
Abstract
Available tools to analyze sequencing data coming from DNA-encoded chemical libraries (DELs) are often limited to in-house methods, which usually rely on strictly looking for the particular DEL structure used. Current methods do not take into account technological errors, such as library codification and sequencing errors, when detecting the sequences. The vast amount of data produced by next-generation sequencing of DEL screens is usually enough to extract the minimum information needed for compound identification. Here, we report a methodology to deconvolute encoding oligonucleotides, thus optimizing the sequencing power regardless of the library size, design complexity, or sequencing technology chosen. tagFinder is a highly flexible tool for fast tag detection and thorough DEL results characterization, which requires minimal hardware resources, scales linearly, and does not introduce any analytical error. The methodology can even deal with sequencing errors and PCR duplicates on single- or double-stranded DNA, enhancing the analytical detection and quantification of molecules and the informativeness of the entire process. Source code is available at https://github.com/jamigo/tagFinder.
Introduction
The interest of DNA-encoded chemical libraries (DELs) for the discovery of novel small molecules is increasing in both pharmaceutical companies and academia, particularly as a source of novel chemical matter. 1 The discovery of bioactive molecules directed to the treatment of diseases associated with targets in which the mechanism of action is not fully understood has always been a challenging field for the pharmaceutical industry and also constitutes a foundational key for many emerging biotech companies. Although classical methodologies in drug discovery have been the major source of compounds that reach clinical phases, the need for new approaches that may interrogate novel interesting biological targets in a rapid, efficient, and secure way is critical for success in future drug identification.
DNA-encoded libraries have emerged as a powerful technology capable of generating billions of compounds in a single library that may be uniquely identified by a DNA sequence associated with each member of the library, thus avoiding the uncertain deconvolution process required in many of the combinatorial approaches utilized in the past. 2 The application of DELs to the hit identification process has provided interesting new chemotypes that have led to the starting points for medicinal chemistry efforts and several clinical candidates.3 –5
DNA codification technology is not limited to the interrogation of difficult targets, and has been demonstrated as a useful tool to assess druggability, to set up multiplexed screenings of several targets in a single assay, and to evaluate the enzymatic activity.3,4,6
DEL technology is based on the creation of large encoded combinatorial libraries through different methodologies, such as pool-and-split chemical-encoded synthesis, DNA-templated or self-assembling library production. The screening is run through affinity selection methodology and allows the parallel testing of large pools of compounds and/or libraries versus several targets. This process requires minimum amounts of both target protein and libraries. The sequences of those compounds that bind to a protein during the affinity selection process are analyzed using PCR amplification and DNA sequencing. The readout gives the number of counts of the tags that match the sequences used to codify the library building blocks (BBs). Due to the specific requirements for DEL sequencing data described later in this article, each research group has developed its own in-house methods for this analysis. Although undocumented pieces of code to perform particular tasks can be found in the literature, 7 to our knowledge there is just one methodological description that covers the entire analysis from raw sequencing data. count 8 is a C code released as is that relies on the direct detection of a small set of fixed tag structures using single-stranded DNA only.
Massive parallel sequencing technologies generate millions of short sequences that are usually part of a much larger sequence. The typical type of analysis involves a series of quality control steps (e.g., sequencing error reduction, base calling, and trimming), followed by a process called aligning or mapping, depending on whether a reference sequence can be used as a template to place those short reads on. The intrinsic technological error of massive parallel sequencing is reported to vary from approximately 0.1% to 1% on the most common technologies available, 9 which implies that all reads of 100-base length could potentially contain one error per read. DEL sequences usually comprise several tens of bases, where the coding region represents a smaller number of these bases (approximately one-third of the total length); hence, the estimated number of reads of 100-base length containing coding errors could vary from 3% to 30%. The coding region is critical for compound identification, but there are other sequence sections that are relevant for the analysis, such as sample barcodes or library identifiers; therefore, the percentage of erroneous reads can be even higher. Although it has not been previously discussed in the literature, direct detection methods should be the fastest and most accurate alternatives for analyzing DEL results, but due to the sequencing error previously estimated, they could potentially miss an important amount of data, depending on the sequencing technology used and the quality of a particular run.
