
Abstract
Allosteric modulators of G protein–coupled receptors have the potential to achieve greater receptor subtype selectivity compared with ligands targeting the orthosteric site of this receptor family. However, the high attrition rate in GPCR drug discovery programs has highlighted the need to better characterize lead compounds in terms of their allosteric action, as well as the signals they elicit. Recently, the use of label-free technologies has been proposed as an approach to overcome some limitations of endpoint-based assays and detect global changes in the ligand-stimulated cell. In this study, we assessed the ability of an impedance-based label-free technology, xCELLigence, to detect allosteric modulation in a neuronal cell line natively expressing rodent M4 muscarinic acetylcholine receptors. We were able to demonstrate that positive allosteric modulation of the endogenous M4 muscarinic acetylcholine receptor can be detected using this technology. Importantly, the allosteric parameters estimated from the label-free approach are comparable to those estimated from endpoint-based assays.
Keywords
Introduction
G protein–coupled receptors (GPCRs) are the largest family of mammalian cell-surface receptors. These receptors are important in the function of all organ systems, with over 30% of marketed drugs interacting with GPCRs to exert their therapeutic effect. 1 However, GPCR drug discovery efforts still suffer from a very high attrition rate, suggesting that much remains to be learned about the complexity of GPCR signaling.
Traditionally, drug discovery at GPCRs has been based on screening compounds using endpoint-based assays, where compounds are defined by their ability to alter a distinct signaling event in a cell. However, the realization that GPCRs adopt different active conformations that recruit or disrupt different signaling components has highlighted the caveats of such approaches as only one signaling event can be measured per assay and often at only one time point.2,3 In addition, many endpoint-based assays require cell lines to express reporter proteins or to overexpress the receptor of interest, and therefore, such manipulations can alter the physiological relevance of a cellular environment.
Recently, the use of label-free technologies has been proposed as an approach to overcome these limitations and to detect real-time global changes in the ligand-stimulated cell.4,5 Label-free technologies have been used in the study of GPCRs, particularly receptor de-orphanization programs, as an unbiased approach that does not discriminate between specific signaling events in a cell.4,5 Moreover, although these approaches require prior knowledge of receptor expression, they can be of use when the G protein coupling preferences of the receptor are unknown. Currently, the two main label-free technologies used for receptor-mediated signaling assays are impedance based and optical based. Impedance-based technologies, such as xCELLigence (Roche Diagnostics GmbH, Mannheim, Germany, and ACEA Biosciences, San Diego, CA) 6 and CellKey (MDS Analytical Technologies, Sunnyvale, CA), 7 use a microelectrode array to measure the changes in impedance of the electrical current applied to a cell layer. On the other hand, optical-based technologies, such as EPIC (Corning, Inc, Corning, NY). 8 and BIND (SRU Biosystems, Woburn, MA), 9 measure the change in refraction index as a function of mass redistribution of the cell layer. Both technologies are sensitive to changes in cell morphology, adhesion, and cytoskeleton reorganization.4,5
