
Abstract
Over the past 20 years, the toolbox for discovering small-molecule therapeutic starting points has expanded considerably. Pharmaceutical researchers can now choose from technologies that, in addition to traditional high-throughput knowledge-based and diversity screening, now include the screening of fragment and fragment-like libraries, affinity selection mass spectrometry, and selection against DNA-encoded libraries (DELs). Each of these techniques has its own unique combination of advantages and limitations that makes them more, or less, suitable for different target classes or discovery objectives, such as desired mechanism of action. Layered on top of this are the constraints of the drug-hunters themselves, including budgets, timelines, and available platform capacity; each of these can play a part in dictating the hit identification strategy for a discovery program. In this article, we discuss some of the factors that we use to govern our building of a hit identification roadmap for a program and describe the increasing role that DELs are playing in our discovery strategy. Furthermore, we share our learning during our initial exploration of DEL and highlight the approaches we have evolved to maximize the value returned from DEL selections. Topics addressed include the optimization of library design and production, reagent validation, data analysis, and hit confirmation. We describe how our thinking in these areas has led us to build a DEL platform that has begun to deliver tractable matter to our global discovery portfolio.
Introduction
Modern drug discovery that is focused on delivering novel, small-molecule therapeutics to treat disease, via either target-based or phenotypic approaches, typically identifies bioactive starting points (“hits”) through the screening of a greater library of molecules. This hit identification (hit ID) process is a fundamental step for the launch of new discovery campaigns and carries with it significant accountability, as it is the initial direction-setter for a project team, ultimately determining the path to the clinic and dictating the chemical characteristics of the moiety tested. In this article, we describe recent advances in hit ID technologies, contemplate the suite of options that are available, and discuss some of the influencing factors when designing a new hit ID strategy for a program in a large pharmaceutical organization such as Pfizer. Furthermore, we delve into the decision to expand our hit ID platform by investing in DNA-encoded library (DEL) technology and share our experiences and current approaches to develop a robust and reliable source of high-quality lead matter for our project teams.
In the 1990s and early 2000s there was a surge in traditional high-throughput screening (HTS). We saw rapid advancements in screening methodologies and automation, including user-friendly, fluorescent readouts, homogeneous assay protocols, miniaturization to standardized 384- and 1536-well formats, and increasingly sensitive image-based detection systems. An increased adoption of combinatorial chemistry and a crusade across the industry to expand compound files through thoughtful exploration of chemical space, often for specific target classes, fueled this expanded screening capacity. The trifecta was delivered with the biological exuberance of the Human Genome Sequencing Project 1 and advances in recombinant DNA, which provided the drive to propose mechanistic hypotheses for new targets to screen. Thus, for a period, the industry screened and waited for emerging metrics to demonstrate that testing of increased numbers of compounds with high chemical diversity would lead to an enhanced ability to identify successful leads. And there are indeed some examples that could be taken to justify this hope; in screens of 2.6–3 million compounds, only a single lead molecule emerged that effectively inhibited the secretion of PCSK9; 2 a single noncatechol hit was validated as a dopamine D1 receptor agonist for cAMP production, 3 and within a single series that was identified, only one compound demonstrated the desired AMPK activator profile. 4 The importance of diversity seems to hold true beyond the Pfizer compound file; for example, Han et al. 5 from Roche describe identifying a singleton hit from a 1 million-compound phenotypic screen that sought inhibitors of hepatitis B surface antigen secretion. Additionally, Forma recently described the identification of tractable hits only after addition of a newly synthesized DEL to an already extensive DEL collection. 6
Overall, however, this golden age of screening appeared to be insufficient to offset the escalating costs of drug development and to solve the challenges of clinical attrition. Recent analyses based on 106 new drugs from 10 pharmaceutical firms calculate that overall investment in discovery and clinical development approaches $2.6 billion for each successful launch, 7 and there is a clear trend for increasing costs in recent years. These costs not only cover an individual project’s discovery and development effort, but also must compensate for the expensive failure of other programs within the clinic where, consistently, efficacy has remained the reason for almost 50% of clinical failures in phase II and phase III since 2008.8,9 Consequently, multiple strategies have been proposed and implemented across the length of the research and development (R&D) pipeline aimed at increasing success in pharmaceutical R&D.10–13 At the earliest drug discovery stages, the evolution of screening strategies in hit ID, hit validation, and lead optimization, and in pharmacokinetic and safety profiling, 14 has increased the breadth of the discovery screening funnel, enhanced its filtering efficiency and quality, and improved compound selection, 15 thus yielding better positioned starting points from which to launch a medicinal chemistry program, and an enhanced ability to identify series with more favorable pharmacology, ADME (absorption, distribution, metabolism, excretion), and safety profiles. The end result is a shorter path to the clinic and an increased confidence that the new asset possesses the chemical and biological credentials to adequately test therapeutic hypotheses in vivo.
Within the realm of hit ID we have seen parallel strategies, the first to make assays more physiologically relevant, moving away from simple reductionist models and “bringing the patient to the dish,” in the hopes of improving translation.16,17 This effort naturally aligns with the resurgence of phenotypic screening, which provides the opportunities to identify completely novel targets and links to disease, to illuminate pathway biology for optimal target selection and capitalize on the potential polypharmacology of test compounds.18–21 Phenotypic programs, however, tend to come with a consequent increase in investment, for example, the cost and time required to build and culture disease-relevant cell models (potentially testing multiple donors), the challenges of hit triage 22 and target deconvolution, the risks of investing in novel, often poorly characterized targets that may emerge, and the liability of potential downstream safety hurdles when proceeding with unknown mechanisms of action.
A complementary approach has been to introduce rapid target-based biophysical screens such as affinity selection mass spectrometry (ASMS),23–25 sometimes known as automated ligand identification system (ALIS)26–28 and DEL selection. 29 Although there are different methodologies for ASMS prosecution (reviewed in Andrews et al. 30 ), all rely on the same basic principle of incubating compound libraries (often >2500 compounds at one time) with a target and then subsequently isolating the target along with bound compounds and using accurate mass analysis to identify the small molecules present. 25
DELs take compound compression log orders of magnitude higher, incubating a target simultaneously with billions of compounds, each encoded with DNA tags. Ligands are identified by next-generation sequencing (NGS) of the DNA tags following separation of the target–ligand complex and the release of bound molecules. Once biophysical platforms are established, they can require considerably less investment per campaign and can provide early insight into druggability as well as tools to build target confidence in addition to providing leads for chemistry efforts. ASMS and DEL can provide advantages in cost, speed, access to chemical space diversity/density, and the ability to tackle varied biological conditions in parallel with the obvious disadvantages of being unable to address function and of generally providing reduced physiological context ( Table 1 ).
Differentiators for Hit ID Technologies.