Due to the absence of literature favoring any particular method for DEL analysis, the validity of already known mapping and aligning methods, like BWA, 10 a fast and efficient Burrows–Wheeler transform–based newer aligner, and BLAST, 11 a thoroughly documented older aligner, was explored because these methods are extensively used in comparative biology and genomics to deal with the known existing sequencing error. This error must be considered in order to reduce as much as possible the false-negative rate, although this effort can unfortunately lead to an important increase of the false-positive rate, which can be even more important. These methods require a reference sequence containing all possible tag combinations, short reads need to be aligned against this reference, and the number of times each reference entry receives an unambiguous match is finally registered. As the input size increases, the entire process ends up being extremely slow and not scalable.
DEL screening is a mature technology used for lead and tool discovery. However, only one example of analytical tool for the identification of molecules coded by DNA has been documented. Here, we report tagFinder, an algorithm for the accurate decoding of DEL hits. This tool is adaptable and scalable to different library designs and sizes, including libraries with different numbers of synthesis cycles, different sequence lengths, single or double strands, and different combinations of codifying and quality control sequences. Therefore, this open-source algorithm could be easily implemented in the analysis workflow of any DEL platform.
Material and Methods
Algorithm Description
tagFinder is a Perl script to process raw sequences from any sequencing platform in the de facto standard FASTQ format. 12 If available, it will use the Seqtk library, 13 which is a fast and lightweight C interface for accessing sequences in the FASTQ format. It will read each read sequence and its corresponding quality string from the input while being aware of the base-calling quality of each sequence base at any time.
Although all information needed by tagFinder can be provided through command-line options, it is recommended to use a configuration file that describes each experiment to be analyzed. This configuration file allows a high degree of customization, as it does not require any fixed sequence structure. It can be used to describe the overhang sequences used to join the tags if any, which sequences were used to start and to end the ligation reactions, or the number of degenerated bases used in the design if applicable. The configuration file can also be used to include a reference to the entire tag library, and to describe any subset of expected (e.g., DNA tags used for library synthesis) or unexpected tags from that library.
Each sequence is then evaluated individually by looking, in both forward and reverse, for specific patterns that locate the compound tags while describing any data inconsistency present. First, too-short reads are filtered so that only potentially valid ones are evaluated. Then, the headpiece and the closing sequence are located, and the latter is stored to detect duplicate reads generated in the PCR step if a degenerated region was included in the experiment design. 14 Once these constant regions are located, the length of the tag section is evaluated. The read would be considered invalid otherwise. Finally, too-long tag sections will be marked for inspection of possible chimera blocks.
Tag sections of the appropriate length are sequentially chopped by each cycle’s lengths. These chopped tag sequences are then individually evaluated against each cycle’s lookup table ( Fig. 1 ). If all the chopped tag sequences are found in their respective lookup tables, the read would be considered a match. If available, the degenerated region on the closing sequence will be evaluated, providing a discrimination power of 4 to the number of bases in the degenerated region. The read will only be counted as a deduped match if the degenerated region sequence is unique; it would be considered a PCR duplicate otherwise.