Most current GPCR-based therapeutics target the orthosteric binding site of these receptors—namely, the site where the endogenous ligands bind. 10 Therefore, a major limitation of these drugs is the occurrence of side effects that can be caused by the drug acting on other subtypes of the target GPCR due to sequence conservation at the orthosteric site. This has led to a focus on the development of allosteric modulators as potentially more selective therapeutic ligands.11–13 Allosteric modulators bind to GPCRs at a site that is topographically distinct from the orthosteric binding site and is frequently less conserved. 14 Allosteric modulators can alter the affinity and/or efficacy of the orthosteric ligand and can sometimes have efficacy in the absence of the orthosteric ligand. Due to these properties, there is potential for allosteric modulators to be not only more subtype selective than current orthosteric drugs but also to maintain the spatiotemporal control of receptor activity. This type of treatment can be beneficial in a number of disease states, particularly ones where current treatments based on orthosteric targeting of GPCRs are suboptimal, such as schizophrenia. 15
Antipsychotics currently on the market for the treatment of schizophrenia are associated with a range of side effects, including movement, cardiovascular, and metabolic adverse effects. 16 Consequently, there have been intensive efforts toward developing novel therapeutics for schizophrenia that have high receptor subtype selectivity and lower occurrence of side effects. The muscarinic acetylcholine receptor (mAChR) subtypes 1, 4, and 5 have all been implicated in schizophrenia, 15 and selective M4 mAChR agonism, in particular, has antipsychotic effects. 17 Xanomeline, an M1/M4 mAChR-preferring orthosteric agonist, significantly improved both positive and negative symptoms in people with schizophrenia compared with a placebo-treated group in a pilot study. 18 However, the presence of gastrointestinal-related adverse effects mediated by xanomeline acting on peripheral mAChRs prevented it from undergoing further clinical development. This paved way for the development of M4 mAChR-positive allosteric modulators (PAMs), with LY2033298 ( Fig. 1A ) being the first to show efficacy in vivo.19,20 In cellular assays, LY2033298 has functional selectivity for the M4 mAChR over other mAChR subtypes in the presence of the endogenous orthosteric agonist, acetylcholine (ACh). 19
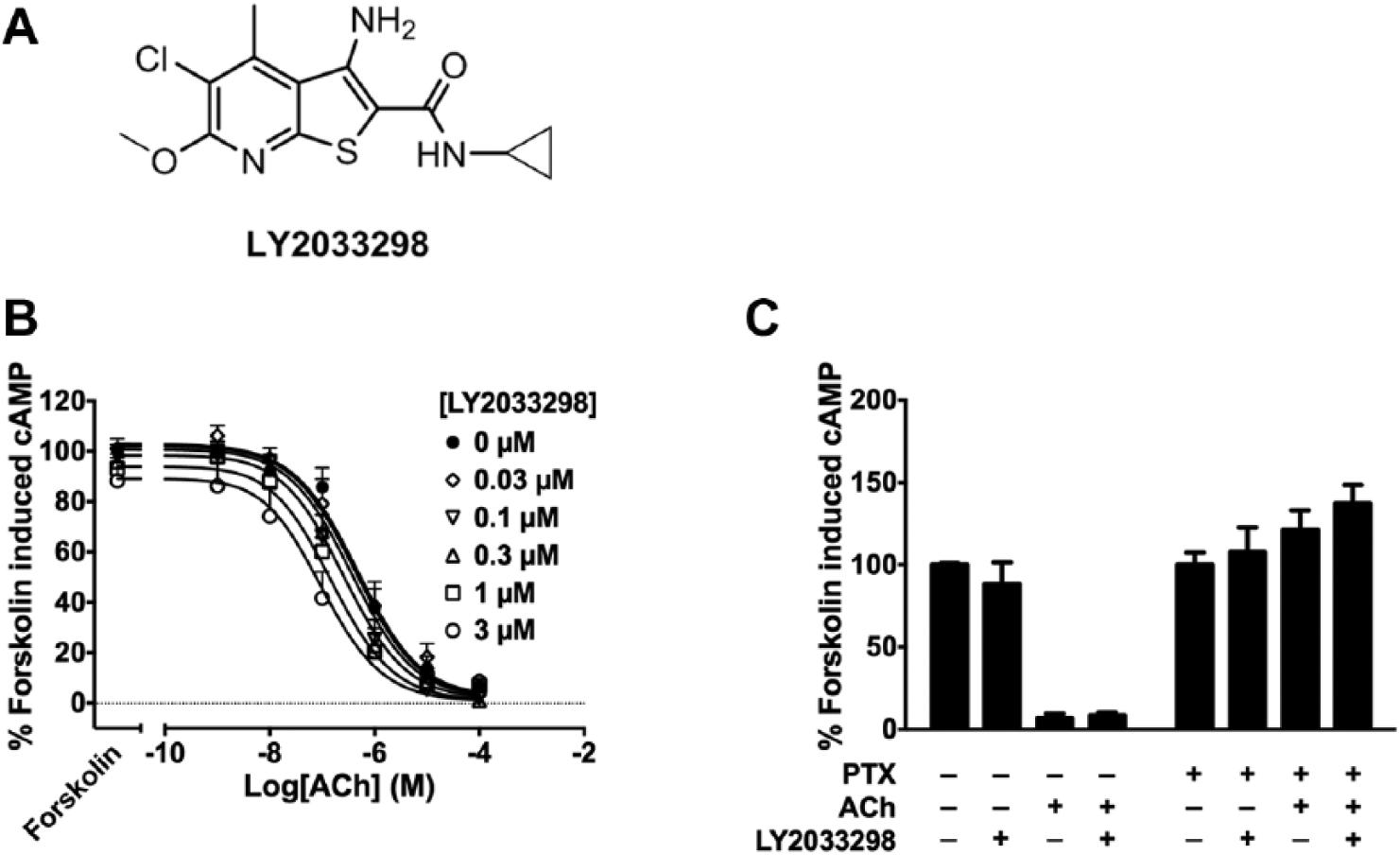
LY2033298 potentiates inhibition of forskolin-induced cAMP by acetylcholine (ACh). (
As label-free technologies detect global cellular changes upon receptor activation, they are ideal systems to evaluate and/or screen for allosteric modulation of GPCRs in native systems endogenously expressing the receptor of interest. Indeed, in the past few years, potentiation of orthosteric ligands by allosteric modulators has been measured using both impedance-based 21 and optical-based 22 technologies. However, these experiments were performed in recombinant cells where the receptors of interest have been heterologously expressed. To date, no study has investigated whether these approaches are sensitive enough to detect and quantify allosteric modulation of endogenously expressed GPCRs.
In the present study, we assessed the ability of an impedance-based technology, xCELLigence, to detect allosteric modulation in a neuronal cell line that natively expresses rodent M4 mAChRs. We demonstrate that the positive allosteric modulation of agonist-induced impedance at the endogenous M4 mAChR could be detected and quantified with this technology. Importantly, the allosteric parameters estimated from the xCELLigence data are comparable to those estimated from endpoint-based assays.
Materials and Methods
Materials
LY2033298 was prepared as a 10-mM stock in DMSO distributed in 100-µL aliquots and stored at −20 °C. Prior to its use, LY2033298 was diluted in the corresponding buffer for each assay.
Cell Culture
NG108-15 cells (American Type Culture Collection, Manassas, VA) were grown and maintained in high glucose Dulbecco’s modified Eagle’s medium without sodium pyruvate supplemented with 10% fetal bovine serum and sodium hypoxanthine, aminopterin, and thymidine (HAT) supplement (Life Technologies, Mulgrave, VIC, Australia). Cells were maintained at 37 °C in a humidified incubator containing 5% CO2.