Layered onto these overarching platform themes we have seen an evolution in the substrate for hit ID campaigns that can render one approach more or less favorable. The desire for truly novel cures for disease drives priorities toward first-in-class targets and novel ways to modulate known pathways/targets that have high confidence in rationale (CIR) but have previously proved intractable across the industry. Hence, for example, we see investment in protein homoeostasis,31–34 RNA targets,35,36 and novel members of gene families that have precedence for druggability, for example, kinases, G-protein-coupled receptors (GPCRs), and solute transporters, 37 where companies can exploit prior medicinal design experience. Furthermore, we see a trend for efficiency gain through the multiplexing of endpoints or targets and a desire for the extraction of more parameters (high-content data) from each screen to increase the richness of the data set and thus the knowledge that can be derived from every experiment.38–40
Typically, each hit ID strategy will begin with a thorough assessment of the extent of target (or disease) knowledge, the competitive landscape, the mechanism of action desired as it relates to laboratory objectives or product profile, and the available reagents or methodologies to test biology rationale and to find target modulators (see Box 1). The competitive landscape and the available chemical equity will often direct the consideration of chemical space that should be addressed. The full Pfizer HTS file of >3 million compounds is viewed internally with high regard in terms of chemical attractiveness, being a curated set acquired from multiple legacy organizations and project-directed synthetic efforts. It is accessible for traditional high-throughput diversity screening in either singleton or compressed formats and also for ASMS, which has the advantage of requiring relatively little assay development (provided the protein is facile to work with), low protein requirements, a rapid turnaround time, low cost, and speedy access to hits for follow-up studies. However, ASMS can be limited in its applicability by proteins with low stability (typically for our needs, stability for 48 h at 4 °C in the presence of 4 % DMSO is desired) and is potentially biased to compounds that have a minimum half-life in the target–ligand complex of ~1 s (which corresponds to a dissociation rate constant [koff] of approximately 0.7 s−1 or slower) due to the time taken for elution from the size exclusion column.25,27
At Pfizer we have a long history of traditional HTS and multiple examples of successful clinical candidates and marketed products (Xeljanz, 41 Slentrol, 42 Maraviroc, 43 PH-797804 [p38 kinase], 44 and PF-05180999 [PDE2A] 45 ). However, the investment into delivering an HTS is frequently high in terms of assay development time and expertise, reagent costs, and timelines, and we have had multiple experiences over the years of HTS campaigns that have had a low return on investment. A low-value HTS has typically been due to running the screen too late in a project life cycle (i.e., when chemical lead matter is already available), running a screen too early in a project life cycle (i.e., when biological understanding has yet to be fully developed), or the screening technology selected failing to deliver valid and viable hits for the mechanism in question. In our minds, there are three key drivers in the decision to use a classical HTS approach to screening: (1) CIR trajectory (or increasing likelihood of biology translation); (2) chemistry need, including the likelihood of equity; and (3) screening precedence (scalability and precedence for technology being successful in the target class). High CIR, such as a compelling case from human biology for causality and functionality in disease; a high chemical need for novel equity, such as an unprecedented target or lack of intellectual property space; and/or a well-precedented screening technology for valid hit ID would all support investment in large-scale HTS, and project teams often proceed with large-file HTS when at least two of these parameters are in their favor. 14 It is also worth remembering that in almost all cases, some kind of high-throughput functional assay is necessary downstream of hit ID methods for orthogonal hit validation and/or to support chemistry structure–activity relationship (SAR) efforts, so the time invested into the development of assays that may be suitable for medium- to high-throughput screening is rarely wasted.
Fragment-based drug discovery (FBDD) has solidified its place in the hit ID toolbox in the past 20 years and has propelled ~30 drugs to various stages of the clinical pipeline. 46 Fragments are generally chemical groups with <20 nonhydrogen atoms, and fragment libraries are typically limited collections (<10,000 compounds) of low-molecular-weight (low-MW) libraries (150–300 Da), which, though small, have the advantage of efficiently exploring chemical space. Fragments can bind to multiple target sites in a variety of poses with high binding energy per atom and may exhibit less steric hindrance or electrostatic repulsion in a binding site compared with drug-like molecules (MW = 500 Da). HTS requires a biochemical or cell-based assay to measure a binding outcome, but these screens are not usually suitable to qualify the weak interactions of fragments and targets; however, since FBDD monitors direct binding to the macromolecule, no such assays are needed at the outset. Biophysical techniques with high detection sensitivity can be used to monitor the weak interactions, including surface plasmon resonance (SPR), microscale thermophoresis (MST), capillary electrophoresis, weak affinity chromatography, biolayer interferometry/ultrafiltration, native MS, and isothermal titration calorimetry, although x-ray crystallography and nuclear magnetic resonance (NMR) tend to afford the greatest throughput in an industrial setting.47,48 Fragment screening generally offers higher hit rates, often on the order of 5%–20% 48 compared with HTS, which is typically <1%; nevertheless, fragment hits are usually weak binders with fast off-rates and must be developed into higher-affinity, larger molecules to become leads, and this is typically the bottleneck in the process. Our experience is that FBDD programs are most successful when structure enabled, and that the application of orthogonal biophysical and biochemical assays is critical for validating hits, prioritizing for x-ray crystallography, and building confidence for medicinal chemistry efforts. 48 As a result, the availability or the high likelihood of generation of a high-quality crystal structure is a prerequisite for us to initiate an FBDD campaign.
DELs have the advantage of being orders of magnitude larger than HTS files, and a campaign of DEL selection and sequencing can also be completed in only a few days to weeks. 49 Additionally, running multiple conditions is a standard approach, for example, with or without known ligands, with binding complex partners, with mutated targets and selectivity targets, with varying target protein concentrations, and so forth, and this can provide unique insight into the molecular mode of action of ligands. However, the compatible chemistries for DEL builds are more restricted since they are performed in water and must not significantly damage the DNA tag. 50 The largest libraries are typically formed by the combination of monomers in three or four cycles of chemistry, which tends to lead to high-MW molecules that can be unattractive starting points for chemistry teams, and it is our experience that the follow-up of hits off DNA requires first a confirmation of true binder identification and then resynthesis that (depending on available resources) can be an activation hurdle for chemistry engagement. In the experimental part of this article, we describe how we have built our internal DEL process to overcome some of these challenges and make our DEL platform a more appealing primary strategy for project teams.