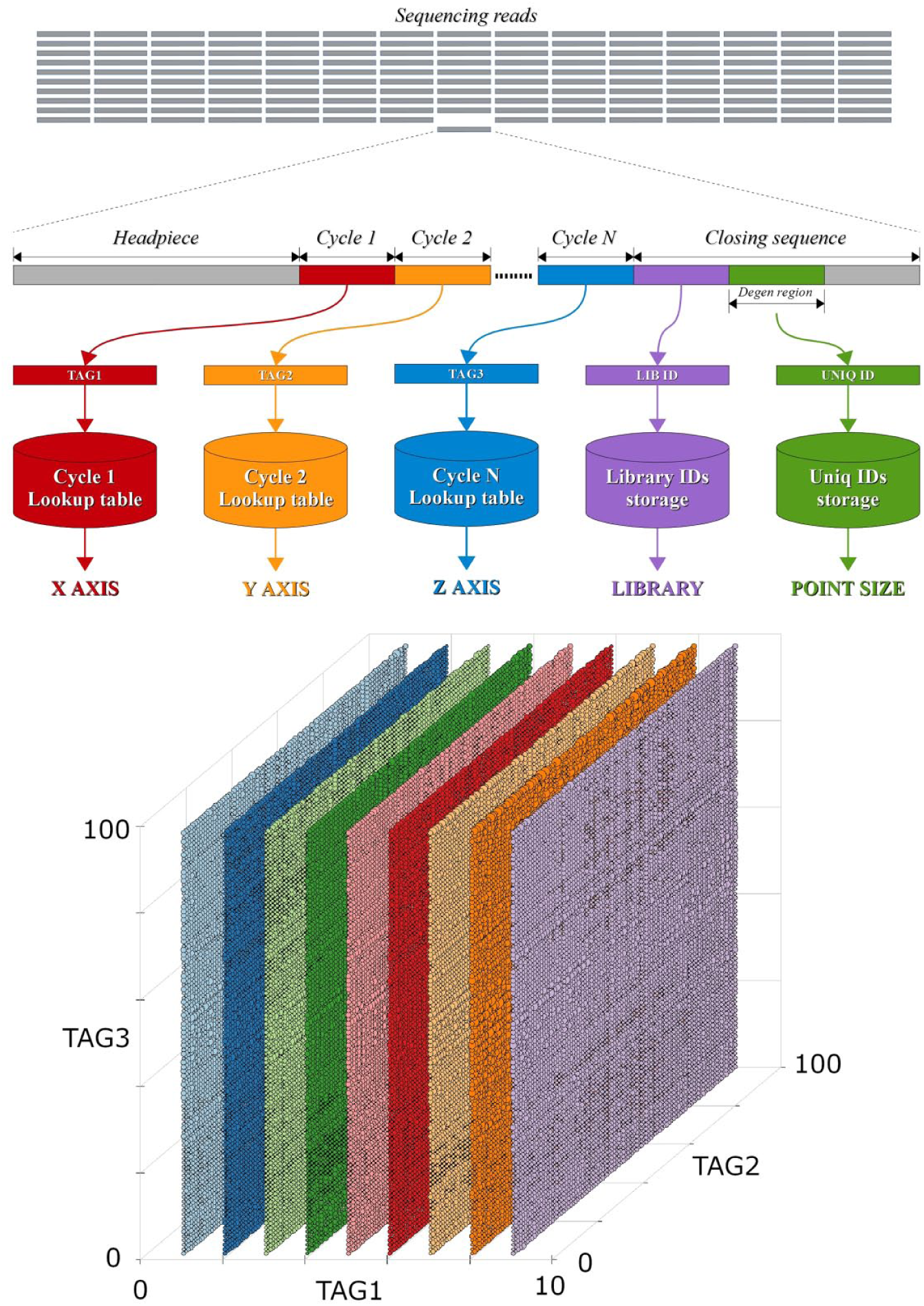
tagFinder algorithm scheme for the identification of DNA-tagged molecules by lookup tables. Representative scheme of the sequencing reads’ data analysis by sequential location of specific patterns and evaluation of the coding regions against each respective cycle’s lookup table. The multidimensional detection and relative quantification of patterns can be visualized in 3D scatter plot of a library containing 86K compounds.
While all the tags are being identified, the number of times each tag appears alone, or in combination with any other, is recorded for data analysis by aggregation. 15 When the identification process finishes, all these counts are compared using averages and standard deviations, providing a method for ranking individual tags and combinations of tags by the number of deviations from the average. This multidimensional pattern detection method allows presenting monosynthons, disynthons, and trisynthons 16 in a 3D plot as planes, lines, or singletons, respectively, and can be of great aid when the false-negative rate increases due to the size of the library. 7
Once all sequences have been read and analyzed, they are written in a tabulated text file containing each tag found, the number of raw counts, the number of deduped counts, their normalization by the number of library members, and the number of deviations from the average (overrepresented dimensions) so that monosynthons, disynthons, and trisynthons can be easily highlighted with a simple column filter. This tabulated file is read by an R script that assigns each cycle to an axis so that each tag combination is a point, and the number of times that particular combination was found determines the point size. Available plots can be histograms for single-cycle display, scatter plots for two-cycle display, and 3D scatter plots for three-cycle display. Displaying tag designs of greater numbers of cycles requires the selection of one, two, or three cycles to be evaluated at a time.