NG108-15 Membrane Preparation
When cells were approximately 90% confluent, they were detached using 2 mM EDTA in phosphate-buffered saline (150 mM NaCl, 16 mM Na2HPO4, 4 mM NaH2PO4) and centrifuged (300 × g, 4 °C, 10 min). The resulting pellets were resuspended in 30 mL of ice-cold buffer containing 20 mM HEPES and 10 mM EDTA at pH 7.4. The cell suspension was homogenized using a Polytron homogenizer (PT 1200 CL; Kinematica, Basel, Switzerland), with three 10-s bursts and 30-s periods of cooling on ice between each burst. The cell homogenate was centrifuged (300 × g, 4 °C, 10 min), and the supernatant was transferred to new tubes and recentrifuged (30,000 × g, 4 °C, 1 h) in a Sorval centrifuge. The pellet was resuspended in 5 mL of buffer (20 mM HEPES and 0.1 mM EDTA, pH 7.4) and briefly homogenized to ensure uniform consistency. The cell homogenate was then separated into 250-µL aliquots and stored at −80 °C. The protein concentration was determined by the method of Bradford.
[3H]N-Methylscopolamine Binding Assay
Saturation binding assay was performed by incubating 5 µg of NG108-15 membrane with increasing concentrations of [3H]N-methylscopolamine (NMS) (85.5 Ci/mmol; PerkinElmer, Waltham, MA) in HEPES binding buffer (20 mM HEPES, 100 mM NaCl, and 10 mM MgCl2, pH 7.4) for 1 h at 37 °C. Binding was terminated by fast-flow filtration onto GF/B-grade filter paper (Whatman, Maidstone, UK) using a Brandel harvester, followed by three washes with ice-cold 0.9% NaCl. Nonspecific binding was defined in the presence of 10 µM atropine, and bound radioactivity was measured in a Tri-Carb 2900TR liquid scintillation counter (PerkinElmer).
cAMP Biosensor Assay
cAMP levels were measured using the bioluminescence resonance energy transfer (BRET) sensor, cAMP sensor using YFP-Epac-RLuc (CAMYEL; American Type Culture Collection). NG108-15 cells were seeded at 2,000,000 per 10-cm culture dish in culture medium and grown overnight. The cells were transfected with 2 µg CAMYEL using polyethylenimine. Cells were seeded into poly-d-lysine (Sigma-Aldrich, Castle Hill, NSW, Australia)–coated 96-well Cultureplates (PerkinElmer) 24 h posttransfection and assayed at 48 h posttransfection. For pertussis toxin (PTX) experiments, cells were treated with PTX 25 ng/mL for 24 h before assaying. Prior to the start of the assay, cells were allowed to equilibrate in Hanks’ balanced salt solution (HBSS) at 37 °C. Under low light conditions, coelenterazine h was added at a final concentration of 5 µM 15 min prior to BRET detection. ACh and LY2033298 were added simultaneously 5 min after coelenterazine h. Forskolin was then added at a final concentration of 0.1 µM after a further 5 min. BRET readings were captured with a LUMIstar Omega instrument (BMG LabTech, Offenburg, Germany) that allows for sequential integration of the signals detected at 475 ± 30 and 535 ± 30 nm, using filters with the appropriate band pass.
ERK1/2 Phosphorylation Assay
ERK1/2 phosphorylation was measured using the Alpha-Screen SureFire phospho-ERK kit (TGR Biosciences, Adelaide, SA, Australia). NG108-15 cells were seeded at 30,000 cells per well into a transparent 96-well plate coated with poly-d-lysine and grown overnight in culture medium with 2.5% fetal bovine serum (FBS). The next day, culture medium was aspirated, and the cells were rinsed with phosphate-buffered saline and incubated for 4 h in culture medium with 0.5% FBS before assaying. For PTX experiments, cells were treated with PTX 25 ng/mL for 20 h before assaying. ERK1/2 phosphorylation time course experiments were initially performed at least twice to determine the time at which the ligands were able to elicit the maximum ERK1/2 phosphorylation response (7.5 min for both ACh and LY2033298). Functional interaction experiments were performed at 37 °C with simultaneous addition of increasing concentrations of ACh in the absence or presence of increasing concentrations of LY2033298. ACh 10 µM was used as a positive control. Ligand stimulation was terminated by removal of medium, and cells were lysed by addition of cold 100 µL SureFire lysis buffer (PerkinElmer) to each well. Lysates were shaken in plates for 5 min at room temperature (RT) prior to transferring 5 µL lysate to a white 384-well Proxiplate (PerkinElmer). Under low light conditions, 8 µL of a 240:1440:7:7 mixture of SureFire activation buffer/SureFire reaction buffer/AlphaScreen acceptor beads/AlphaScreen donor beads was added to each sample well. Plates were incubated in the dark at 37 °C for 1 h and read with a Fusion-α plate reader (PerkinElmer) using standard AlphaScreen settings.
Cellular Impedance Assay
The label-free technology, xCELLigence Real-Time Cell Analyzer (RTCA) single-plate (SP) instrument (Roche Diagnostics GmbH and ACEA Biosciences), was used to measure changes in cellular impedance over time, which was defined as the cell index variable. Prior to the start of the assay, background cell index readings of each well of the 96-well E-plate (Roche Applied Science and ACEA Biosciences) were taken with culture medium in the absence of cells. This background reading was subtracted from all subsequent cell index values measured after addition of cells. NG108-15 cells were then seeded at 30,000 cells per well in culture medium with 2.5% FBS and grown for 16 h at 37 °C. Medium was then manually aspirated and replaced with culture medium with 0.5% FBS. The E-plate was inserted into the RTCA SP device station for both the cells and the system to equilibrate for 4 h at 37 °C. Immediately after simultaneous additions of increasing concentrations of ACh in the absence or presence of LY2033298, cell index values were obtained at 15-s intervals for 90 min. Data were baseline corrected with cell index values from the time point immediately prior to ligand addition and normalized to vehicle-treated wells. Importantly, DMSO concentration was maintained at a constant 0.05% across the plate. Data from the first 20 to 30 min following ligand addition were used to calculate the area under the curve (AUC) of the peak. For PTX experiments, AUC was calculated from the first 30 min immediately after ligand addition.
