
Abstract
Modified messenger RNAs (mRNAs) hold great potential as therapeutics by using the body’s own processes for protein production. However, a key challenge is efficient delivery of therapeutic mRNA to the cell cytosol and productive protein translation. Lipid nanoparticles (LNPs) are the most clinically advanced system for nucleic acid delivery; however, a relatively narrow therapeutic index makes them unsuitable for many therapeutic applications. A key obstacle to the development of more potent LNPs is a limited mechanistic understanding of the interaction of LNPs with cells. To address this gap, we performed an arrayed CRISPR screen to identify novel pathways important for the functional delivery of MC3 lipid-based LNP encapsulated mRNA (LNP-mRNA). Here, we have developed and validated a robust, high-throughput screening–friendly phenotypic assay to identify novel targets that modulate productive LNP-mRNA delivery. We screened the druggable genome (7795 genes) and validated 44 genes that either increased (37 genes) or inhibited (14 genes) the productive delivery of LNP-mRNA. Many of these genes clustered into families involved with host cell transcription, protein ubiquitination, and intracellular trafficking. We show that both UDP-glucose ceramide glucosyltransferase and V-type proton ATPase can significantly modulate the productive delivery of LNP-mRNA, increasing and decreasing, respectively, with both genetic perturbation and by small-molecule inhibition. Taken together, these findings shed new light into the molecular machinery regulating the delivery of LNPs into cells and improve our mechanistic understanding of the cellular processes modulating the interaction of LNPs with cells.
Introduction
Lipid Nanoparticle Delivery of mRNA
The portfolio of modalities in the pharmaceutical toolbox has expanded significantly over the past 5 years, and an important part of this portfolio is the use of modified messenger RNA (mRNA) to exploit the host cell protein machinery for the localized production of a protein that drives therapeutic effect. 1 However, the successful delivery of mRNA remains challenging, as these molecules are large, negatively charged, and therefore often require specialized formulation that not only facilities transport across the cell membrane but also importantly releases the cargo into the cytoplasm of the cell for translation. 2 Lipid nanoparticles (LNPs) are the most clinically advanced system for nucleic acid delivery, with the first RNA-based medicine delivered via an LNP receiving Food and Drug Administration approval in 2018. 3
Despite this success, LNPs are immunogenic at high doses, and the development of safer and more potent LNPs is an area of unmet need for the field of nucleic acid delivery. Underpinning this is the requirement to gain a deeper understanding of the molecular mechanism of LNP delivery to facilitate rational design of new formulations.4–6 We therefore wanted an unbiased way to identify genes that either positively or negatively modulate the productive delivery of MC3 lipid-based LNP encapsulated mRNA (LNP-mRNA). With this information, we aimed to both increase basic understanding around delivery and potentially identify cellular targets that can be modulated with small molecules in a co-dosing strategy to improve localized delivery efficiency.
There is precedence for the idea that both genetic and small-molecule perturbations can modulate the effective delivery of LNPs,7–10 but these have largely been focused on targeting only the endocytic pathway and using formulations encapsulating small interfering RNA (siRNAs) or antisense oligonucleotides, not mRNA. To this end, we decided to leverage the power of functional genomics and apply our new arrayed CRISPR platform for the unbiased detection of genes from within the druggable genome that can modulate delivery. By defining our phenotype as “productive delivery,” we will probe not only the endocytosis of LNP particles but also the release and subsequent translation of the mRNA cargo. Therefore, we are likely to identify genes not only involved directly in endocytosis and trafficking but also those involved in host cell translation.
CRISPR Technology
The power of precise genome editing with CRISPR/Cas9 and related technologies for functional genomics has been well stated and extensively reviewed.11–13 In brief, Cas9 endonuclease is targeted to precise genomic locations by a guide RNA (gRNA) composed of a variable CRISPR RNA (crRNA) and constant trans-activating RNA (tracrRNA) that, upon dimerization, bind to Cas9. This cr:tracrRNA:Cas9 complex is able to scan the genome for the crRNA complement and protospacer adjacent motif sequence, upon which the nuclease catalyses a double-strand DNA break (DSB). This DSB is either faithfully repaired, and thus liable to cleavage again, or unfaithfully repaired by insertion or deletion of nucleotides (indels), which most commonly results in a frame-shift mutation and subsequent loss of gene function. 14
Although the mechanism of gene editing remains consistent, libraries for CRISPR screening are formatted in two fundamentally different ways, referred to as “pooled” and “arrayed.” In pooled libraries (almost exclusively lentivirus based), gRNAs are synthesized and cloned in a pool and subsequently infected into cells en masse (i.e., all gRNA lentivirus), targeting all genes together in one flask. Deconvoluting screen outputs to correlate gene and phenotype requires next-generation sequencing, which limits pooled screening to those whose desired phenotype is selectable, primarily growth-related phenotypes, for which the depletion or enrichment of a given gRNA in the population is used as a method of selection. In contrast, in arrayed CRISPR libraries, the gRNA are already deconvoluted into individual wells such that in a single well, only a single gene will be targeted. Libraries are available in numerous formats, including chemically synthesized (synthetic), plasmid-based in vitro transcribed, and arrayed lentivirus. However, synthetic libraries are currently gaining traction in the field because of their ease of use, reproducibility, high efficiency, and compatibility with screening platforms. Most critically, the use of synthetic libraries is enabled by commercial manufactures improving synthesis methods and providing genome-wide (GW) libraries as off-the-shelf products. That said, it is likely that arrayed lentivirus libraries will meet their niche application when arrayed screening moves into harder to transfect models such as primary cells, for which efficient lipid delivery of synthetic RNA could be challenging.
The use of pooled CRISPR libraries for target discovery screening is well established,15–17 with institutes such as the Broad and the Wellcome Sanger Institute having delivered GW screens across several hundred cell lines.18,19 Arrayed CRISPR is a much newer paradigm, with only a handful of publications demonstrating the potential of this powerful approach and with only small subset libraries.20–23 While GW arrayed CRISPR screening presents numerous challenges, not least the large upfront investment in libraries, equipment, and expertise to carry out cell-based phenotypic screens at scale, it does open the door to the full power of high-content microscopy and phenotypic screening.
