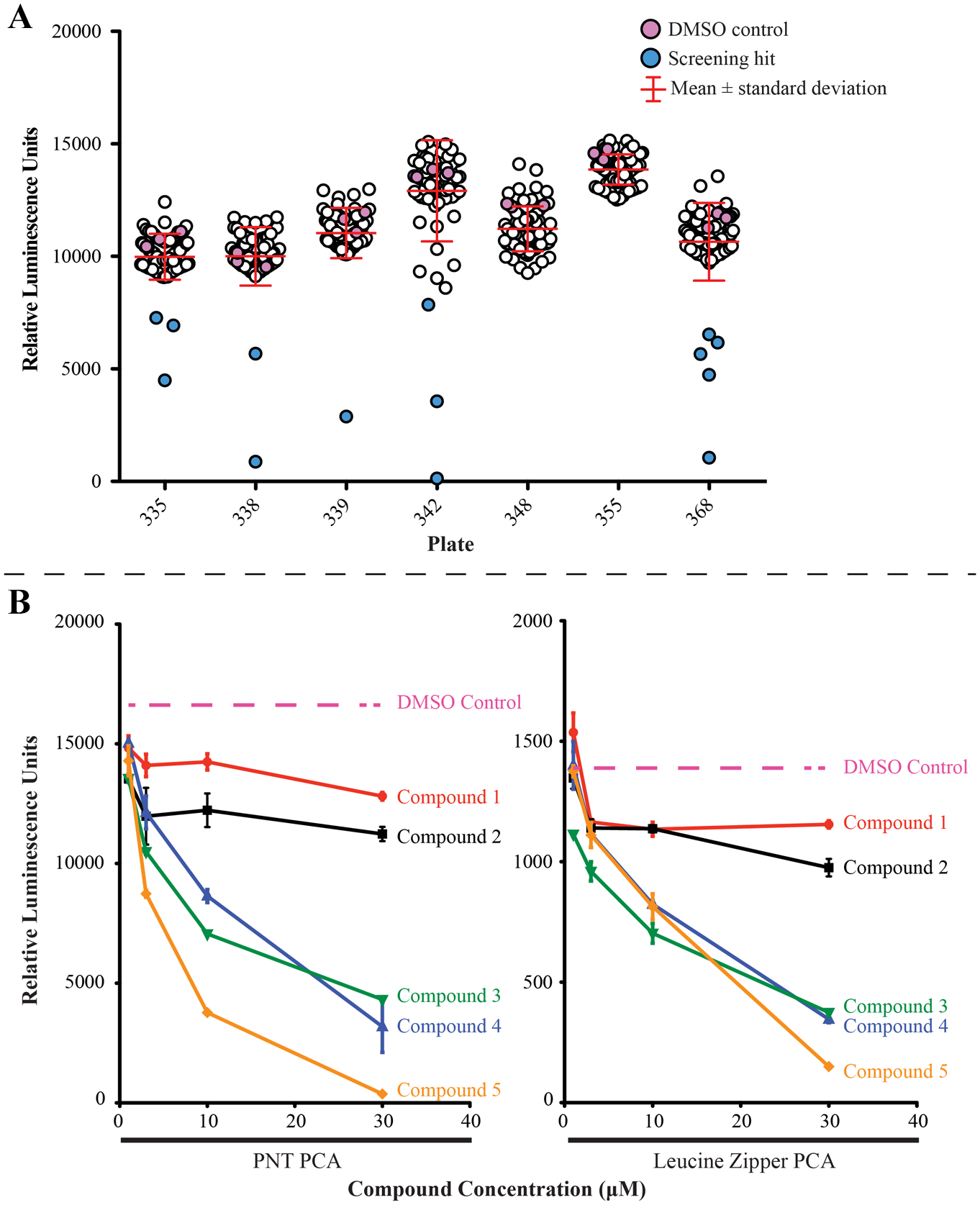
(
Secondary testing of screening hits was performed in the PNT domain PCA and the control leucine zipper PCA systems ( Fig. 3B ). Compounds were retested in duplicates at 1, 3, 10, and 30 µM, and every well was examined under the microscope. All compounds showing evidence of precipitation or obvious cell toxicity, demonstrated through cell rounding or death, were excluded for further validation. Out of 179 compounds retested, 83 did not decrease the luminescence in a concentration-dependent manner and were likely artifacts of the initial screens. All 96 of the remaining compounds that showed a concentration-dependent decrease of luminescence in the PNT domain PCA exhibited the same pattern with the leucine zipper PCA. Thus, changes in luminescence were likely due to factors other than inhibition of the PNT domain association.