Data Analysis
Data from the xCELLigence system were analyzed and extracted with RTCA software 1.2.1.1002 (ACEA Biosciences). Data were analyzed with GraphPad Prism 6.00 (GraphPad Software, La Jolla, CA). Allosterism can be quantified by applying the operational model of allosterism to the ACh and LY2033298 interaction data to obtain the best-fit curve data, equation (1), 23 where Em is the maximal effect of the pathway; [A] and [B] are concentrations of the orthosteric agonist and the allosteric modulator, respectively; KA and KB are the equilibrium dissociation constant of the orthosteric agonist and allosteric modulator, respectively; τA and τB are operational measures of the respective signaling efficacies of orthosteric agonist and allosteric modulator that incorporate receptor expression levels and efficiency of stimulus-response coupling; α is the cooperativity factor of the allosteric effect of the modulator on orthosteric agonist binding affinity, whereas β is that of the signaling efficacy; and n is the transducer slope factor linking occupancy to response.
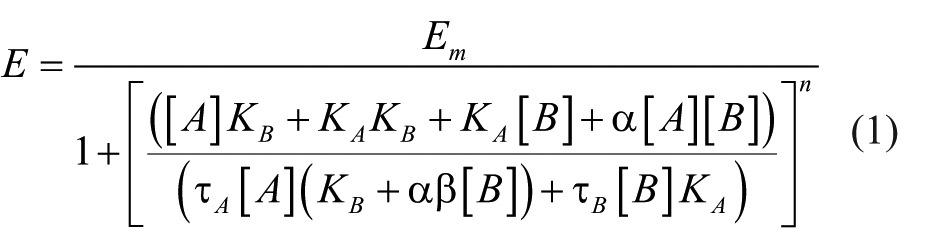
All affinity, potency, and cooperativity values were estimated as logarithms 24 and statistical comparisons between values were by one-way analysis of variance using a Tukey’s multiple-comparison posttest to determine significant differences.
Results and Discussion
We have previously characterized LY2033298 ( Fig. 1A ) as a potent M4 mAChR PAM of ACh affinity and function in cells overexpressing human M4 mAChR. 20 In the same study, we found both LY2033298 agonism and potentiation of the ACh response (cooperativity) were markedly reduced when tested in NG108-15 cells that natively express rodent M4 mAChR. Although these results could be due to the species variability described for LY2033298, 25 they could also be a consequence of different M4 mAChR expression levels in the different cell lines. Thus, we initially assessed M4 mAChR expression levels in NG108-15 cells by [3H]NMS in membrane saturation radioligand binding experiments (data not shown). The Bmax obtained in these experiments was 0.17 ± 0.04 pmol/mg of membrane protein (n = 4), which is markedly lower than that of human M4 mAChR heterologously expressed in FlpIn Chinese hamster ovary (CHO) cells (1.1 ± 0.2 pmol/mg 26 ).
To compare the allosteric properties of LY2033298 in NG108-15 cells using the xCELLigence system, we first characterized the effects of the PAM using two classical endpoint-based assays. M4 mAChR predominantly couples to Gαi proteins, the activation of which inhibits production of cAMP by adenylyl cyclase, as well as promoting ERK1/2 phosphorylation further downstream in the signal transduction pathway. We explored the ability of LY2033298 to modulate ACh-mediated inhibition of cAMP accumulation using a BRET sensor, cAMP sensor using YFP-Epac-RLuc (CAMYEL).27,28 Briefly, the CAMYEL sensor has yellow fluorescent protein (YFP; acceptor) and Renilla luciferase (Rluc; donor) on either termini of the Epac1 protein. Binding of cAMP to Epac1 induces a change in conformation, increasing the distance between YFP and Rluc, resulting in a decrease of the BRET signals. Increasing concentrations of ACh alone resulted in a concentration-dependent inhibition of forskolin-induced cAMP accumulation ( Fig. 1B ). Coaddition of LY2033298 caused a potentiation of ACh-mediated inhibition of forskolin-induced cAMP accumulation ( Fig. 1B ).
ERK1/2 phosphorylation was analyzed using the Alpha-Screen SureFire Phospho-ERK1/2 assay, based on the transfer of oxygen radicals from the donor to the acceptor beads, which detect total or phosphorylated ERK1/2, respectively. ACh induced ERK1/2 phosphorylation in a concentration-dependent manner ( Fig. 2A ). As seen with the cAMP BRET assay, costimulation of LY2033298 potentiated ACh response, although the potentiation was greater in the ERK1/2 phosphorylation assay. In terms of allosteric agonism, while LY2033298 alone only weakly inhibited cAMP accumulation, it showed significant agonism in ERK1/2 phosphorylation (p < 0.05; one-way analysis of variance [ANOVA] with Dunnett’s multiple-comparisons test; Figs. 1C , 2B ).