Incorporating the factors described above, Table 1 provides a summary of many differentiators (based on both practical and fundamental scientific principles) that may serve as a useful reference to inform the choice of hit ID strategy. One final factor to bear in mind is that there is inevitably a constraint within any organization that comes with considering a portfolio of programs in a holistic manner and fitting them within the capacity of the available capital resources, flexible budget, and colleague expertise. We acknowledge that in an ideal situation, parallel approaches to hit ID will often lead to novel insights through integration of complementary data sets, an advantage that is well described by Leveridge et al.; 51 however, in a world of restricted budgets and pressures to deliver clinical candidates with lean investments, we often find that we are required to limit the number of arms to our hit ID strategies and prioritize resources accordingly. With this in mind, we sought to enhance the impact of our DEL platform in order to fully exploit its unique combination of attributes.
Materials and Methods
General
DELs were prepared using a split-and-pool methodology at HitGen Ltd. (Chengdu, China) following a general strategy similar to that described in Kung et al. 52 Bromodomain 1 of BRD4 (cat. 6x-His-tev-BRD4-1(44-170)) was purchased from XTAL Biostructures (Natick, MA). The magnetic affinity beads used were either Neutravidin SpeedBeads (GE Healthcare, Piscataway, NJ) or His-Tag DynaBeads (Thermo Fisher, Carlsbad, CA).
DEL Selection and Data Analysis
DEL affinity selection and chemoinformatic analysis was conducted essentially as described in Chen et al.
53
For
Bead-Assisted Ligand Isolation
BRD4 containing a His-Tag (0.75 µM nominal concentration) was incubated with a Cy5-conjugated analog of PFI-1 54 for 30 min in a total volume of 40 µL of assay buffer (50 mM HepesNa, 150 mM NaCl, 0.01% Tween 20, 1 mM TCEP, pH 7.4). Following this equilibration step, the samples were transferred to wells of a PCR plate that contained prewashed His-Tag DynaBeads. The beads were resuspended and allowed to incubate at room temperature for 10 min. Magnetic separation was performed with a ring magnet (Alpaqua Engineering, Beverly, MA). The supernatant was aspirated to a receiver plate, and the protein was eluted from the beads in assay buffer supplemented with EDTA (10 mM). Following transfer of the eluate to the receiver plate, the fractions were evaluated in an Infinite M1000 plate reader (Tecan Group, Männedorf, Switzerland).
Bead-Assisted Ligand Isolation–Mass Spectrometry
Experiments were performed according to the protocol above using JQ1 55 as a titrant with the following exceptions: the BRD4 protein concentration was increased to 1.0 µM, and after removal of the supernatant fraction, the beads were resuspended in high-performance liquid chromatography (HPLC)-grade water (20 µL). Bound compounds were eluted from the beads by adding an equal volume of 90% acetonitrile containing 1% formic acid. The samples were analyzed on a Sciex 6500+ triple quadripole mass spectrometer fitted with a LeadSampler 1 autosampler (Sound Analytics, Niantic, CT) and Agilent (Santa Clara, CA) 1290 HPLC system. As needed, the samples were diluted with 50% acetonitrile to bring them within the dynamic range of quantitation. Data were processed with LeadScape Analyst 1.7 (Sound Analytics).
Multiparameter Optimization Score Calculation
Our DEL multiparameter optimization score (MPO_score) is weighted heavily by MW. For every compound in the analysis, individual molecular properties are assigned a score between 0 and 1 according to the descriptions below. Calculation of the MPO score is accomplished according to eq 1, where the sum of cLogP, aromatic ring count, and hydrogen bond donor count scores are multiplied by the MW score to provide a parameter ranging between 0 and 3. The individual and total scores are calculated as follows:
MPO_score calculation:
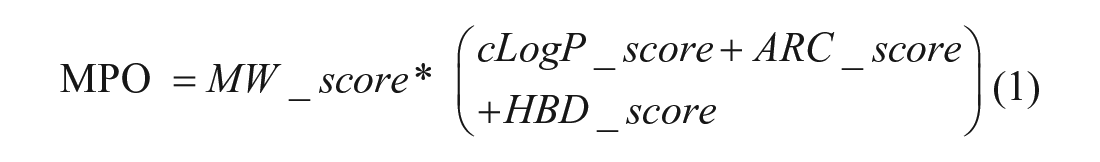
Selecting Hit ID Approaches

Results and Discussion
Implementing a DEL Platform for Hit ID: Starting Point and DEL Design
DELs provide an exciting opportunity to build diverse chemical collections unbiased by previous organizational therapeutic areas of focus and promise to enable the search for small-molecule ligands to first-in-class targets of academic and industrial importance. The concept of encoding small molecules was first introduced in 1992 by Sydney Brenner and Richard Lerner 56 and was subsequently reduced to practical routines that include the DNA encoding of multistep library synthesis and hit ID via affinity selection paired with high-throughput sequencing.57,58 Early demonstrations garnered much attention, for example, the identification of small-molecule ligands to tumor necrosis factor α, 59 and in the same year, Praecis Pharmaceuticals reported their encoded library technology (ELT) industrialization of split and pool synthesis, along with the discovery of various Aurora A and p38 MAP kinase inhibitors from an 800 million-member DEL. 49 A summary of the subsequent evolution of ELT at GlaxoSmithKline (GSK; née Praecis) can be found in Arico-Muendel. 29 Following the Praecis acquisition by GSK, there was an explosion of ELT-based biotech companies (X-Chem, HitGen, Vipergen, Ensemble Therapeutics, and Philochem) and a rapid uptake of DEL by other pharma (e.g., Roche, Novartis, Lilly, AstraZeneca, and Pfizer). A description of different approaches to compound selection and detection is provided in Chan et al. 60 and Salamon et al. 61 provides a good overview with multiple examples of chemical tools successfully identified from DELs.
Over the past decade, Pfizer has strategically aligned with partners to explore the promise of ELT: Ensemble Discovery, 62 X-Chem, 63 and HitGen. 64 Through the most recent partnership with HitGen, we leveraged our organizational expertise in both parallel medicinal chemistry and external collaborations to establish and apply DNA-compatible chemistry using our corporate building block collection to deliver DELs containing designed warheads that possess lead-like properties. Coupling this intuitively advantaged starting position with the speed of DEL hit ID campaigns is an attractive approach to accelerate our delivery of clinical assets. In addition to the benefit of speed, the ability to employ multiple selection conditions in parallel has been demonstrated to provide qualitative assessments of selectivity 65 and affinity 66 through the inclusion of an antitarget or varying protein concentrations, generating a rich biological profile for each compound in the screen. This profile enables the prioritization of compounds for follow-up that possess the desired combination of biological attributes. However, while there is often a clear path to nominating excellent in vitro tools, their optimization into viable drug leads remains a formidable hurdle. Embracing this challenge, molecular property optimization of DEL products has become the cornerstone to our DEL design philosophy, and while not a new concept, its implementation has received little attention in the literature.