In addition to the secondary cellular screening, 13 of the 96 compounds were purchased and tested for binding in vitro to the PNT domain using NMR spectroscopy. These compounds were chosen as they had exhibited a concentration-dependent response in the PNT domain PCA prior to the leucine zipper PCA counterscreen and were commercially available. The 15N-HSQC spectra of 15N-labeled A93D-ETV643–125 were recorded upon progressive titration with each compound. Amide 1HN and 15N chemical shifts are highly sensitive to even subtle structural changes accompanying ligand binding. 38 In no case were any chemical shift perturbations observed, indicating no detectable binding ( Suppl. Table S3 ).
Development of a Yeast Two-Hybrid Assay to Screen for Inhibitors of PNT Domain Association
The yeast two-hybrid assay involved several plasmids in auxotrophic yeast strains ( Fig. 1B ). The bait plasmid pIS586 encoded a fusion between the POU DNA-binding domain and the A93D-PNT domain, the prey pIS591 encoded a fusion between the NLS-B42 AD and the V112E-PNT domain, and the prey control pIS580 lacked the PNT domain. To validate the assay, we showed that the yeast strain ISY361+/+ containing the “bait + prey” plasmids indeed grew in the absence of His in the medium, whereas the “bait + control” strain ISY361+/– lacking the V112E-PNT domain showed little to no growth ( Fig. 2B ). Growth was restored upon addition of His to the latter, indicating that lack of growth was specifically due to lack of HIS3 gene expression.
High-Throughput Screening Using the Yeast Two-Hybrid Assay
The compounds from the PoPPI, Sigma, Biomol, Selleck, Prestwick, and Microsource libraries were screened in the yeast two-hybrid assay. The effects of the compounds on yeast cell growth were displayed as histograms for each library, with representative examples in Figure 4A . In general, the libraries showed a narrow growth distribution range, with most compounds affecting growth by less than 10%. The 214 compounds from the Sigma, Biomol, Selleck, Prestwick, and Microsource libraries that showed >50% growth inhibition were selected for secondary screening. No compound from the PoPPI library caused >50% growth inhibition. Instead, the 21 compounds that showed >20% growth inhibition in the PoPPI library were selected. Z′-factor calculation was carried out on each plate with the cells without addition of compound acting as positive controls and media as negative controls. The average Z′ factor was 0.67 ± 0.29 (n = 66 plates).