To overcome our limited mechanistic understanding of the molecular machinery critical for the productive delivery of LNP-mRNA in cells, we describe here the application of an arrayed CRISPR screen to identify mechanisms modulating the productive delivery of LNP-mRNA ( Fig. 1 ). To this end, we have developed and validated a robust, high-throughput screening–friendly phenotypic assay and integrated it with pathway and multivariate analysis. We screened the druggable genome (7795 genes) and validated 44 genes that either increased (37) or inhibited (14) the productive delivery of LNP-mRNA. Many of these genes clustered into families involved with host cell transcription, protein ubiquitination, and intracellular trafficking. We show that both UDP-glucose ceramide glucosyltransferase (UGCG) and V-type proton ATPase (ATP6V) can significantly modulate the productive delivery of LNP-mRNA, increasing and decreasing, respectively, with both genetic perturbation and by small-molecule inhibition.
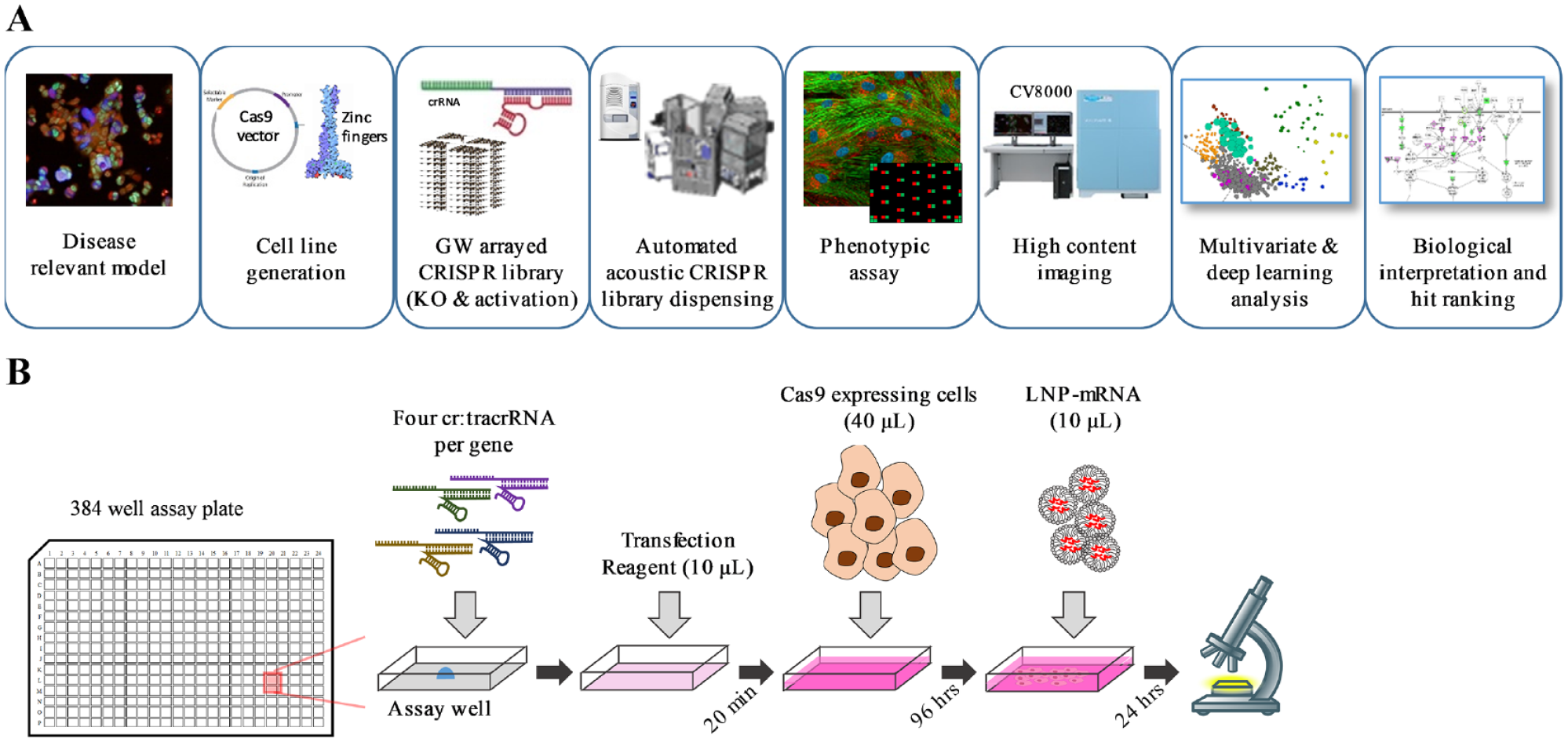
Overview of screening platform and assay format. (
Materials and Methods
Cell Line Generation
NCI-H358 cells from AstraZeneca Global Cell bank were grown in RPMI-1640 media, 10% (v/v) fetal calf serum, 1x GlutaMAX (“complete media”), and routinely passaged 1:3 twice weekly using TrypLE Express to dissociate cells. For screening, cells were cultured in the presence of 100 U/mL penicillin-streptomycin. All reagents were obtained from Thermo Fisher Scientific. An inducible SpCas9-wt fused to a self-cleaving enhanced green fluorescent protein (T2A-EGFP) was stably integrated into cells. In brief, H358 cells were seeded into six-well plates and forward transfected with transfer DNA and CompoZr (CTI-1KT; Merck, Kenilworth, NJ) using FuGENE HD (Promega, Madison, WI) at a ratio of 1:2 following the manufacturer’s instructions. Media were exchanged 24 h posttransfection (hpt), and on day 8, antibiotic selection with 400 µg/mL G418 (Thermo Fisher Scientific, Waltham, MA) was initiated. On day 26, Cas9-T2A-GFP expression was induced for 24 h with 100 ng/mL doxycycline (Thermo Fisher Scientific), and all cells underwent fluorescence-activated cell sorting (FACS; FACS Jazz, BD Biosciences, Franklin Lakes, NJ) to isolate the high- and medium-expressing GFP population for onward expansion. Cells were selected for a further 21 d with 400 µg/mL G418 before being cryopreserved as per standard protocols. A negative control (no DNA) was used to confirm when antibiotic selection had been successful.