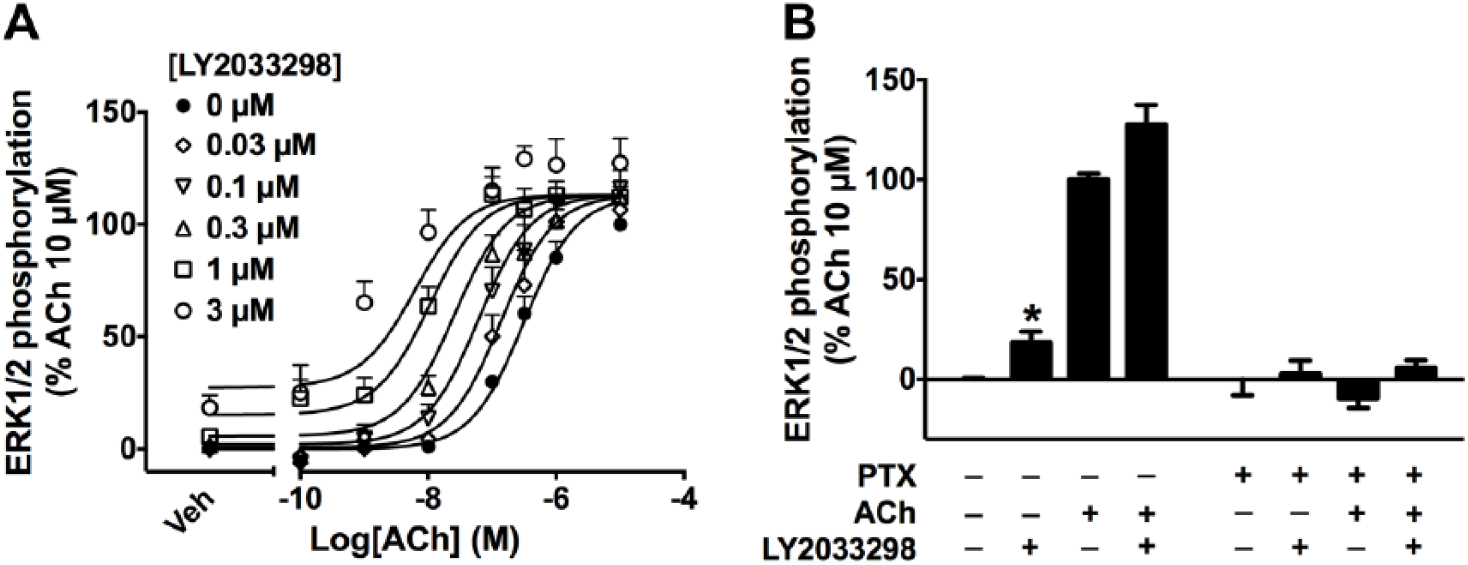
LY2033298 potentiates acetylcholine (ACh)–induced ERK1/2 phosphorylation. (
The allosteric effect of LY2033298 was quantified by fitting concentration-response curves to the operational model of allosterism, equation (1), 23 to yield the allosteric parameters shown in Table 1 . The ability of LY2033298 to potentiate the ACh-mediated response (denoted as functional cooperativity, αβ) in the ERK1/2 phosphorylation assay was significantly higher than that estimated from the cAMP BRET assay (αβ, 127 vs. 12, respectively; p < 0.01; one-way ANOVA with Tukey’s multiple-comparisons test). ACh and LY2033298 efficacy (τA and τB, respectively), although not significant, were estimated to be marginally higher in ERK1/2 phosphorylation than the cAMP BRET assay (τA, 231 vs. 147, respectively; τB, 0.66 vs. 0.14, respectively).
Operational Model Parameters for Functional Interaction between ACh and LY2033298 at the M4 Muscarinic Acetylcholine Receptor (mAChR).
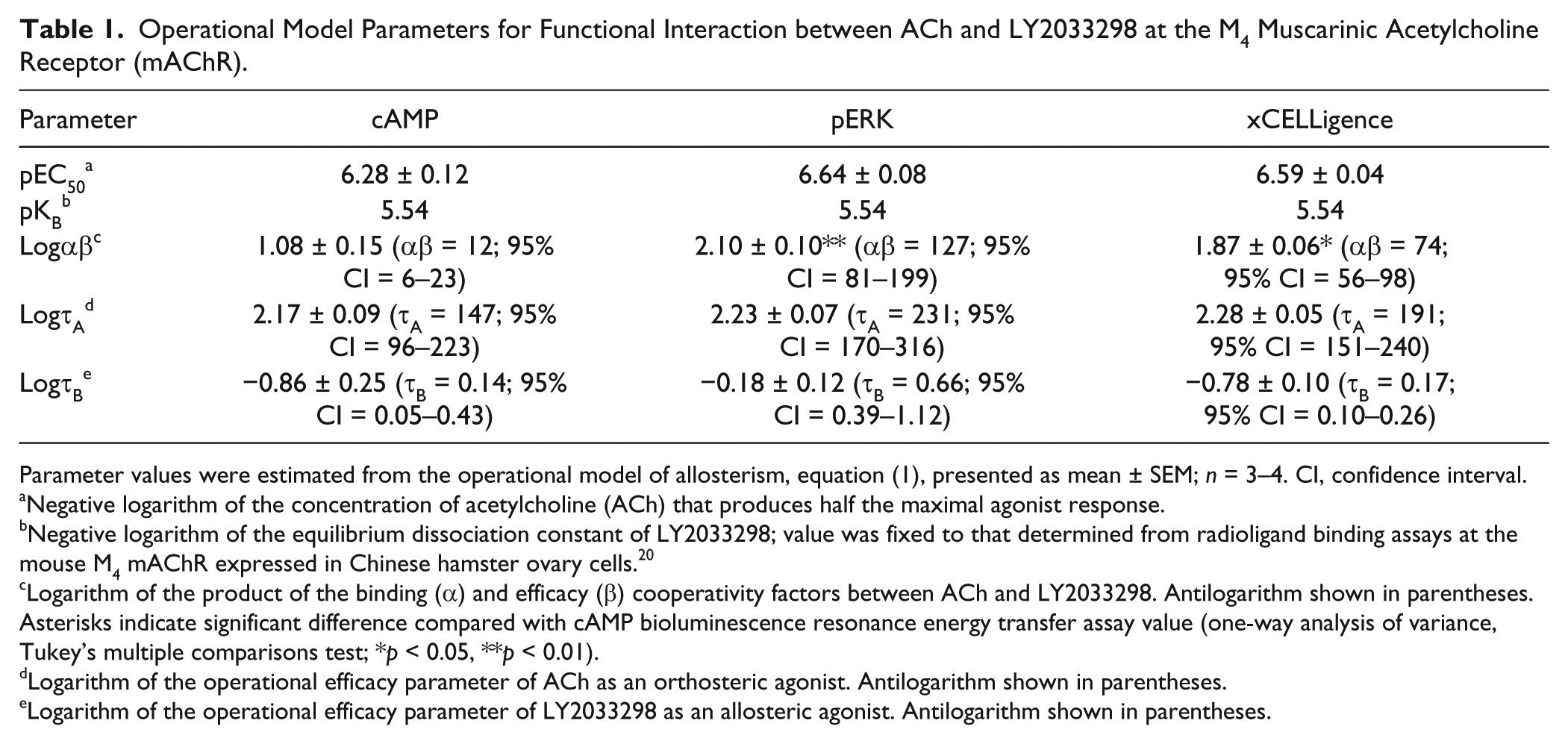
Parameter values were estimated from the operational model of allosterism, equation (1), presented as mean ± SEM; n = 3–4. CI, confidence interval.
Negative logarithm of the concentration of acetylcholine (ACh) that produces half the maximal agonist response.
Negative logarithm of the equilibrium dissociation constant of LY2033298; value was fixed to that determined from radioligand binding assays at the mouse M4 mAChR expressed in Chinese hamster ovary cells. 20
Logarithm of the product of the binding (α) and efficacy (β) cooperativity factors between ACh and LY2033298. Antilogarithm shown in parentheses. Asterisks indicate significant difference compared with cAMP bioluminescence resonance energy transfer assay value (one-way analysis of variance, Tukey’s multiple comparisons test; *p < 0.05, **p < 0.01).