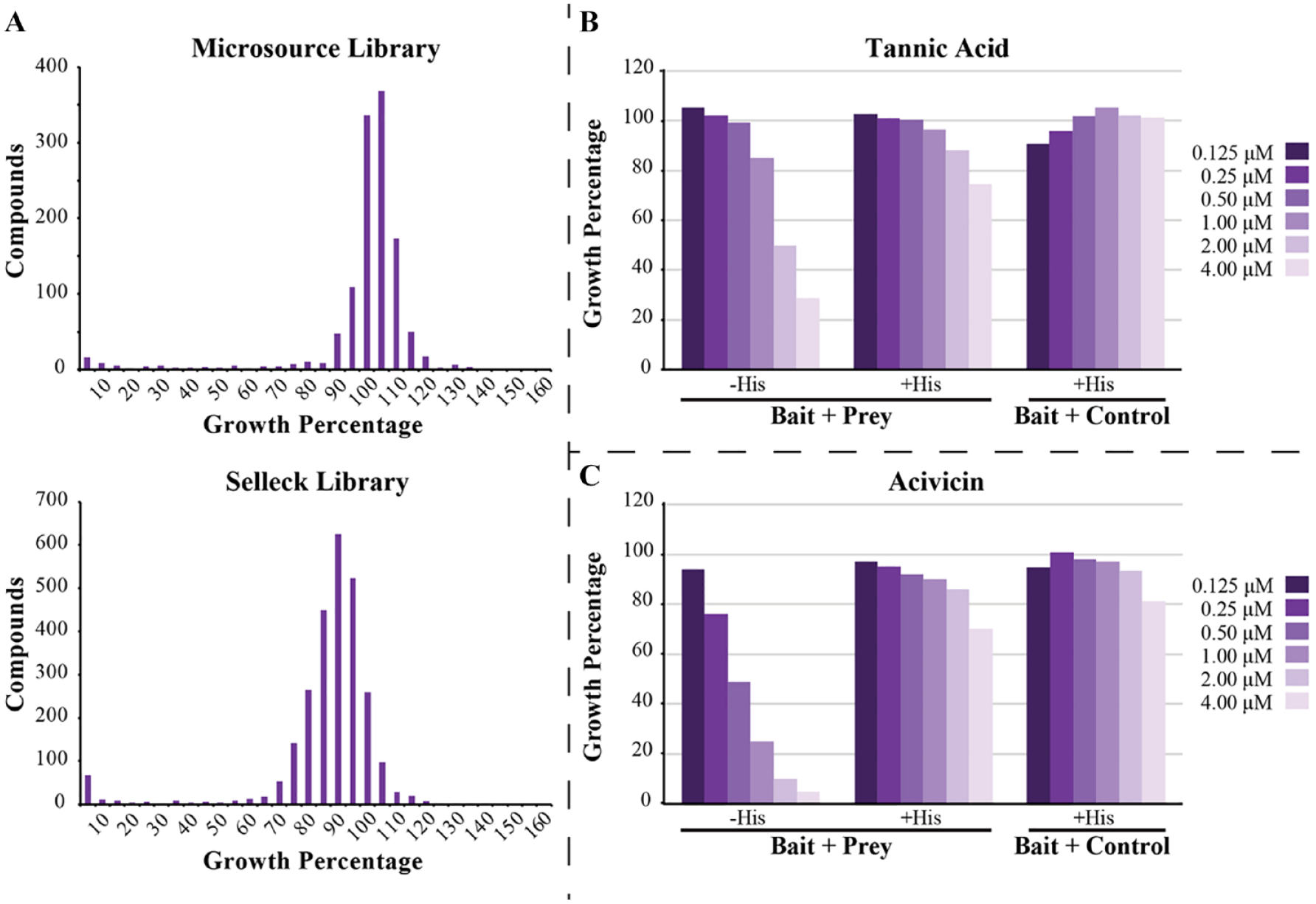
(
Secondary screening was carried out using multiple concentrations of the hits from the primary screen. Several compounds, such as tannic acid ( Fig. 4B ) and acivicin ( Fig. 4C ), showed a decrease of cell growth in the “bait + prey” strain that could be restored with the addition of histidine and exhibited no decreased cell growth in the “bait + control” strain. It was subsequently recognized that acivicin is a glutamine analog and can inhibit γ-glutamyltransferase, 39 which is needed for histidine biosynthesis. While such compounds are false positives of the two-hybrid screen, they do validate that a growth response should be observed if a compound interferes with histidine biosynthesis through inhibition of PNT domain association.
In total, three compounds (paromomycin, tannic acid, and sanguinarine) caused a decrease in cell growth while having no reported impact on the biosynthesis of histidine. Thus, they were purchased for final testing utilizing NMR spectroscopy. NMR-monitored titrations were carried out with both 15N-labeled A93D- and V112E-ETV643–125. In all cases, no amide 1HN-15N chemical shift perturbations we observed, indicating that the three compounds do not detectably bind to the monomeric PNT domains in vitro ( Suppl. Table S3 ). The reasons underlying their effects on the yeast two-hybrid screen are currently unknown.
Virtual Screening for Inhibitors of ETV6 PNT Domain Self-Association
Having found no inhibitors in the cell-based screening assays, we decided to perform virtual screening. Virtual screening for ligands predicted to bind the ETV6 PNT domain self-association interfaces was performed using the BUDE algorithm with a high-performance computer cluster. More than 160 million total poses of ~8 million ligands from the ZINC8 library, averaging 20 conformers per molecule, were tested. Virtual docking of these molecules was initially directed toward 15 × 15 × 15 Å search boxes centered on the A93 and V112 interfacial regions of the PNT domain ( Fig. 5 ).