Preparation of Individual gRNA and gRNA Library
Synthetic two-part gRNA (cr:tracrRNA) supplied by Horizon Discovery (Cambridge, UK) was used for all experiments. Individual tubes of crRNA were resuspended to 10 µM in the presence of an equimolar concentration of tracrRNA in 10 mM Tris.pH7 to provide a final duplex concentration of 10 µM, assuming a complete 1:1 dimerization of crRNA to tracrRNA. Throughout, “pool” refers to four individual cr:tracRNA combined at an equimolar ratio. The druggable genome library (GC-004650-E2/004670-E2, Horizon Discovery) containing 7795 genes with four crRNA targeting each gene was supplied across 104 plates (320 genes per plate and one crRNA per well). Plates were arranged such that four crRNA for any given gene were in the same well position across four sequential plates. Library plates (n = 104) containing 0.1 nmol crRNA were resuspended by the addition of 20 µL duplex buffer (5 µM tracrRNA, 10 mM Tris.HCl) and incubated for 60 min at room temperature (RT), followed by automated pooling of sets of four plates into a single plate and aliquoting into 384-well acoustic low-dead volume plates (LP-0200, Labcyte, San Jose, CA). This resulted in 26 library plates each containing 5 µM of duplexed cr:tracrRNA comprised of four different crRNA targeting the same gene at different loci.
Reverse Transfection of Synthetic gRNA
NCI-H358-Cas9 cells were seeded at 2.75 × 105 cells/cm2 and grown for 3 d prior to induction of Cas9 with doxycycline (100 ng/mL) addition 24 h before reverse transfection. Duplexed cr:tracRNA was acoustically dispensed with an Echo 555 (Labcyte) into 384-well assay plates (Cell Carrier Ultra, PerkinElmer, Waltham, MA) followed by the addition of transfection solution (10 µL serum-free RPMI-1640, 0.6 % [v/v] RNAiMAX, Thermo Fisher Scientific). After a 20-min incubation at RT, 40 µL cell suspension was dispensed into assay plates (4000 cells/µL) and incubated at 37 °C, 5% CO2.
Transferrin Reporter Assay and Hoechst Staining
Transferrin-647 (T23366, Thermo Fisher Scientific) prepared in serum-free RPMI-1640 was prewarmed to 37 °C and added to cells (final 10 µg/mL) and incubated for 15 min at 37 °C, 5% CO2, before fixation with 4% (w/v) paraformaldehyde (PFA) for 20 min. Cells were washed three times with phosphate-buffered saline (PBS) and stored at 4 °C. For nuclei staining, cells were incubated with modified block/permeabilizing buffer (0.2 % [v/v] Triton-X100, 2% [w/v] bovine serum albumin, PBS) containing 10 ∝g/mL Hoechst (Thermo Fisher).
High-Content Imaging and Analysis
Images were acquired using a Cell Voyager 7000 (CV7000, Yokogawa Inc., Tokyo, Japan) with a 20× objective and no image binning or compression; for each 384-well plate, a total of nine field-of-views were captured. Images were imported into Columbus (PerkinElmer) image analysis software for classical image analysis. In brief, cell nuclei were detected from Hoechst staining and cell cytoplasm from the diffuse background Hoechst signal. Intensity, texture, and morphology (area, width:length, and roundness) features were detected for cell nuclei to enable gating of a “healthy” population of cells based on nuclei morphology and used for all downstream analysis. LNP-mRNA productive delivery was determined with three endpoints based on image analysis of mCherry expression and defined as (1) the total amount of mCherry expressed (integrated intensity) on a per cell basis (total expression per cell), (2) the average mCherry expressed (mean intensity) on a per cell basis (mean expression per cell), and (3) the total amount of mCherry expressed in the whole well assay well, independent of cell number (total expression per well). For transferrin reporter assays, the negative and positive populations were gated based on both total cell transferrin-657 intensity and presence of positive puncta using a cell texture metric and the percentage of cells negative for the receptor quantified.
LNP Preparation and Characterization
5′ methoxyuridine mCherry mRNA (L-7203, TriLink Biotechnologies, San Diego, CA) was formulated into LNPs using the microfluidic setup described in detail elsewhere. 24 In brief, stocks of lipids were dissolved in ethanol and mixed in the weight ratio of 20:1 lipid L mRNA to obtain a lipid concentration of 12.5 mM (1.85 mg/mL). The aqueous and ethanol solutions were mixed in a 3:1 volume ratio using a microfluidic apparatus NanoAssemblr (Precision NanoSystems, Vancouver, BC, Canada) at a mixing rate of 12 mL/min. LNPs were dialyzed overnight in PBS using Slide-A-Lyzer G2 dialysis cassettes (Thermo Fisher) with a molecular weight cutoff of 10 kDa. The size of the LNPs was measured by dynamic light scattering using a Zetasizer Nano ZS (Malvern Instruments, Malvern, UK), and the concentration and encapsulation of mRNA were determined using the RiboGreen assay. The final ratio of lipids by molar percentage was 50:38.5:10:1.5 (MC3:DSPC:PEG-DMG 2000) with a lipid to RNA mass ratio of 20:1.