Affinity Selections
Avi-tagged proteins were used at 1 μM concentration in selection buffer and immobilized on streptavidin beads. The beads were then incubated with 5 nmol of a single DNA-encoded library. The total amount of the beads was washed to remove unbound molecules, and those that remain bound to the protein were eluted by heat denaturation. Three rounds of affinity selection were made to enrich the elution with the encoded molecules that potentially bind to the target.
Sequencing
Sequencing libraries were prepared following the Fusion Method for Ion Amplicon Library protocol. Quality control was done by Agilent 2200 TapeStation. The Ion Chef System was used for template preparation, and the run was done in the Ion Torrent semiconductor system. Both the Ion PGM and Ion Proton platforms were used to obtain lower and higher sequencing yields, respectively.
Benchmarking
tagFinder’s performance was measured on two different double-stranded DNA experiments: the first one including a library of 86,436 compounds (86K) with a 5 N degenerated region and enough sequencing power to detect them all, and the second one including a library of 6,156,288 (6M) compounds with a 9 N degenerated region. In order to include count in the comparison, all input sequences starting with the closing sequence’s reverse complement were reverse complemented in advance, because it is designed to deal with single-stranded DNA only ( Fig. 2 ).
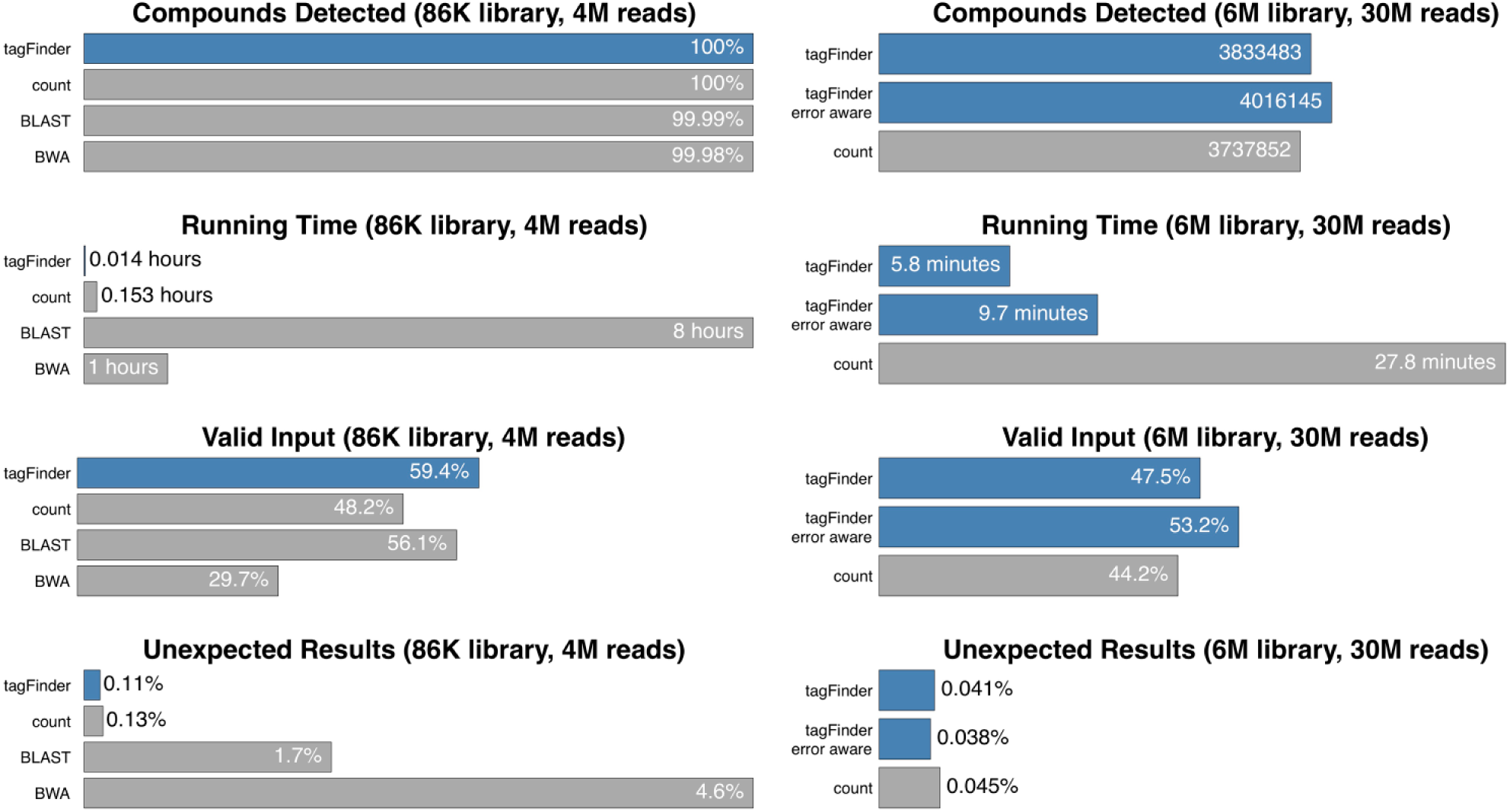
Benchmarking of DNA-tag decoding methods. Summary of the performance of available alignment methods (BWA and BLAST) and count algorithm vs tagFinder, which outperforms them all by not only providing a higher detection power and a lower error rate, but also showing lower running times and being capable of processing more input data.
Regarding quality, in the first experiment (86K-compound library) direct methods detected 100% of the expected compounds with a rate of unexpected results of 0.1%, while alignment methods detected 99.99% of the expected compounds, although with higher unexpected rates. Regarding quantity, tagFinder was able to use almost 60% of the input data for the analysis, which is 10% more than count did and 3% more than alignment methods did, even if they are designed to recover as much data as possible.