Logarithm of the operational efficacy parameter of ACh as an orthosteric agonist. Antilogarithm shown in parentheses.
Logarithm of the operational efficacy parameter of LY2033298 as an allosteric agonist. Antilogarithm shown in parentheses.
We then confirmed the contribution of Gαi proteins to M4 mAChR-mediated signals in NG108-15 cells. In cells treated with PTX, which prevents the coupling of Gαi proteins to GPCRs, the inhibition of forskolin-induced cAMP accumulation mediated by either ACh (100 µM) or ACh + LY2033298 (3 µM) was completely abolished, confirming the involvement of Gαi proteins in these responses ( Fig. 1C ). This was also observed in the ERK1/2 phosphorylation assay, where the signals mediated by either ligand were abolished in PTX-treated cells ( Fig. 2B ).
Next, we investigated the ACh response in NG108-15 cells using the xCELLigence label-free system. This system measures the change in cellular impedance using microelectrodes lined on the bottom of the wells of a 96-well plate (E-plate). As the cells adhere to the microelectrodes, the local ionic environment changes, which leads to an increase in impedance. 6 Therefore, the change in cell adhesion and/or morphology is reflected in a change in impedance.4–6 Of note, the DMSO content was kept constant across the plate, as DMSO itself changes cellular impedance. 4
Immediately after addition of ACh, there was a sharp increase in impedance, which reached a maximum within 10 min ( Fig. 3A–E , black lines). There was a concentration-dependent increase in the maximum impedance reached by ACh ( Fig. 3F , filled circles). The return to baseline produced a more gradual slope that decayed slowly back to the baseline ( Fig. 3A–E ).