Optimization
Stability experiments confirmed that libraries were stable at 8 °C for the period of time required for multiple screening runs, and preduplexing libraries with tracrRNA to a final concentration of 5 µM enabled fast acoustically dispensing of 250 nL per 384-assay well, 20 min per plate. The inevitable desiccation of this 250 nL duplex in the assay plate did not affect performance and therefore enabled us to separate the library dispensing protocol and the reverse transfection protocol into two sequential phases. Unlike other two-part systems, there was no requirement for a high-temperature annealing step. For reverse transfection, a small panel of transfection reagents was benchmarked, and RNAiMAX in serum-free media was found to be efficient for many cell lines and the optimal concentration of 0.6 % (v/v) determined (data not shown). A low volume is preferable to ensure cr:tracrRNA is at an appropriate concentration for the formation of transfection complexes, and an optimized volume of 10 µL per 384-well plate was a compromise between introducing variation from low-volume dispensing on liquid handlers and yet not to dilute. The addition of a cell suspension in complete media did not affect delivery efficiency and removed additional liquid-handling requirements of doing so in serum-free media.
Primary Screen, Hit Confirmation, and Normalization
The druggable genome library was acoustically dispensed into 384-well assay plates using an Echo and chilled plate storage docked onto a standard Star6 automation platform (HighRes) to enable automated dispensing and 8 °C storage of the library and assay plates. Dispersed positive (PLK#1 pool) and negative (nontargeting control [NTC] pool) controls were acoustically dispensed (n = 32 each, per plate) across all assay plates. Off-board control (OBC) plates containing titrations of cr:tracrRNA of our phenotypic controls Polio-like Kinase 1 (PLK1) and Transferrin Receptor Complex (TFRC) and LNP-mRNA were included at the start and end of the assay run to enable additional assay quality control. Reverse transfection by the sequential dispensing of transection solution and doxycycline-induced cell suspension was as described. All plates were processed as a single batch of 32 plates (26 library, 2 OBC, and 4 blank plates) and then incubated in a rotating incubator at 37 °C, 5% (v/v) CO2. Cryopreserved LNP-mRNA was prepared fresh into complete media at 2 µg/mL and dispensed (10 µL) onto library plates at 96 h posttransfection (hpt), followed by a further 24 h incubation and then fixation with 4% (w/v) PFA and Hoechst staining. Assay plates were stored at 8 °C prior to image acquisition on a CV7000 (Yokogawa Inc.). Images were analyzed in Columbus (PerkinElmer) as described and data imported into Genedata (Basal, Switzerland) for normalization and quality control. For cell number, data were normalized to negative (NTC, 0%) and positive (PLK1, –100%) controls. For LNP-mRNA delivery endpoints, data were normalized on a per-plate basis to the library population, defined as “neutral.” Hit thresholds were based on mean ± standard deviation (SD). 25 Hit confirmation was conducted as per the primary screen but at a smaller scale, with increased replicates and the inclusion of a high-dose LNP-mRNA dispersed control. Cell number data were normalized as above, and LNP-mRNA delivery data were normalized as fold change from the NTC population.
Network Analysis
STRING v11 was used for both network analysis and Gene Ontology (GO) enrichment on confirmed hits only. 26 Edges illustrate confidence, with a minimum required interaction score of 0.7 or higher. For GO and Reactome statistical enrichment, the druggable genome was used as background reference. As the primary screen used a single-shot approach, in which each gene is tested only in a single well (i.e., n = 1), we only undertook network and GO analysis on those hits that were validated in the subsequent hit confirmation assay.
Compound Treatment
Small-molecule inhibitors with log IC50 >7 were identified from internal compound collections for ATP6V (A1, A2, and A3) and UGCG (U1, U2, and U3). Cells were seeded as described except for the absence of RNA and RNAiMAX reagent. At 96 h after seeding, cells were co-treated with compound (10-pt dose response) and LNP-mRNA, 20 and 40 ng/well, followed by 24 h of incubation and fixation, imaging, and analysis as described.
Results
Assay Platform Build and Arrayed CRISPR Screen Optimization
NCI-H358 cells were engineered to express Cas9 (NCI-H358-Cas9). We initially generated cells with constitutive expression of Cas9 using a standard lentivirus vector (Merck Millipore, Burlington, VA); however, we observed a difference in phase contrast morphology and LNP delivery efficiency with respect to parental cells (data not shown). We subsequently switched to a doxycycline-inducible Cas9 expression system with a T2A-GFP fused to the Cas9. Four NCI-H358-Cas9 cell lines were generated in parallel to one control line (no vector). Cell lines were generated in duplicate and during FACS sorting were gated for high and medium GFP expression (a surrogate marker for Cas9); antibiotic selection for both remained the same. At day 21 post-FACS, we profiled Cas9 expression in response to doxycycline using GFP as a surrogate marker for Cas9 and observed a correlation between the original FACS-gated populations (high and medium) and current Cas9 expression ( Fig. 2A ). Comparison of morphology and kinetic growth rate by phase contrast confirmed that all four lines retained a parental phenotype ( Suppl. Fig. 1A ). However, we observed that in cell lines selected for high GFP expression, a small population of cells expressed Cas9 in the absence of doxycycline induction ( Fig. 2B ), potentially indicating the misinsertion of the Cas9 cassette. We confirmed the incorporation of the Cas9-T2A-GFP cassette by immunofluorescent staining for Cas9 and the antibiotic selection gene neomycin phosphotransferase (NPT; Suppl. Fig. 1B ). Finally, we compared the editing efficiencies of all four cell lines using standard TFRC and PLK1 control genes. The transferrin assay is a quick way to understand the editing efficiency with a functional endpoint because of the binary nature of the receptor that, once knocked out, prevents any uptake of a fluorescent probe. PLK1 enables a label-free way of estimating editing efficiency as successful gene knockout leads to rapid cell death. Editing efficiency was comparable across all four lines, with cell line C slightly outperforming the others. This cell line was taken forward for screen optimization ( Fig. 2C ).