Apart from a default exact detection method, an error-aware running mode has been included in tagFinder to deal with sequence mismatches in order to overcome the basal sequencing error. On a larger library comparison (6M compounds), tagFinder, and this error-aware mode in particular, was confirmed as the most accurate detection method described to date, being able to detect 4,016,145 expected compounds (number of DNA tags used for library synthesis) while lowering the false-positive rate to 0.038% (5751 unexpected counts in 15 million total counts). tagFinder’s default mode is able to process 3.3% more reads and detect 2.5% more compounds than count while being five times faster, and its error-aware mode is able to process 9% more reads and detect 7.4% more compounds than count while still being three times faster. Furthermore, tagFinder needed 413 MB to store all results, while count needed 4.5 GB, representing a 10 times reduction in storage requirements.
Results and Discussion
tagFinder Is an Efficient Decoding Tool for the Identification and Quantification of DNA-Tagged Molecules
Universal methods to detect and quantify molecules coded with DNA from sequences generated by DEL screens are needed, so there was room for the development of a new direct method for tag detection to fulfill the specific requirements of sequencing data from any DEL screen. Its blueprint does not introduce any analytical error, performs fast, has minimal hardware requirements, allows linear scalability in terms of length and number of sequences, and generates negligible error rates. It is sequencing platform independent and able to deal with raw unprocessed sequencing results, without the need of preprocessing them in any way, directly in untreated FASTQ files. Other FASTQ processing tools in the field, like count or FASTAptamer, 17 try to simplify the process by not reading the quality string and reading sequences only, but the base-calling qualities can be used to create a minimum base quality threshold that would improve the entire analysis confidence.