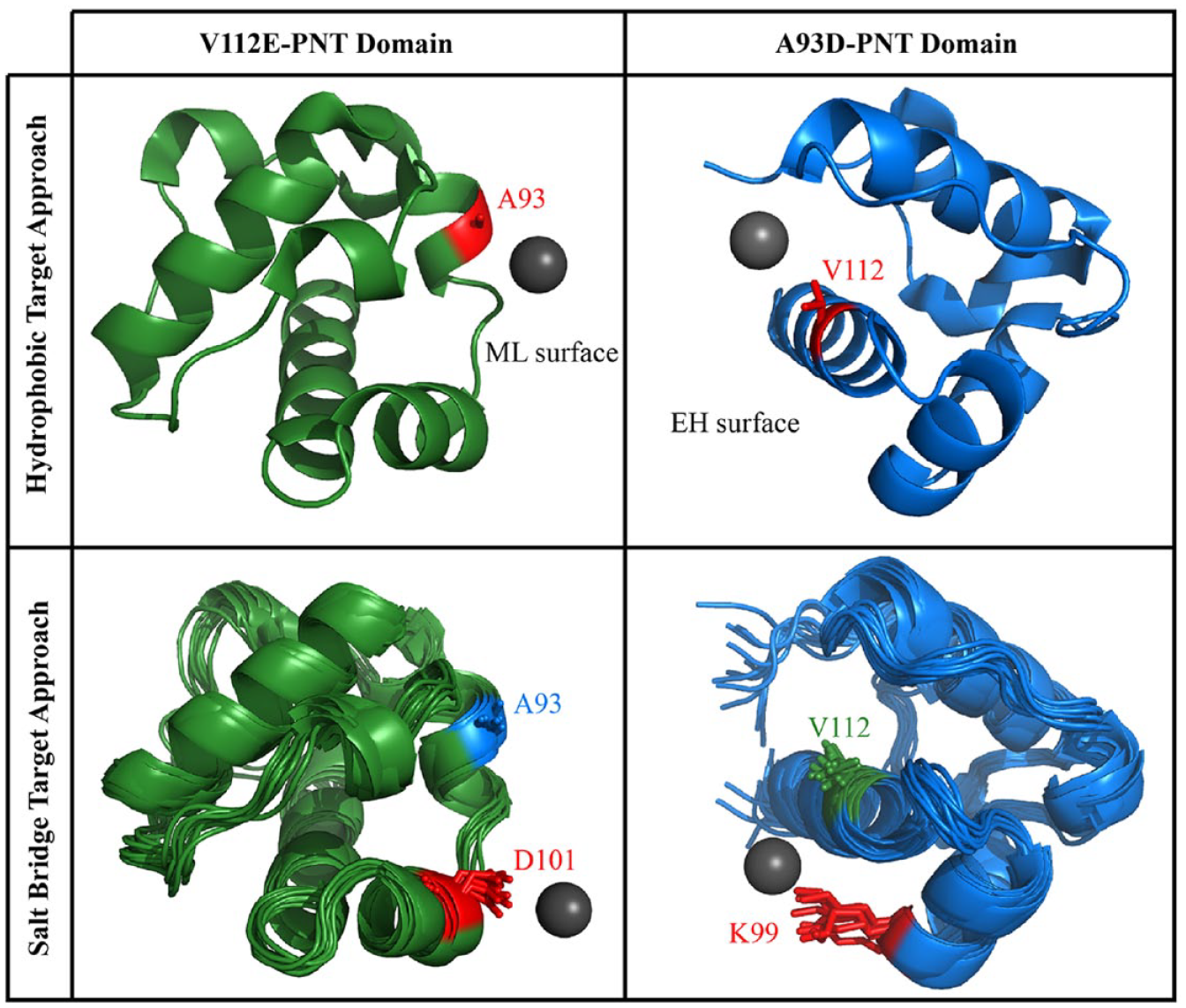
General overview of the BUDE screening. The origin for the starting position of the ligand in the virtual screen is indicated by the gray sphere, and the search space was centered around this point. To target the hydrophobic interfaces (upper), PNT domain structures were taken from (PDB: 1LKY). To target the flanking salt bridge (lower), 10 structures from a 100 ns MD simulation were chosen.