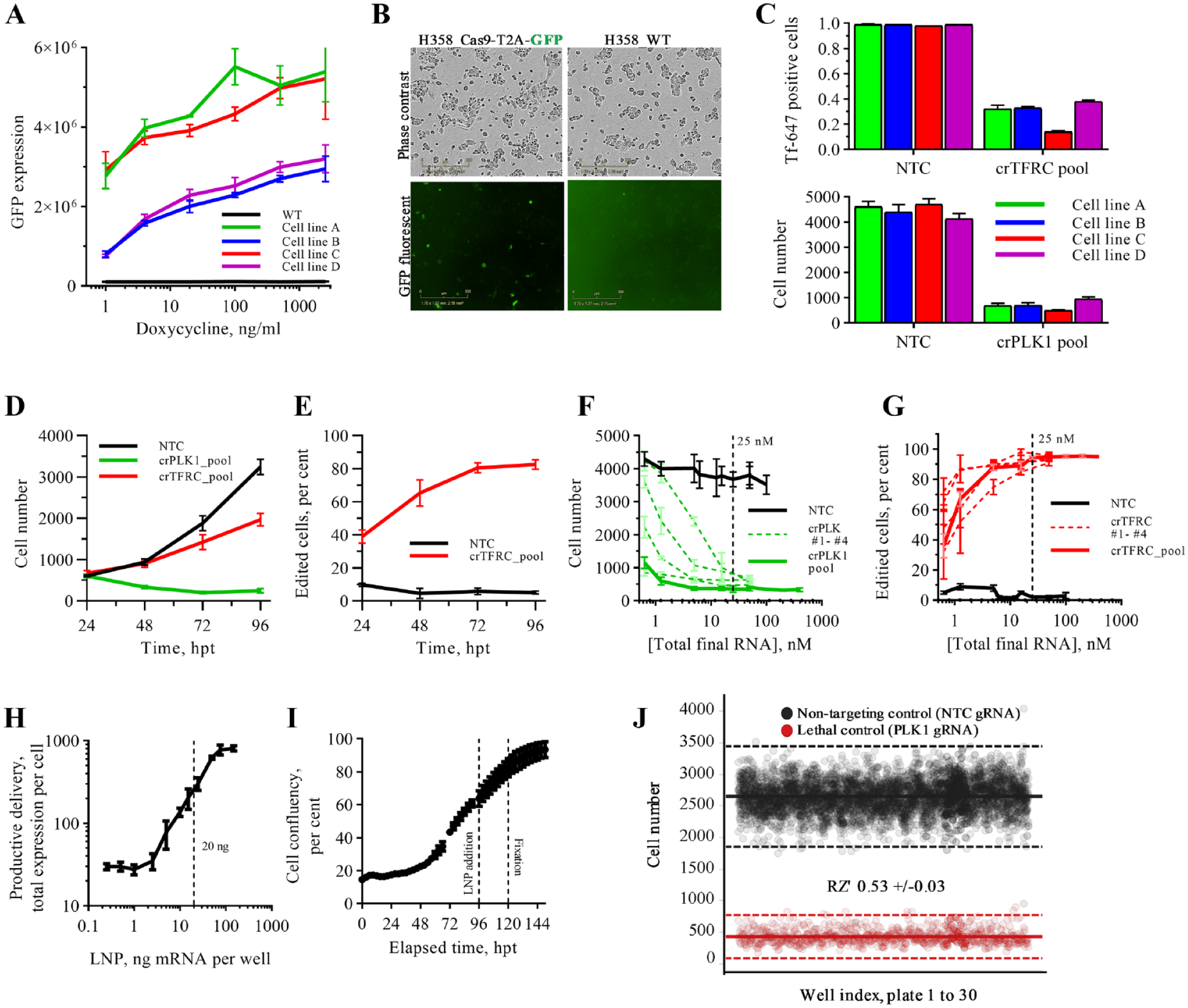
Screen optimization. (
The generation of robust high-quality data from large-scale arrayed CRISPR screening is nontrivial. To this end, we conducted extensive optimization of crRNA library preparation, method of cellular delivery, and execution of the phenotypic assay on automated robotic equipment (see the Materials and Methods section). It was important to simplify the end-end assay process to as few steps as possible to minimize the potential to introduce error and to maintain the integrity of reagents. A schematic of the optimized workflow is depicted in
Figure 1B
. To identify the best assay conditions for our screen, we first performed time-course experiments in which we found that that phenotypic effects of gene knockout of our chosen positive controls (TFRC, PLK1) were evident at 24 hpt, and editing efficacy did not increase beyond 72 h (
Libraries were initially supplied as one crRNA per well, equating to 104 plates per screen. However, to minimize the resource and reagent cost required per screen, we sought to pool all four crRNA per gene into a single well, and so we quantified the effect this would have on editing efficiency and potential toxicity. Dose-response data were obtained for individual and pooled cr:tracrRNA targeting PLK1 and TFRC using the optimized experimental conditions. A pooled concentration as low as 5 nM (i.e., 1.25 nM of each cr:tracrRNA) was sufficient to provide >90% editing for both control genes, and for the best individual cr:tracrRNA individually, we observed ~75% knockout efficiency at 0.6 nM (
Validation of the optimized screen workflow was obtained through completion of a 30-plate run using striped positive (PLK1) and negative (NTC) controls. The average robust Z-score (RZ′) was 0.53 ± 0.3, which was greater than our internal criteria of 0.4 for single-shot screening ( Fig. 2J ). The major variation observed was driven by edge effects as the library format necessitated the use of all 384 wells per plate.
Dose optimization of LNP-mRNA delivery confirmed a linear relationship, and a submaximal dose of 20 ng LNP-mRNA was taken forward for screening ( Fig. 2H ). This enabled a bidirectional screen in which we can identify genes that either increase or decrease the productive delivery of mRNA in the same screen. To control for assay performance across the screen (i.e., whether the cr:tracrRNA was effectively delivered to cells, causing an edit and a phenotype manifest), we opted to use PLK1 as a dispersed positive control.