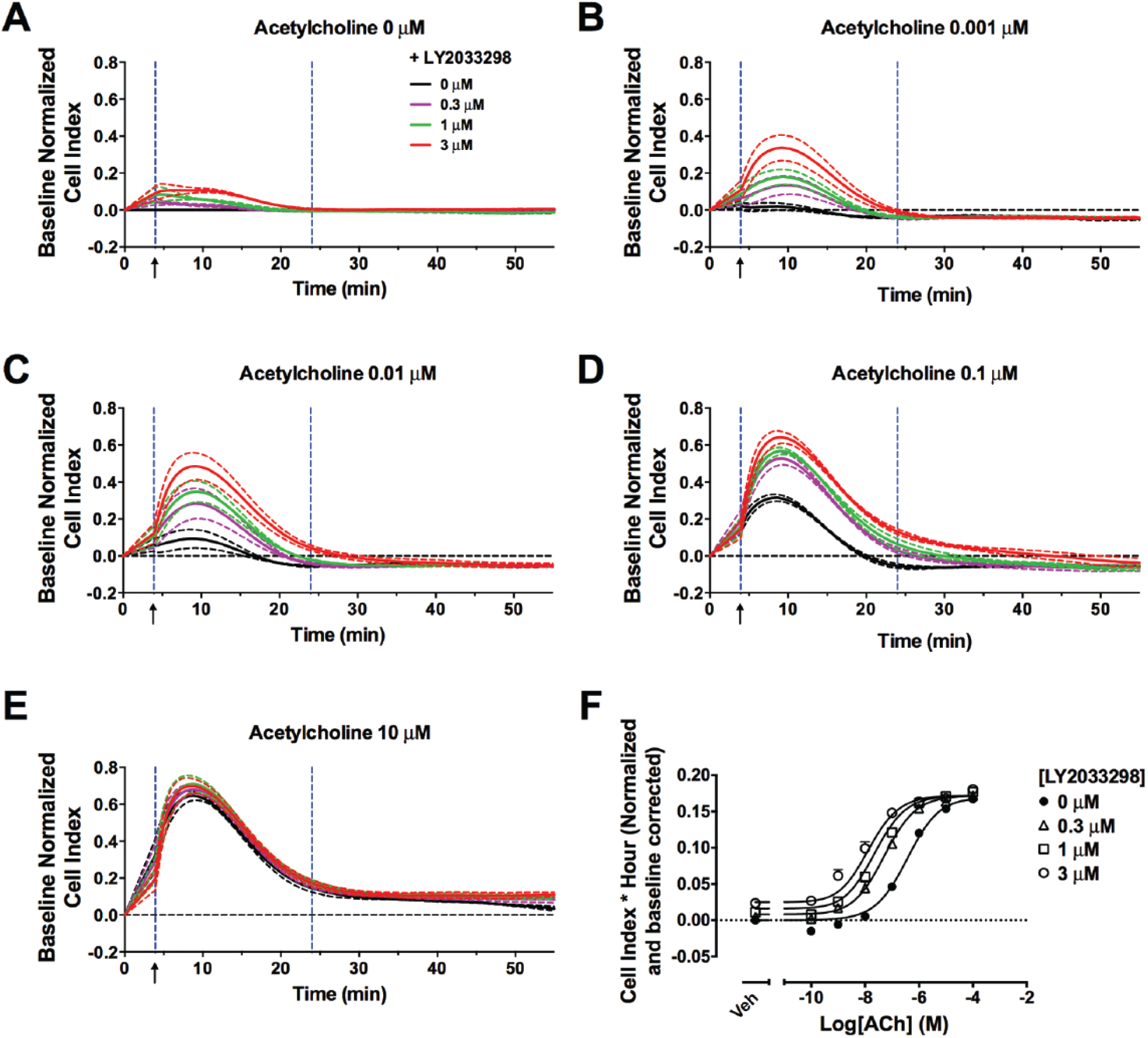
Positive allosteric modulation of acetylcholine (ACh) by LY2033298 can be detected with xCELLigence. Change in impedance induced by ACh (
As shown in the impedance traces ( Fig. 3A–E ), coaddition of LY2033298 potentiated an ACh-mediated response, and this potentiation reached a limit at the highest concentrations of modulator. Of note, increasing concentrations of the PAM increased the peak height, although not the width, of ACh-mediated impedance.
Concentration-response curves were obtained by calculating the AUC of the impedance peaks, and allosteric parameters were estimated through analysis of the data with equation (1) ( Fig. 3F and Table 1 ). The potency of ACh in all three assays (cAMP inhibition, ERK phosphorylation, and cellular impedance) was similar ( Table 1 ). This is in contrast to a previous study with the C5a receptor where the potency of the agonists tested was consistently lower in label-free assays compared with endpoint-based assays. 29 The potentiation of ACh-mediated impedance by LY2033298 using the label-free approach was similar to the cooperativity estimated for ERK1/2 phosphorylation and significantly higher than that for the inhibition of cAMP accumulation (αβ, 74 vs. 12, respectively; p < 0.05; one-way ANOVA with Dunnett’s multiple-comparisons test). However, LY2033298 agonism (τB) as estimated by xCELLigence was similar to that by the cAMP BRET assay (τB, 0.17 vs. 0.14, respectively).
Interestingly, in cells treated with PTX, the impedance change induced by ACh (100 µM) was significantly impaired ( Fig. 4 ). These results suggest that the overall changes in morphology of NG108-15 cells induced by ACh are predominately Gαi protein dependent. Interestingly, upon PTX treatment, the LY2033298 (3 µM)–induced impedance, both in the absence and in the presence of ACh, fell into the negative range, which was a phenomenon not seen in cells with intact Gαi protein activity ( Fig. 4 ). These results suggest LY2033298 may be able to activate signaling pathways that are PTX insensitive. Although these data need further exploration, our cAMP data ( Fig. 1C ) show that in the presence of PTX, LY2033298 does not elicit changes in cAMP, suggesting that the unmasking of a Gαs-mediated response is highly unlikely.