After running BUDE targeted to each PNT domain hydrophobic interface, the top 500 compounds predicted to have the best binding energies were selected for short MD simulations. The simulations were carried out to determine if the compounds would theoretically remain associated with the PNT domain over a 10 ns sampling. The coordinate RMSDs of each ligand throughout the simulation versus its initial, midpoint (5 ns), and endpoint (10 ns) poses were plotted. Based on these RMSD plots, ligands were scored as “excellent,” “good,” “mediocre,” or “bad” ( Fig. 6 ). For the A93D PNT domain, 33 and 101 compounds were deemed excellent and good, respectively, and the V112E PNT domain had 42 and 73 compounds that were deemed excellent and good, respectively. In parallel, every compound in the top 500 was visually analyzed for common structural motifs and suitable interactions.
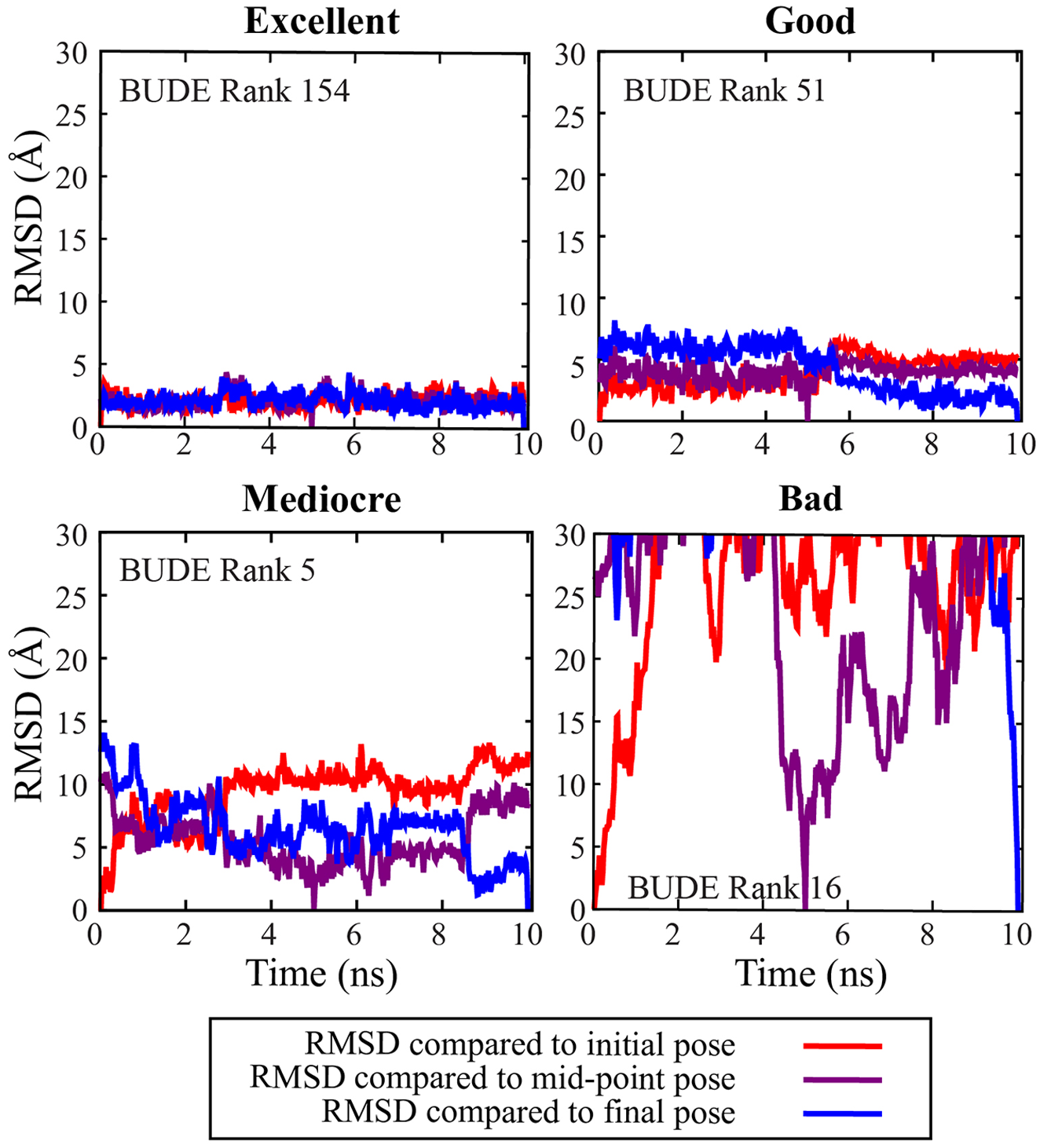
The coordinates of selected ligands over the course of 10 ns MD simulations were compared with the starting point (red), midpoint (purple), and endpoint (blue). Ligands were deemed as “excellent” (low RMSDs), “good” (RMSDs < 10 Å), “mediocre” (RMSDs > 10 Å), or “bad” (RMSDs > 30 Å, indicating dissociation).