Primary Screen Using a Druggable Genome-Scale Library of Synthetic gRNAs
The primary screen was conducted using the optimized assay conditions as summarized in
Off-board controls (first and last assay plates) confirmed that over the 32-plate screen, the dose linearity of LNP-mRNA remained unchanged, meaning that a twofold increase on the first plate was comparable with a twofold increase on the last plate ( Fig. 3A ). Cell number was consistent across the screening run ( Fig. 3B ), and performance of the screen controls showed a mean assay performance of RZ′ 0.59 across all 26 library plates ( Fig. 3C ). A cell health threshold of 50% modulation (increase or decrease) of cell number was used to exclude genes eliciting a significant growth or lethal phenotype. LNP-mRNA productive delivery was determined with three endpoints: total expression per cell, mean expression per cell, and total expression per well. LNP-mRNA productive delivery primary hits were defined as those genes resulting in greater than 3 SD change from the library population for any one of the three endpoints ( Suppl. Fig. 2 ). All of these endpoints could be confounded by different factors (cell size, shape, number, etc.), and so they were used independently to generate an inclusive list of genes to take into hit confirmation. Therefore, it is expected that a subset of genes will be false-positives; hence, pathway mapping of putative hits was not performed on primary screening data.
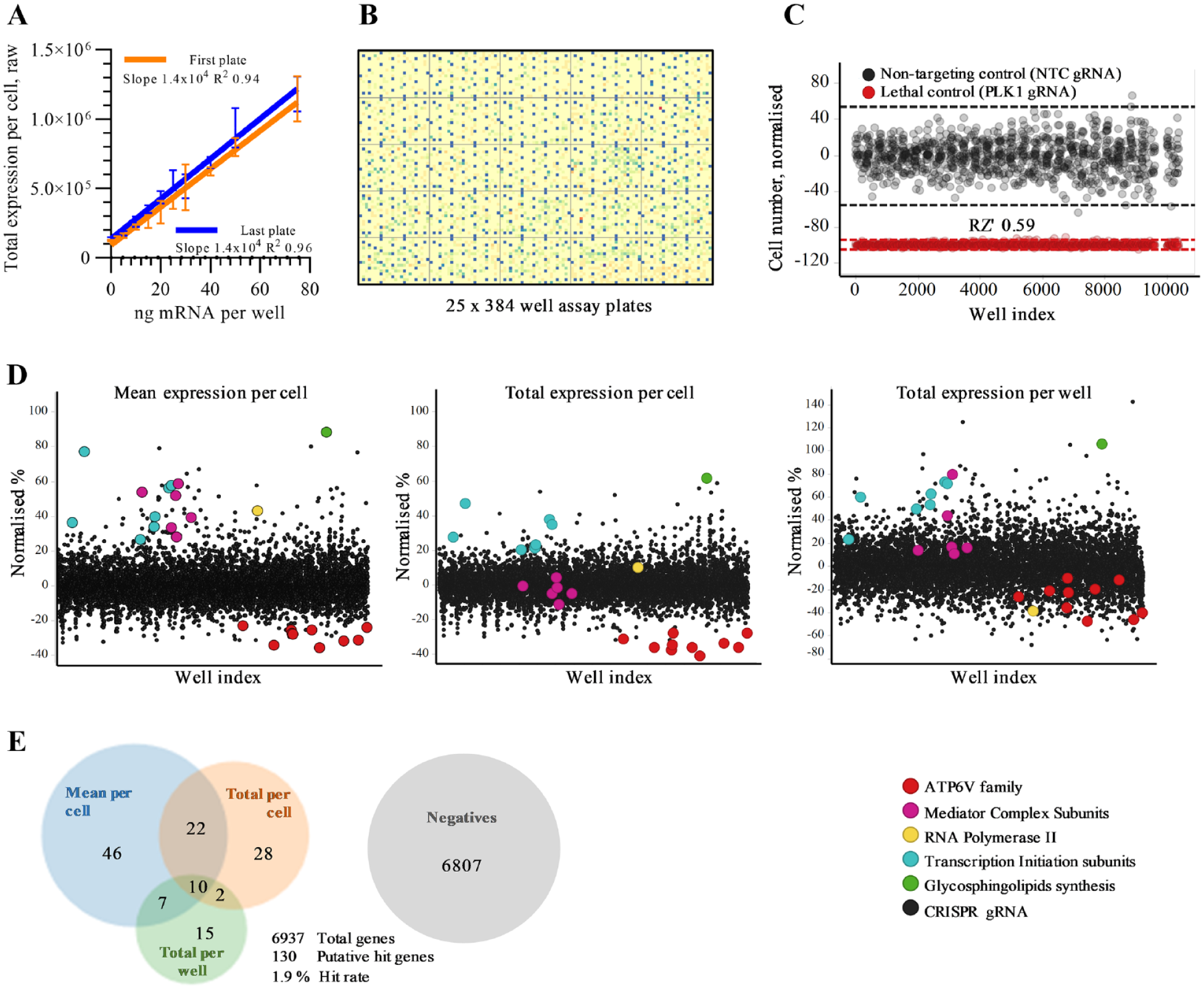
Primary screen. (
Hit scatter plots for all three endpoints illustrate a tight distribution of data, with primary hits being identified across the screen and in all three endpoints. In Figure 3D , the colors show highly represented families of genes from the primary hit list. There were a total of 130 primary hits (from the 6937 genes that passed quality control, for a 1.9% hit rate) that either increased or decreased productive delivery of LNP-mRNA ( Fig. 3E ).