At first glance, it is an attractive thought to raid the corporate building block collection to create large combinatorial crosses for synthetic designs that employ three or four cycles of chemistry. Such approaches have been described to produce DEL collections of unprecedented size, with the largest report of 40 trillion discrete library members. 67 While a staggering scale, our assessment of designs that employed four cycles of chemistry indicated that much of the designed matter possessed an MW far above what is considered ideal for drug-likeness. Additionally, our early project team experience found that truncations of high-MW DEL hits lost activity and yielded poor SAR. We have developed new (to DEL) chemistries that feature carbon–carbon bond-forming transformations, targeting more drug-like space with a higher sp3 fraction.68–70 Incorporating this opportunity, our efforts have focused on library builds that employ chemistries that improve drug-like properties and limit the warhead MW by incorporating only two or three building blocks. Even so, library designs can easily exceed a billion compounds, a count that creates an incredible computational burden to enumerate. We have described a pragmatic solution to this problem, 74 where properties are determined for representative sublibraries and these subsets are used to guide design. Even with these approaches, the complete combinatorial cross created through split-and-pool synthesis requires further diligence to ensure that the finalized library faithfully represents the property space of the original design.
Reagent Validation
Critical to the success of any hit ID platform is the quality of the reagents used in the primary screening assay; in the context of DEL discovery, the validation of these materials is no exception. Of course, it must be appreciated that the effort expended to characterize protein reagents comes at the expense of swiftness, which is a desirable characteristic of DEL. While an application has been described that leverages the technology as an unbiased approach to prioritize targets for ligandability, 75 the philosophy does not extend well to targets that have a high CIR for their modulation. In these instances, the probability of clinical success takes priority, and our success as a hit ID platform is measured in the ability to deliver tractable starting points to these programs, regardless of the apparent doability.
In contrast to HTS, where usefulness of a reagent (e.g., highly specific ligand or substrate) can be greatly influenced by the design of an assay that will tolerate poor folded fractions or crude tissue extracts, affinity selection is driven by the concentration of the protein under study and thus places an absolute requirement on the material’s folded fraction. Anecdotally, it has been our experience that for enzymatic and receptor targets, a cavalier approach that includes limited validation risks the generation of unpredictive results, which at best constitute a low quantity of actionable ELT signals and at worst signals that are misleading due to the affinity selection of library components by the misfolded protein. In the former case, the ligandability of the target has not adequately been investigated, and in the latter, considerable resources are expended in the pursuit of false-positive signals.
With this in mind, we typically focus our reagent validation efforts to quantitatively assess the active site content of our protein reagents, both before and after immobilization on affinity resin, as this metric predominates any changes to apparent rate or signal size in a biochemical assay that may occur when enzymes are immobilized. Such changes may result from, for example, a loss of translational freedom in three-dimensional space or an incompatibility of detection reagents with salmon sperm DNA or salt concentrations used in the selection buffer. One such experiment with BRD4 is shown in Figure 1A , where a considerable reduction in a fluorescence polarization (FP) signal window was observed for the protein after immobilization. While the pharmacology appeared to reproduce for immobilized protein, we could not confidently conclude that the protein functionality was preserved for the immobilized reagent.
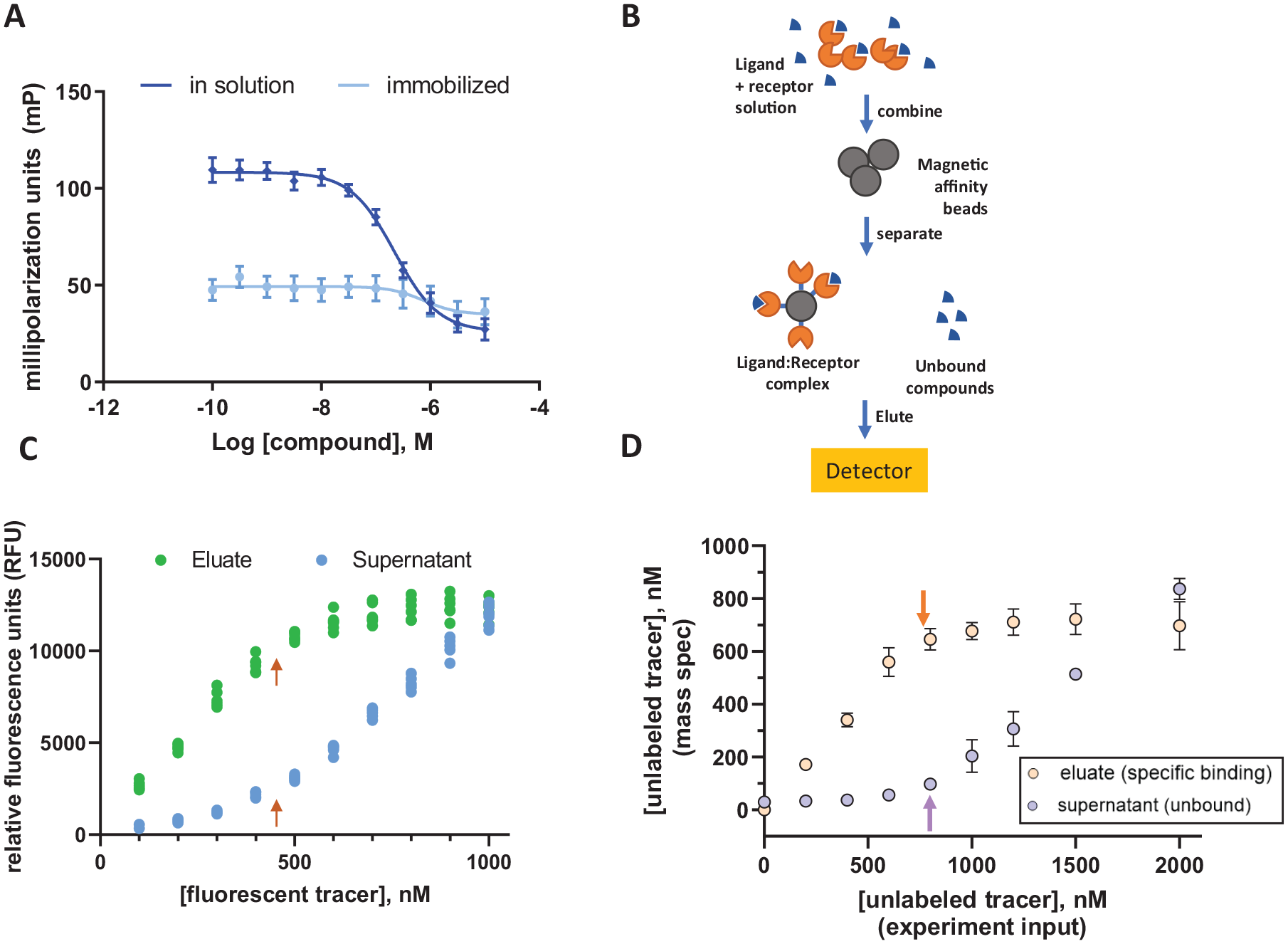
Overcoming ambiguity in target validation with BALI and active site titration. (
To rectify this, we developed an affinity selection platform that we refer to as bead-assisted ligand isolation (BALI), which, when appropriately configured, can allow for the quantitative assessment of active site content via the titer of an immobilized receptor sample. The design of the experiment, outlined in Figure 1B , is analogous to the DEL selection approach, whereby captured ligand is detected from the sample after a separation from the bulk solution using an affinity matrix. For low-affinity ligands, this method provides a qualitative assessment of ligand capture, but when the receptor concentration exceeds the ligand KD by a factor of approximately 10 or more, binding becomes stoichiometric and can be leveraged to quantitatively titer the active sites in the sample.