tagFinder defines each experiment with a flexible configuration file containing specific information regarding the structure of the entire sequence of the library and the individual compounds. Individual reads are then sequentially evaluated by looking for those specific patterns, filtering for short reads, locating the constant regions (e.g., headpiece and closing sequence), and filtering for long reads. Tag sections of the appropriate length are then individually evaluated against each respective cycle’s lookup table, and if found would be considered a match (
Analysis of All Components and Identification of Streptavidin Binders from a 6-Million-Member DEL
The performance of tagFinder was tested by analyzing a library containing 167 BBs in cycle 1, 192 in cycle 2, and 192 in cycle 3, identified by DEL-B tags, 14 for a total of 6K theoretical tag combinations ( Fig. 3A ). Our double-stranded library was built by using the pool-and-split methodology, which allows full combinatorial mixing of all BBs at each cycle. Purification by liquid chromatography–mass spectrometry in the pools during library production and quality control by quadrupole time-of-flight analysis was performed after each ligation step and each single chemical reaction. A total of 4,016,145 unique molecules were detected by sequencing our library with the Ion PI chip, which resulted in 65% of the theoretical tag combinations after the decoding. Partial identification of compounds was expected because DEL composition is highly dependent on the efficiency of chemical reactions, which ultimately determines the abundance of synthesized molecules in the library, and because it depends on the sequencing power given by the maximum number of reads available (30 million in our sequencing run).
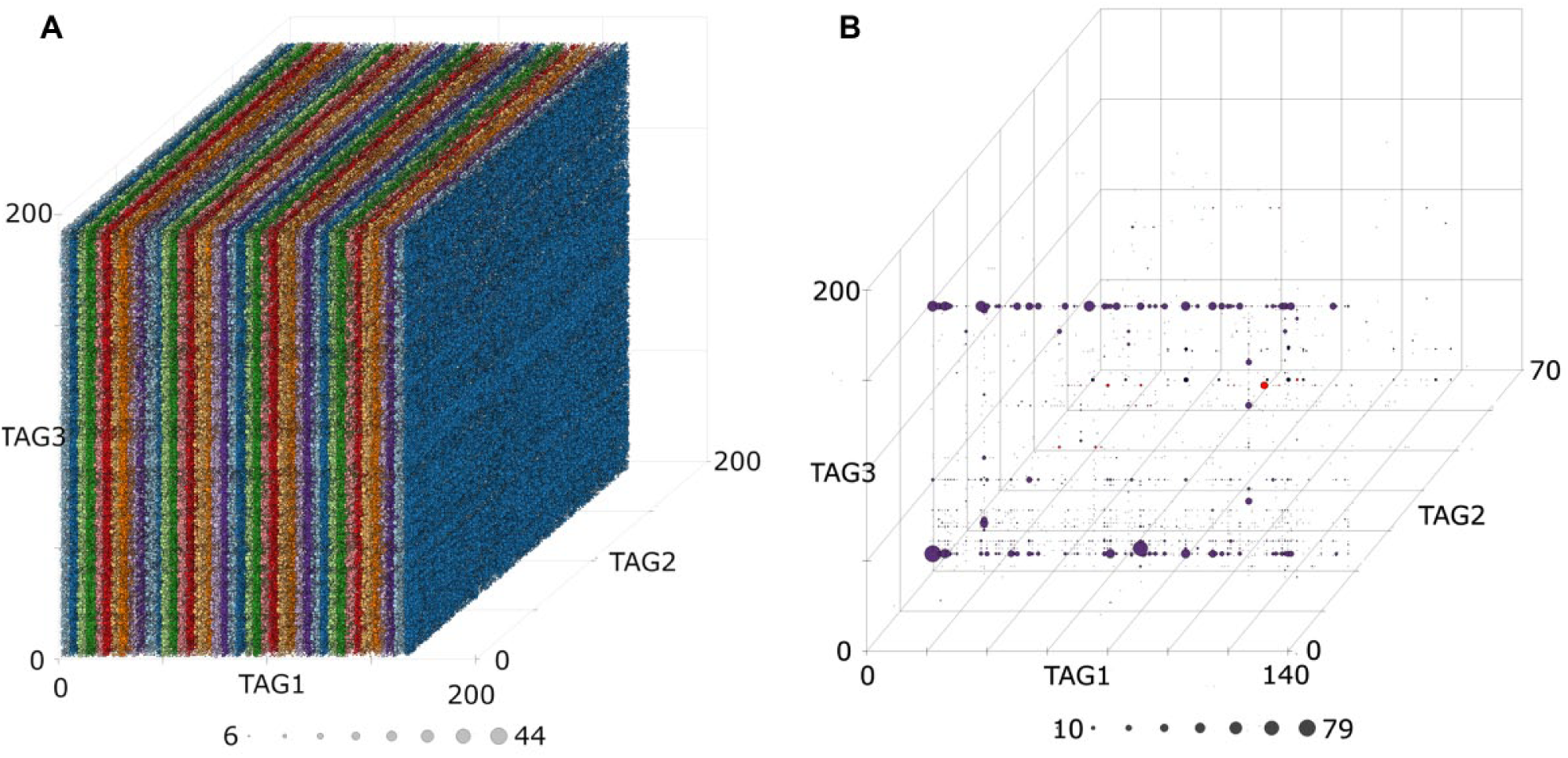
Analysis of all components and identification of streptavidin binders from a 6-million-member DEL. (
An affinity selection experiment of this library against streptavidin yielded some BBs and a series of chemotypes that remain bound, detecting 10,294 different compounds. Our novel analysis strategy reveals efficient identification of sequences, while highlighting an interesting plane and several linear trends, which challenges the performance of existing methods for the analysis of DELs ( Fig. 3B ).