Compounds selected for testing by NMR spectroscopy and the screening assays had excellent or good scores in MD simulations, appeared to interact closely with the PNT domain, were commercially available, and represented a range of chemical motifs to increase diversity. Preference was given to compounds that were predicted to bind in multiple conformers. Several additional compounds that had a BUDE ranking >500 were also selected due to possessing certain structural motifs, such as carboxylates or protonated amines, that could potentially interact with complementary motifs of the PNT domain. Collectively, this resulted in 35 and 17 compounds predicted to bind the hydrophobic interfaces centered around A93 (ML surface) and V112 (EH surface) of the V112E- or A93D-PNT domain monomers, respectively ( Suppl. Fig. S2 ).
A second screening was subsequently carried out to target the interfaces around K99 and D101. These residues form an intermolecular salt bridge ( Fig. 5 ). Furthermore, to help account for protein dynamics, an ensemble of protein structures taken at 10 ns steps of 100 ns MD simulations were used for docking. To accelerate the process, we also focused on only the top 200,000 compounds from the first BUDE screen and their respective conformers. In total, ~4 million poses of ligands were tested for docking to the 10 different A93D- or V112E-PNT domain structures. The resulting top 500 candidates were visually inspected, and 10 compounds with chemical diversity were selected for testing ( Suppl. Fig. S3 ).
Experimental Testing of the Virtual Screening
A total of 60 candidate compounds from two BUDE screens were purchased for further testing. All compounds were used for NMR-monitored titrations with samples of 15N-labeled A93D- or V112E-ETV643–125, as appropriate. None were found to bind with any detectable affinity as evidenced by the lack of amide 1NH-
15
N chemical shift perturbations (
(
Discussion
In this study, we established three complementary approaches to screen for potential inhibitors of ETV6 PNT domain polymerization. Unfortunately, we did not succeed in discovering any small molecules that bound the PNT domain in vitro or disrupted its self-association in vivo. Nevertheless, valuable insights were gained that should facilitate future studies aimed at modulating PPIs with systems similar to ETV6.
The mammalian and yeast cell assays were designed for unbiased high-throughput screening of compounds in a cellular context, with a readout that is dependent upon disruption of a heterodimer model of the ETV6 polymer. However, in the PCA assay, screening hits can arise not only from inhibition of heterodimerization, but also from perturbation of split luciferase reconstitution or luciferase activity, as well as due to cytotoxicity or inhibition of cellular proliferation. Therefore, the leucine zipper PCA was critical as a control assay to determine whether any reduction in luminescence was due to the latter confounding effects. Similarly, in the yeast screening assay, a control vector and the addition of histidine to secondary testing of screening hits for growth rescue were used to eliminate any compounds that caused cell death or inhibition of the histidine biosynthetic pathway.
It is worth emphasizing that the ETV6 A93D- and V112E-PNT domains heterodimerize with low nanomolar affinity.18,19 This certainly presents a challenge in discovering potentially rare compounds in chemical libraries with comparable affinities to effectively disrupt this tight PPI. A mitigating strategy might involve the initial use of PNT domain variants with additional mutations to weaken their association and thereby reduce the stringency of the assay. Such variants may be inspired by detailed biophysical analyses of the ETV6 PNT domain interfaces. 40 In addition, it would be beneficial to use cell-based “up” assays that positively select for disruption of PNT domain association or employ a readout that increases, rather than decreases, with this result.