We conducted the BALI experiment with BRD4 employing the same fluorescent tracer used in the FP experiments such that the concentration of the captured tracer was determined by measuring the fluorescent intensity in a plate reader after elution from the beads. The results from the experiment are shown in Figure 1C as the fluorescent intensities of the supernatant (unbound) and eluate (bound) fractions of the binding reaction. In these data, a clear accumulation of tracer in the BRD4 eluate fraction was observed up to 500 nM, after which a further increase of the tracer dose did not lead to increased retention of the compound in the eluate. Similarly, analysis of the supernatant fraction demonstrated the concomitant absence of tracer in samples with a substoichiometric dose of the compound until the dose approached the equivalence point, after which further increases in tracer led to linear accumulation in this unbound fraction. Taken together, these data indicate the availability of a 500 nM equivalence of competent binding sites, which is in good agreement with the nominal value of 750 nM estimated from the reagent provider using the Bradford assay, a method that can possess considerable uncertainty when the test sample and reference do not have closely matched structures or relative amino acid composition.
We immediately recognized the elegance of this experimental design; the workflow was straightforward to execute in a microplate format without specialized liquid handling or washing procedures, and it eliminated weeks of laboratory work from the target validation process. We were curious to extend the method to other targets but were limited by the fact that each target would require a fluorescent probe, the design of which requires considerable a priori knowledge for fluorophore attachment, that is, known high-affinity ligands and a structural or SAR understanding of exit vectors. To address this, we sought to determine if MS could serve as a detection approach to characterize the capture of unlabeled ligands and eliminate the need to attach a fluorophore. We were delighted that we were able to repeat the experiment with excellent fidelity when we titrated BRD4 with unlabeled ligand and detected it via MS (
This MS detection-based technique has since become the foundation of our target validation procedure as it obviates assay development and uncertainty that arises from apparent fluctuations in activity. For targets that do not possess any chemical matter of sufficient affinity to enable these studies, qualitative assessments for weak-affinity ligands (KD up to 10 µM) have been sufficient to demonstrate the ability of immobilized proteins to enrich such ligands and thus indicate a high probability of a successful screen. In the case of targets without small-molecule ligands, similar studies of equivalency for the capture of protein or nucleic acid binding partners have also sufficed to indicate a reagent’s suitability for progression to a DEL selection campaign.
On the Interexperiment Reproducibility of DEL Selection
When transferring plate-based assays between laboratories, it is a commonly accepted practice to repeat an experiment at a new site that has been conducted previously at the site of origin to demonstrate an equivalence of results between the two locations. We used a similar approach for the transfer of DEL selection capabilities from our partner’s lab in China to Pfizer’s U.S. lab (Groton, CT), and we sought to repeat a screen whose results had translated to confirmed off-DNA ligands. Anecdotally speaking, one would expect the straightforward binding assay that underlies DEL selection to reproduce with a reasonable degree of fidelity, and this has been described, in a limited capacity, for a panel of intraexperimental replicates.
76
However, this report lacked any description of data that pertained to a confirmed hit, and in the absence of a cubic plot to understand the relative change of the experimental fingerprint in that study, we were not able to extract a meaningful understanding of repeatability. As such, we were unable to benchmark expectation of experimental reproducibility and, for a lack of better metric, used the rediscovery of the line feature that provided the confirmed off-DNA hit as an indicator for success (
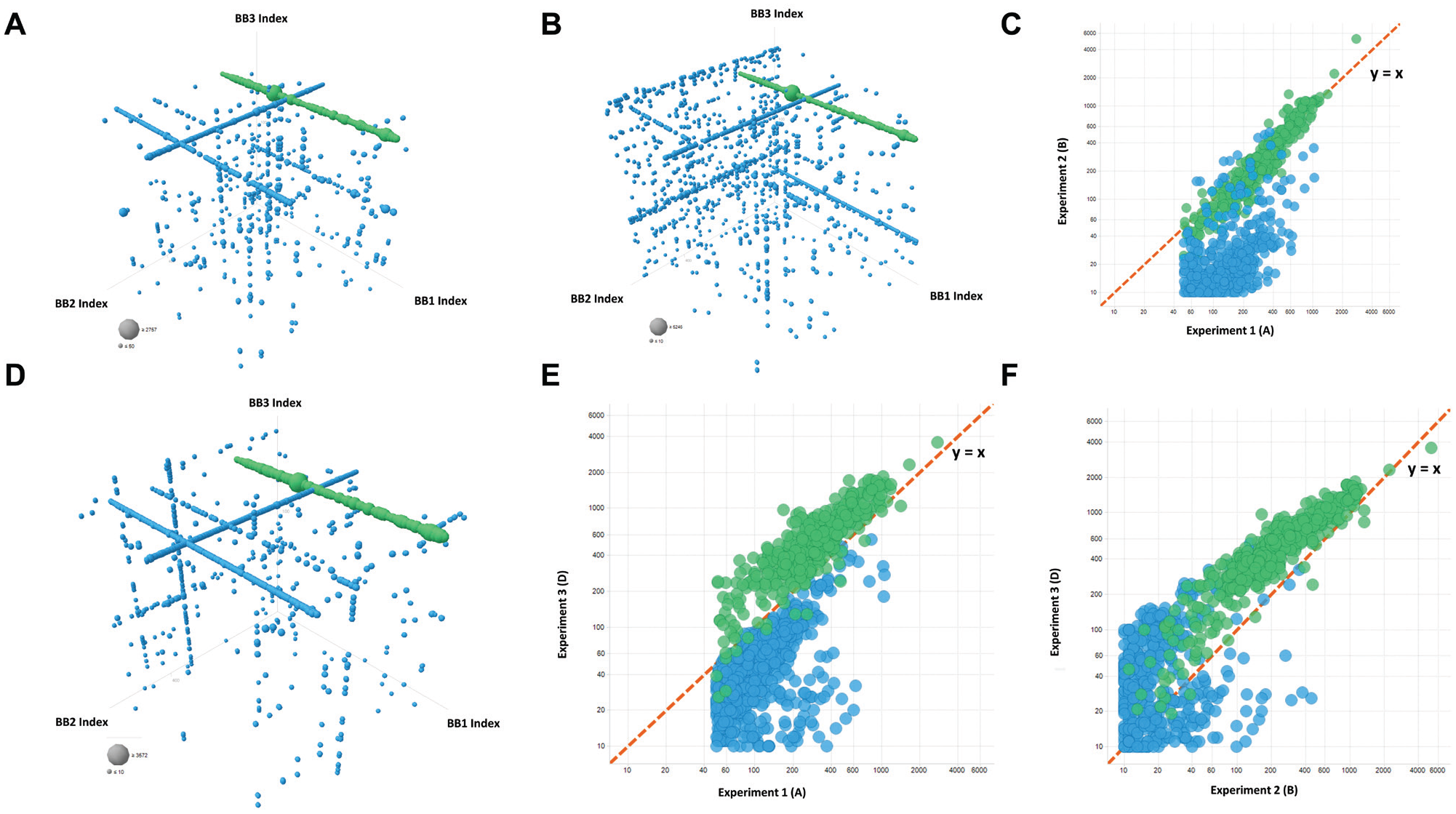
Assessing the interexperimental reproducibility of DEL selection results. To assess and transfer DEL selection capabilities between sites, we repeated a DEL selection that provided a strong line feature that had given rise to small molecules that were confirmed as ligands to the target off DNA. In
Following this successful replication by Pfizer scientists, we repeated the same selection experiment a third time in the United States, 8 weeks after returning from China. The cubic plot for this Groton experiment is shown in
Figure 2D