Hit Confirmation
A total of 130 genes from the primary screen identified as putative hits for either increasing or decreasing productive delivery of LNP-mRNA were taken into hit confirmation. Genes were rescreened in the same assay format as for the primary screen but using new library plates (four cr:tracrRNA pooled per well), new cell banks, and LNP-mRNA formulation. Each gene was screened at n = 3, and the reduced screen size (three plates total) enabled the addition of a high-dose LNP-mRNA control (40 ng per well) to be used as a positive control (
Fig. 4A
). Data were normalized to dispersed NTC controls per plate, and the activity threshold was defined as 2 SD from the mean (
Suppl. Fig. 3
). Population analysis showed a clear distinction between the 20 and 40 ng LNP-mRNA dose with 2 SD hit thresholds (dotted lines), effectively dividing these populations in both the total and mean expression per cell endpoints (
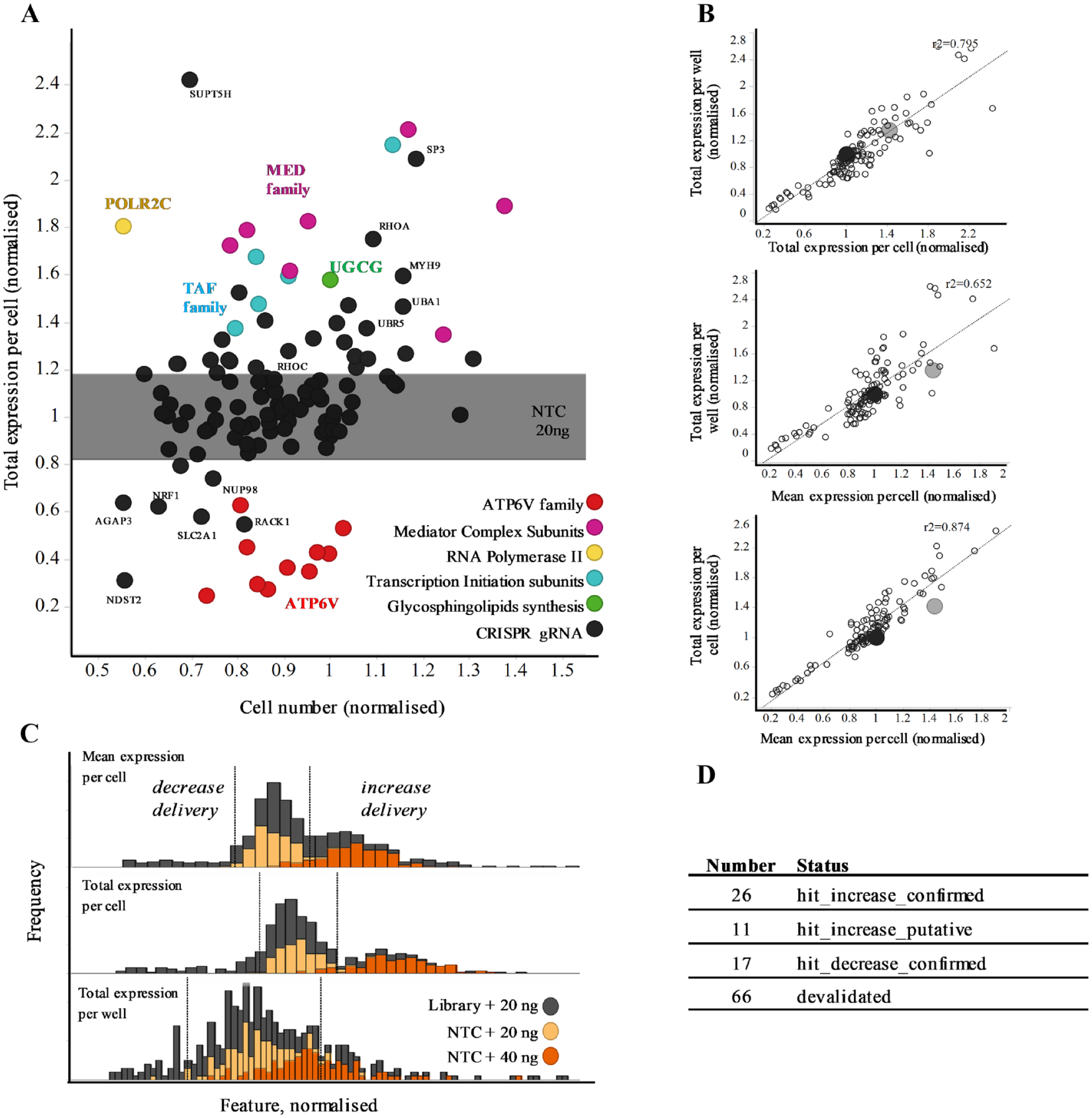
Hit confirmation. (
Network and Gene Ontology Analysis of Confirmed Hits
The confirmed hit gene list was taken into STRING v11 for network analysis, with hits that increased or decreased productive LNP-mRNA delivery analyzed independently. For genes whose loss increased productive LNP-mRNA delivery, four network clusters were identified with high confidence (>0.7; Fig. 5A ). Two clusters dominated the network analysis, and both were centered on host cell transcription: the Mediator complex (MED), a complex that functions as a transcriptional co-activator, and TATA-box binding protein associated factors (TAF). GO analysis supported this dominance, with 8 of the 19 biological processes with a false discovery rate (FDR) of p < 5 × 10−5 relating to this host cell transcription ( Suppl. Fig. S4 ). A small but statistically relevant cluster, associated with the protein ubiquitination pathway (FDR, p = 2.03 × 10−5), contained a mixture of E3 ligases (CUL1, UBR5, and UBA1) and associated proteins (SKP1). Finally, an additional cluster mapped with high significance to the Reactome term “RHO GTPase Effectors” (p = 1.2 × 10−4) and contained genes clearly related to cytoskeletal organization and intracellular trafficking, including RhoA and RhoC. Interestingly, beta-Catenin (CTNNB1) was a central node to three out of four clusters. The significance of this finding is still unclear but could reflect the key role that this protein plays in regulating the Wnt signaling pathway.