Identification of Hits from Multiplexed DEL Affinity Selection Screens
The use of complex DEL inputs, such as large pools of libraries containing different chemical structures at different proportions, can lead to errors when analyzing any screening dataset. A series of affinity selection experiments against a number of pharmacologically relevant proteins, including epigenetic domains, protein–protein interactions, and transcription factors, were performed. Using a pool of libraries varying in BB chemical structures and size, and each one codified with closing sequences as unique identifiers, accurate identification of binding patterns, such as planes, lines, and singletons, is achieved ( Fig. 4 ). Some examples of the identified hits from multiple targets were synthesized off DNA, in the same manner as in Clark et al., 14 and subjected to further biochemical analysis to confirm their activity. Consistent with the DEL screen data, a significant number of compounds were found to bind the target proteins by biophysical assays and activity was confirmed by biochemical assays (data not shown). These data illustrate the versatility of our strategy to analyze multiplexed DEL affinity selection screens efficiently.
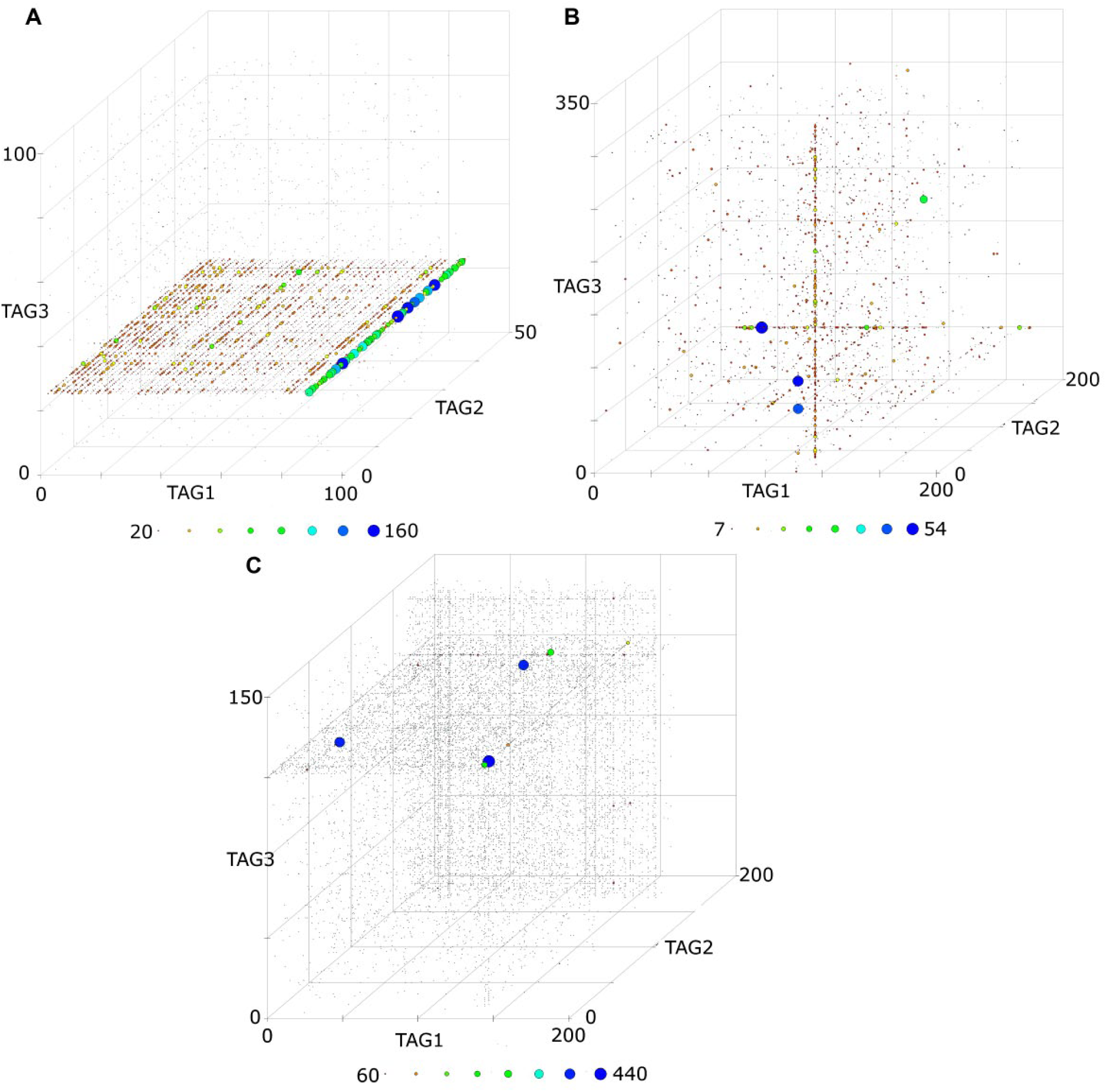
Identification of hits from multiplexed DEL screens. Representative examples of 3D displays of planes, lines, and singletons detected from an affinity selection of a mixture of 11 libraries. The figure shows the patterns identified with three different libraries with 1.2 million compounds (
Comparison of DNA-Tag Decoding Methods
Alignment tools are not preferred to analyze DEL data due to their specific limitations. The alignment process adds a small yet noticeable error to the already known sequencing error, and the scalability with library sizes is exponentially unaffordable in terms of the computing resources required. Although they can still be used with small libraries sequenced on faster but less accurate technologies, they are currently not indicated as the sequencing accuracy continues to improve in time.