, again demonstrating the robust enrichment of our hit feature (green line). Pleasingly, comparison of this run to the previous iterations from Chengdu revealed an overall recapitulation of the experiment with well-preserved correlation for the confirmed hit feature, albeit deviation from the unity line due to increased signal within the Groton experiment (
On Intraexperimental Variability and the Comparison of Between-Sample DEL Selection Variability
In the most common form of industrialized ELT, it is not uncommon for a collection of DELs to total many billions of compounds. It has been demonstrated by others that the productivity of DEL selection can be enhanced by combining multiple DELs of different origins together, and the affinity selection of putative ligands occurs from a single sample of the library pool. Rather than a single affinity selection step, these hit discovery campaigns are conducted as an initial selection followed by iterative reselections using the previous cycle’s output as input for the next experiment; the campaign is terminated when the total compound count of the selection eluate (determined by PCR-based quantitation of DNA) is sufficiently small to allow for adequate sampling by NGS. 76 We sought to investigate the intersample but intraexperimental reproducibility of DEL selection. This study was built into our initial DEL selection experiment in Groton, where we conducted three parallel selections using the same treatment (i.e., DEL pool dose, sample volume, and target dose).
The three selection campaigns were each composed of an initial round of affinity selection (using an input of 1.5 × 1015 total copies of DNA-encoded molecules), followed by a single cycle of reselection. The selection outputs ranged from 2.0 to 3.7 × 109 total copies per sample. A quarter of each output sample was processed for sequencing, and the NGS run was allocated to each sample proportionally based on the total population ratios of the campaign eluates.
Given the interexperimental concordance we had observed in the previously described studies, we expected that the affinity selections would be reproducible between samples. Hence, we were intrigued by the nearly twofold intraexperimental variance between selection eluate populations of this study.
The relative portioning of the samples for NGS was conducted to balance the sequencing runs and generate approximately equal signal reads for hit compounds as rationalized in
Figure 3A
, where each selection eluate is represented as a circular diagram scaled relative to the total population size of the sample. In this model, we assume that the affinity selection of hits yields an approximately equal quantity of hit molecules for each sample, defined by the “true hits” inner circle, with varying amounts of background signal. The quarter portion processed for analysis is indicated by the darkly enhanced region. We consider the primary variable between samples to be the efficiency of washing during each round of selection that arises from a variable degree of compound carryover; this nonbinding fraction constitutes the background signal. If this is correct, then we expect that the magnitude of true hit read signals would be approximately equal between the three replicates. The outcome of this experiment for the individual DEL described in
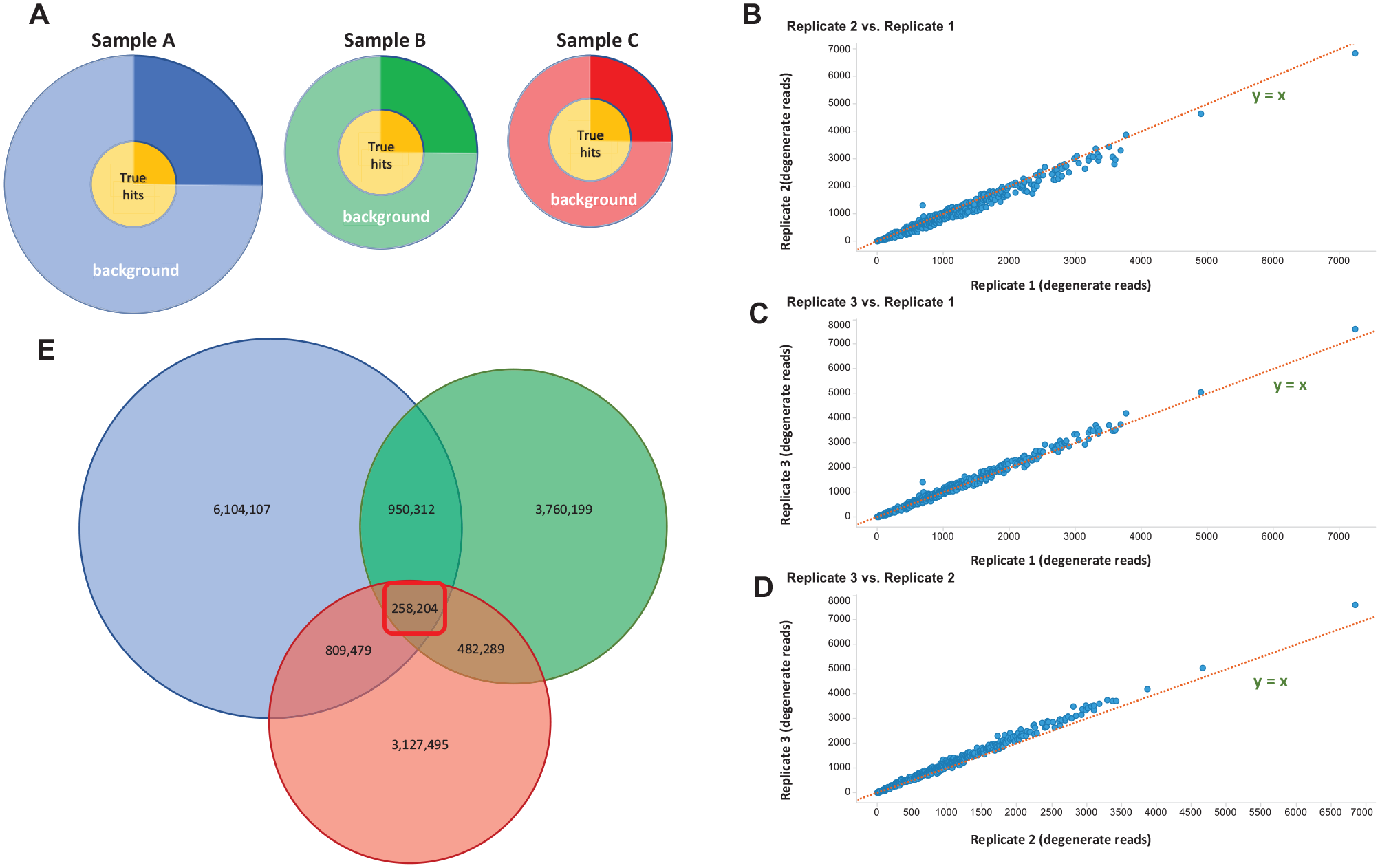
Intraexperimental repeats demonstrate a high degree of repeatability. Within the experiment that provided the data in Figure 2D, two additional intraexperimental repeat samples were included to probe the within-experiment but between-sample variability of DEL selection. (
Finally, concerning this experiment, the frequency of compound appearance from this individual DEL in each of these three samples is shown in Figure 3E . Here, we see the common compounds between all three samples totaling 258,204 compounds, and excitingly this number includes all 436 compounds observed in the data set of 440 total possible compounds in the hit line feature. Conversely, very large portions of the individual samples were observed only once. This indicates to us a high reproducibility of DEL selection for true hits, and for efforts seeking to mine the lower discovery threshold of the technique, the inclusion of experimental replicates may considerably help to bolster confidence in weak signals.
Incorporation of Drug-Likeness into the Hit-Calling Process
In the context of small-molecule drug discovery, there are many physicochemical properties of a compound, aside from its inherent affinity for the target, that constitute its credentials of being a good starting point for the development of a clinical candidate. Attributes such as MW, lipophilicity, the number of aromatic rings (sp3 fraction), polar surface area, permeability, and clearance are all critical determinants that contribute the overall disposition of a chemotype. The speed at which hits can be translated into viable assets that can be used to adequately test therapeutic hypotheses in animal models of disease has a strong dependence on the position of this starting point. Consequently, the decision to begin optimizing an individual series is multifaceted and therefore affinity alone is unable to accurately assess the value of each series found within a hit discovery campaign.