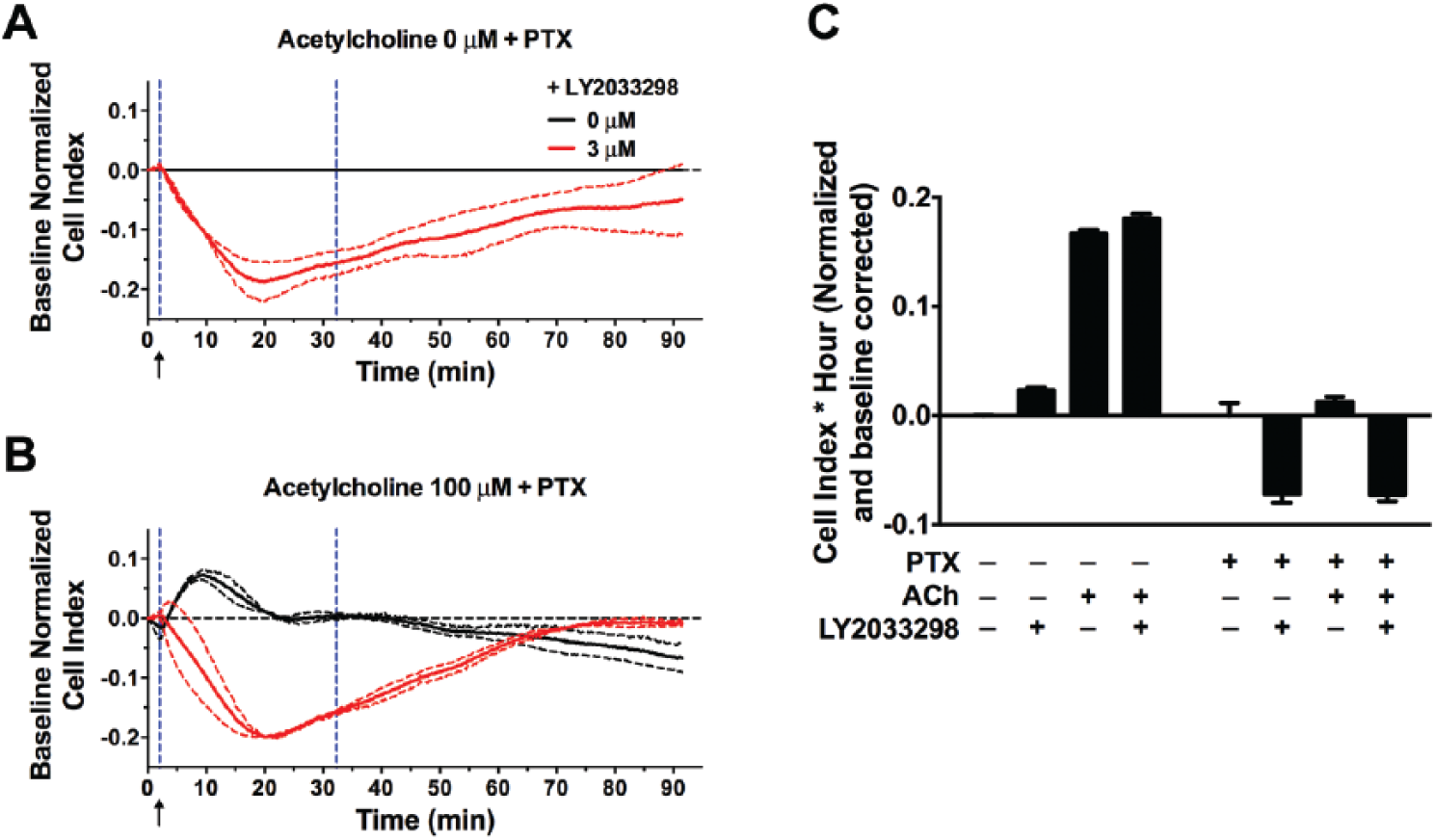
Overall change in impedance induced by acetylcholine (ACh) is predominately Gαi protein dependent. (
Label-free technologies on their own do not allow the determination of the precise intracellular pathways activated by a particular receptor or whether the different signaling pathways are modulated by an allosteric ligand. The deconvolution of label-free traces into specific signaling cascades can be achieved using pharmacological inhibitors. However, it is worth noting that activation of intracellular signaling cascades is cell type dependent, and as such, the interpretation of impedance traces is highly restricted to a particular cell system. Previous publications that have deconvoluted label-free traces into signaling pathways have also highlighted its cell type dependency. Stallaert et al. 30 used xCELLigence profiles in combination with pharmacological inhibitors to cluster and classify different β2-adrenergic receptor (β2AR) ligands in HEK293S cells overexpressing this receptor or in rat aortic smooth muscle cells endogenously expressing β2AR. While the clustering of ligands remained unaltered, the impedance profiles of the ligands were markedly different. Schröder et al. 31 dissected the signaling patterns of several GPCRs with differential G protein coupling preferences using dynamic mass redistribution (DMR). This study also revealed a cell type dependency of the DMR response. Taking these studies into consideration, we would anticipate that in a different cell line, the impedance traces would change but the allosteric modulation would remain. Indeed, in preliminary studies using CHO FlpIn cells overexpressing the M4 mAChR, the impedance traces were different from those observed in NG108-15 cells; however, LY2033298 allosteric modulation could be readily observed and quantified (data not shown).
Our results show that the potentiation of an orthosteric ligand by an allosteric modulator can be detected and quantified by xCELLigence in a neuronal cell line that endogenously expresses the receptor of interest. To show this, we have used a prototypical GPCR with known G protein coupling preferences. However, our data also show that this approach is also amenable to screen for allosteric modulators in the absence of information about G protein coupling preferences.
In addition to allosterism, label-free approaches have previously been used to investigate another GPCR signaling paradigm, termed biased agonism or functional selectivity, where ligands that bind to the same GPCR preferentially activate one signaling pathway over another, leading to diverse physiological outcomes. 30 Both types of GPCR behaviors provide additional scope for the development of novel potential treatments with improved target selectivity and reduced occurrence of side effects. 3 The ability of impedance-based technology to detect and quantify allosteric and ligand-biased signaling in native environments can substantially aid the characterization of novel GPCR ligands.
Footnotes
Acknowledgements
LY2033298 was a generous gift from Christian C. Felder (Eli Lilly & Co., USA).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the National Health and Medical Research Council of Australia (NHMRC) (program grant APP1055134) to A.C. and P.M.S.; M.C. is a Monash fellow; A.C. and P.M.S. are principal research fellows of the NHMRC.