Direct detection methods have proven to perform much faster than alignment-based tools. Their main drawback is that anything not fitting the design definition is discarded, although the vast amount of reads provided by massive parallel sequencing technologies plus the increasing sequencing accuracy favor these methods. In order to minimize this issue, tagFinder not only detects expected compounds, but also improves the whole process by inspecting those discarded reads. Therefore, it is able to determine whether a read is discarded because of its length (too short or too long), its low sequencing quality, or the constant regions, such as the headpiece and closing sequence, not being appropriately read. Additionally, it is also able to detect tag chimeras, which is a feature that can be of great aid when creating and testing new tagging designs. Tag chimeras may appear while chemically building a library due to unexpected DNA-tag binding, but they can be detected by inspecting tag regions looking for tags not necessarily in their expected design position.
A thorough description of a methodology for the efficient identification of DNA tags independent of the sequencing technology chosen has been provided. It is more accurate than previously described methods requiring less computing resources by showing an important decrease of existing running times while reducing the overall error. Its high degree of customization allows the identification of libraries with a different number of codification cycles, different sequencing lengths, and the discrimination of degenerated regions to identify the occurrence of unique sequences based on the specific combination of codifying and degenerated sequences. Therefore, this algorithm could be easily implemented in the analysis workflow of any DEL platform for the fast, efficient, and accurate detection of codifying DNA tags.
Footnotes
Acknowledgements
We want to thank the entire team working on DNA-encoded libraries within Eli Lilly for all the work and data provided to achieve this publication, and the management for all the support. We also want to thank Grant Vaught for his critical review of the paper and all the comments to improve the quality and overall understanding.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.