While screening DELs and proposing hits from the resultant data using signal amplitude alone as a metric for value had proven to be a straightforward exercise, the physicochemical properties of resultant hits prioritized in this manner for initial DEL projects often made them disadvantaged versus those arising from other hit ID technologies that sourced chemical matter from our historical chemical collection. To improve this and enable data-driven decision-making, we have integrated multiparameter optimization (MPO) scoring, a concept often used to guide the medicinal chemistry optimization of leads (thoroughly described in Wager et al. 79 ). In applying this to DEL, we use an MPO score heavily biased by MW that also includes three additional physical chemical properties (calculated partition coefficient [cLogP], aromatic ring count, and hydrogen bond donor count) as a visualization and sorting tool to highlight compounds that have the best balance of signal and favorable properties. This allowed us to select chemical series and individual compounds with the best balance of drug-like properties for hit confirmation activities.
An example of how this is applied is shown in
Figure 4
. Here, a line feature is shown for hits arising from a selection campaign with bromodomain 1 of the BRD4 protein. An MPO score is generated for the designed compound represented by the DNA tag to provide a drug-likeness metric where increasingly positive values correlate with better property values. In this analysis, the size of each data point in the cubic plot indicates the relative amplitude of signal for each compound, and color in a typical stoplight scheme from low (red) to high (green) is used to visualize the MPO score value. Taking in the entire feature (

Demonstrating the identification of hits with better physicochemical properties through the visualization and filtering of multiproperty optimization scores. Approaching a DEL data set, one’s initial intuition is to focus on compounds that generate the largest signal. While this can direct you to compounds that may possess the highest affinity or yield during library synthesis (be it the targeted product or side product), these compounds may not make the best starting points for development. We have implemented a strategy that colorizes the data points in a cubic plot based on an MPO score calculated from multiple physicochemical properties. (
Application of On-DNA Resynthesis and BALI-MS to Improve the Productivity of DEL Hit Confirmation
In this final section, we address our effort to resolve a poor conversion rate of NGS-identified hits to confirmed ligands off DNA, a result that further complicated the realization of value expected from the technology. An often underappreciated challenge in the application of DEL hit ID technology is the complexity of the chemistry that arises from the monomer diversity employed in the construction of industrial DEL collections. Even with high stringencies for productive rehearsal yields under library synthesis conditions, these preliminary tests with representative upstream intermediates do not capture the spectrum of reactivity that can be encountered during production of a DEL according to the split-and-pool paradigm. As such, the combination of incomplete yields at different steps as well as the propensities for side product formation and subsequent diversification in downstream reactions means that for any single given barcode, a multitude of chemical species can be present. Following that logic, the DNA-recorded synthesis of DELs is best thought of as a recipe for a mixture of compounds rather than a single, discrete chemical entity. 80 We refer to this combination of possible entities as the “hit tree.” Given this, it is quite possible that signal variability within a feature in a DEL selection data set could arise through affinity of the designed compound (a structure–activity result), or possibly the propensity to react through alternate pathways to produce high-affinity side products. In this latter case, variation in signal amplitude could relate to the designed library compound as a structure reactivity relationship and could be exemplified by cases such as a reduced reactivity of amines (primary > secondary), relative steric hinderance of reactive groups (e.g., α-monosubstituted vs α,α′-disubstituted carboxylic acids), electron donating versus withdrawing effects, and so forth.