To this end, we explored the use of a yeast two-hybrid assay where the expression of HIS3 and LacZ reporter genes is dependent upon PNT domain heterodimerization. However, this assay as implemented was not useful for the discovery of inhibitors because potential hits in small-molecule screens would prevent growth on medium lacking His, and loss of LacZ expression, which could also be produced by various alternative effects unrelated to inhibition of the PNT domain interaction. Ideally, the screening assay could be modified to include a reporter such as CAN1 or URA3, where inhibitors of the interaction would prevent activation, allowing detection by growth on canavanine or 5-fluoroorotic acid, respectively. 41 Alternatively, yeast-based strategies could also be used, such as the repressed transactivator system, where the interaction of bait and prey fusions causes repression of gene expression, and inhibitors of the interaction cause gene activation, which is detected by growth of yeast on selective media. 42 However, this system is currently limited to use with Gal4 DNA-binding domain fusions, and we were unable to detect interaction of PNT fusions in yeast using the Gal4 DNA-binding domain linked to the PNT domain.
In parallel to cellular assays, we undertook a targeted virtual screening approach to search for compounds that bind the known polymerization interfaces of the ETV6 PNT domain. Although BUDE has been successful in identifying inhibitors of PPIs, 26 and numerous potential ligands were predicted to bind the ETV6 PNT domain in silico, none were found to do so when tested in vitro with NMR spectroscopy or in vivo with cellular assays. However, common motifs that were enriched may serve as starting structures for rational design or further high-throughput screening experiments.
We recognize that this target is particularly challenging since the ETV6 PNT domain polymerizes with high affinity through two relatively large flat interfaces. These comprise complementary central hydrophobic patches ringed by hydrophilic and charged residues with relatively flexible sidechains. Tightly bound ligands typically occupy depressions or cavities on the protein surface, but these features are absent with the PNT domain. However, in the case of targeting the intermolecular salt bridge, the virtual ligands seemed to be predicted to preferentially bind to certain PNT domain structures taken from short MD simulations. This flexibility of the protein may expose conformations better suited for small-molecule interactions.
It is unfortunate that some of the best hits from the virtual screen were not commercially available. Experimental testing of these compounds, as well as more in silico hits, might yield success. Indeed, a “similarity paradox” has been described where minor chemical modifications of otherwise similar molecules can render them active or inactive. 43 This paradox may suggest that, in our efforts to test a diverse set of compounds that sampled chemical space, we missed a high-ranking compound with affinity for the ETV6 PNT domain. Such a large-scale screening approach may be effective, as many PPI inhibitors are found through traditional cellular screening only after screening in excess of hundreds of thousands of compounds and carrying out structure–activity relationship studies to optimize initially detected weak-binding leads. 44
Fragment-based drug design and disulfide tethering, combined with combinatorial chemistry, are two approaches that could also be undertaken as the next steps in developing an inhibitor against PNT domain polymerization.45–47 Inspiration can also be found in the design of cyclic peptides and helix peptide mimetics that weakly bind the SAM domains of the Ship2 and EphA2 receptors.48,49 In all cases, virtual screening could be used to narrow down potential chemical motifs of interest for a more targeted screening approach. Collectively, such a multipronged strategy may lead to the discovery of a potentially new class of therapeutics that prevent the polymerization of chimeric oncoproteins resulting from a wide array of ETV6 chromosomal translocations.
Supplemental Material
sj-pdf-1-jbx-10.1177_2472555220979599 – Supplemental material for A Multipronged Screening Approach Targeting Inhibition of ETV6 PNT Domain Polymerization
Supplemental material, sj-pdf-1-jbx-10.1177_2472555220979599 for A Multipronged Screening Approach Targeting Inhibition of ETV6 PNT Domain Polymerization by Chloe A. N. Gerak, Si Miao Zhang, Aruna D. Balgi, Ivan J. Sadowski, Richard B. Sessions, Lawrence P. McIntosh and Michel Roberge in SLAS Discovery
Footnotes
Acknowledgements
We thank Mark Okon for assistance with NMR spectroscopy, Stephen Michnick for providing starting plasmids for the split luciferase PCA, Andrew Wilson for the PoPPI library, and Poul Sorensen for advice on ETV6 translocations.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by funds from the Canadian Cancer Society Research Institute to I.J.S., L.P.M. and M.R., and from the Canadian Institutes of Health Research (CIHR) to L.P.M. C.A.N.G. held graduate scholarships from CIHR and the University of British Columbia. We thank the Advanced Computing Research Centre at Bristol University for the provision of high-performance computing.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.