With this in mind, we took keen note of an early description of the impact that on-DNA resynthesis had on the productivity of off-DNA hit confirmation
80
by removing an assumption of DEL warhead identity and, rather, critically assessing the complexity of compound mixtures actually made from the individual monomer combinations under the library synthesis conditions. We have now established a strategy for on-DNA hit resynthesis and testing shown in
Figure 5
. To demonstrate this workflow, we returned to the study with BRD4 and examined compounds in this line feature with a good balance of signal and properties (
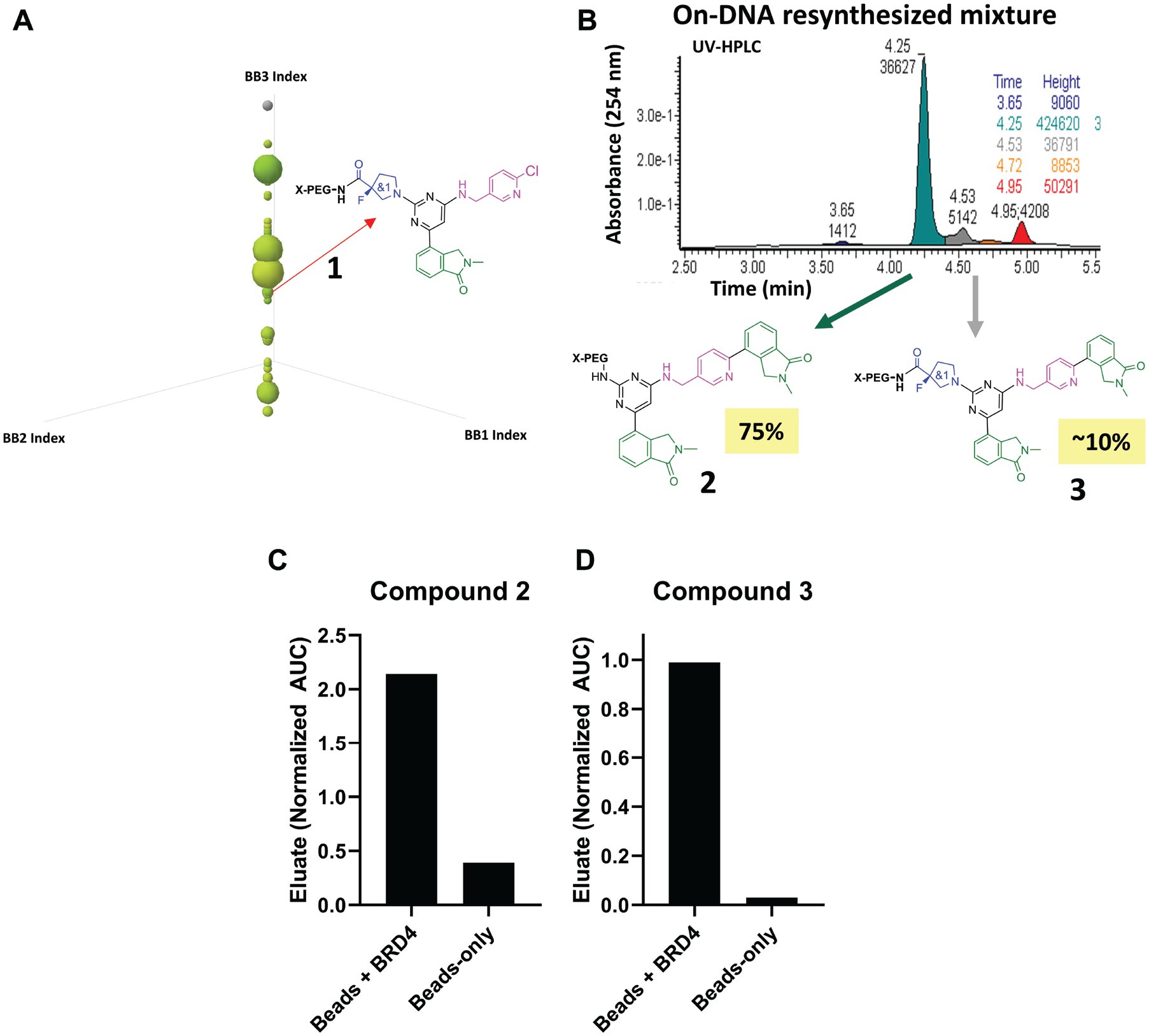
Demonstrating the value of on-DNA resynthesis and BALI-MS analysis to DEL hit confirmation. (
The UV-HPLC trace for the on-DNA resynthesis of
While telling with respect to which compounds could be attached to the DEL barcode and be present to bind to BRD4, the identity of the true ligand(s) responsible for the DEL selection campaign signal is not substantiated from resynthesis alone. In the original description,
80
cleavable linkers were employed during resynthesis as a means to liberate on-DNA synthesis products from the minimal hp-DNA handle, but the exact structure of the linkers used was not disclosed. During our initial investigation, we found the top candidates from established solid-phase synthesis approaches to be unsatisfactory for use because they either did not efficiently cleave or led to a multiplicity of products, the latter result being unsatisfactory as it further diversifies the product mixture. Acknowledging that our LC-MS/MS platform for analyzing on-DNA chemical products had the necessary sensitivity to quantify submicromolar concentrations of on-DNA products, we extended our BALI-MS experimental design to enable the identification of chemical structures within the on-DNA compound mixture that bind to the target when the compounds are interrogated with the same affinity selection conditions used in the DEL hit ID campaign. An example of the workflow that we established on this premise is shown in
Concluding Remarks
As hit ID technologies have evolved, affinity selection techniques including ASMS and DEL have become increasingly popular options for project teams due to the combined reduction of investment, the clarity of their results, and the speed with which they can interrogate large swaths of chemical space. Over the last 4 years, we have established a DEL hit ID platform that, through the incorporation of the above-described approaches, has begun to deliver value to our portfolio as a reliable source of actionable chemical matter. Through the assay transfer and repeatability studies detailed above, we became confident in the robustness of DEL selection as a binding assay, and we have learned to trust the data from multifaceted campaigns as a source of rich biological information that, when collected in an appropriate manner, can prioritize the most interesting compounds for initial follow-up. In our experience, under the circumstances of limited resources and an increasing pressure to deliver for first-in-class targets, not all hit ID approaches can be pursued in parallel for every project, and thus in this article we have outlined the aspects that we consider when designing a hit ID campaign. Ultimately, it is the most appropriate pairing of technology with project biology and chemistry that will efficiently deliver clinical assets that affect the lives of patients. This realization of positive impact on the experience of those suffering from disease drives us to continually evaluate and improve our hit ID toolbox in the hope that an enhanced ability to identify actionable chemical equity will deliver disease-modifying and curative medicines to those who need them in the not too distant future.
Footnotes
Acknowledgements
The authors wish to acknowledge Adam Gilbert, David Israel, Rob Stanton, Sylvie Sakata, and all members of the Pfizer/HitGen DEL team for their support.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: T.L.F., W.B., M.E.F., B.K., J.I.M., A.S.R., H.Z., M.-C.P. are employed by Pfizer Inc.; their research and authorship of this article were completed within the scope of their employment with Pfizer Inc. Q.C. and X.L. are employed by HitGen Inc.; and their research and authorship of this article were completed within the scope of their employment with HitGen Inc.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.