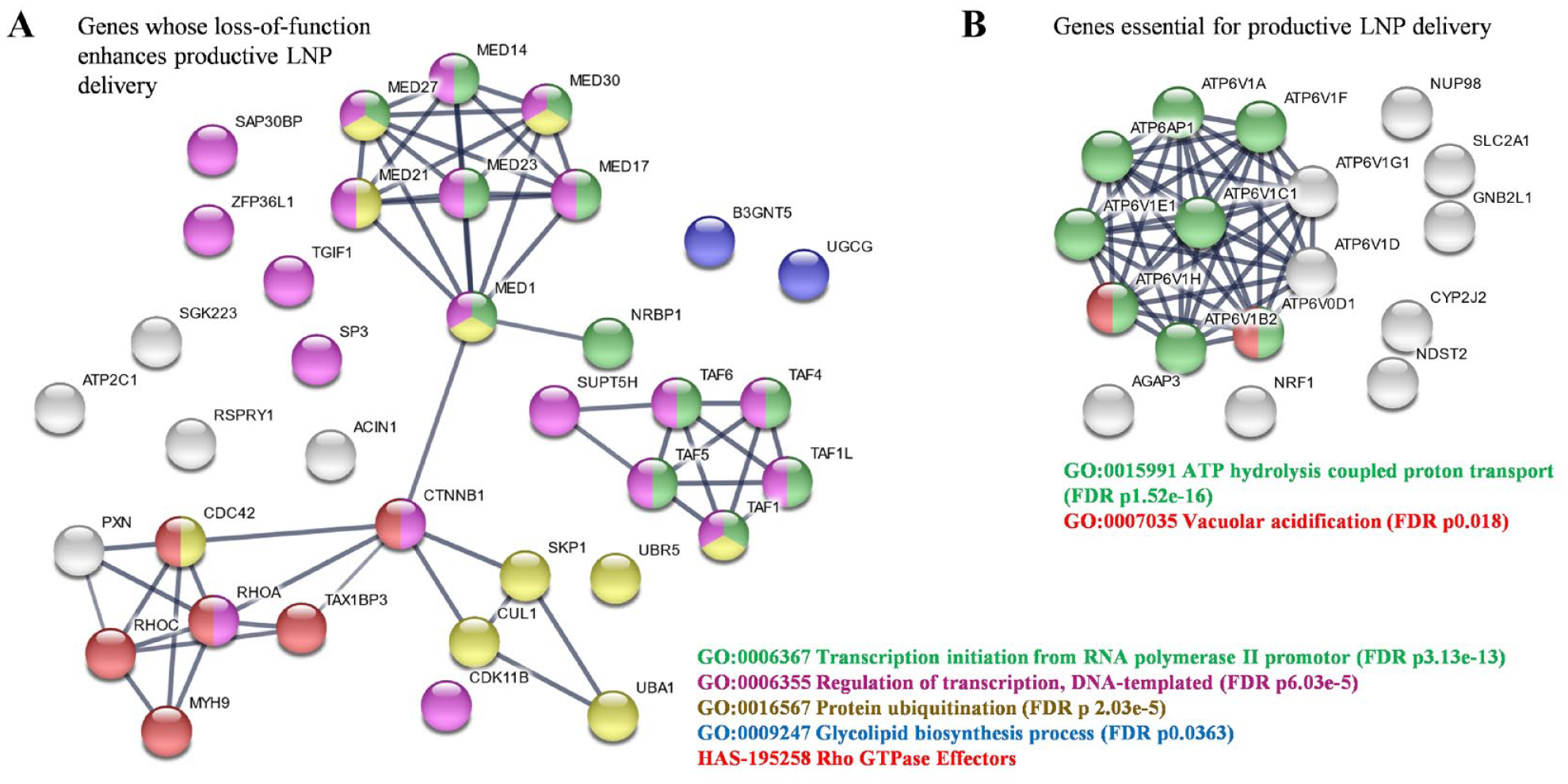
STRING pathway analysis of confirmed hits. Confirmed hits were processed through STRINGv11 for network and Gene Ontology analysis, with increased and decreased hit lists analyzed independently, both using the background of druggable genome for statistical relevance. (
Although not clustered into a network, two functionally related proteins were grouped by the GO term “glycolipid biosynthesis”: UGCG and UDP-GlcNAc:BetaGal Beta-1,3-N-acetylglucosaminyltransferase (B3GNT5). These two proteins play central roles in the glycosphingolipid (GSL) synthetic pathway, a group of membrane lipids that cluster on small, intracellular “lipid rafts.” 27 As UGCG catalyzes the first step in GSL synthesis, and because membrane lipid composition has a pivotal role in both vesicle recognition and intracellular trafficking, we decided to focus on further validating the role of UGCG in LNP-mRNA delivery.
For genes whose loss decreased LNP-mRNA delivery, the network analysis was simpler, with a dominance of genes involved in ATP hydrolysis-coupled proton transport related to vesicle acidification forming a discrete cluster ( Fig. 5B ). A total of 10 subunits of the V-type proton ATPase subunit (ATP6V) were identified to near abrogate the delivery of LNP-mRNA. As acidification of endosomes is an essential step for maturation and processing through the endocytic compartments, the identification of genes belonging to the proton pump that are essential for acidification of endosomes lends validity to these findings. Other hits such as the Receptor for Activated C Kinase 1 (RACK1/GNB2L1) gene also strongly inhibited productive delivery, as well as the N-deacetylase and N-sulfotransferase 2 (NDST2) and Solute Carrier Family 2 member 1 (SLC2A1), all through pathways likely independent of vesicle acidification.
Validation of UGCG and ATP6V as Key Factors in Mediating Productive LNP-mRNA Delivery
To build confidence in UGCG as a gene that enhances the delivery of LNP-mRNA, we deconvoluted the gRNA pool into the four constituent crRNAs and tested them singularly alongside siRNA. Each of the four cr:tracrRNA, which targeted independent sites within the coding region of the gene, resulted in enhanced delivery of LNP-mRNA. Consistent with previous observations, the strongest guide within the pool has a dominant effect, and pooling does not have an additive or synergistic effect ( Fig. 6A ). In comparison, siRNA-mediated knockdown of UGCG provided a comparable effect to gene knockout by CRISPR; however, the siRNA was less well tolerated and resulted in a slight reduction in cell number. Counterscreening the same panel of crRNAs and siRNA in NCI-H358-WT (i.e., no Cas9) resulted in only an enhancement of productive mRNA delivery with siRNA treatment. This confirmed that the phenotype observed in the screen was a result of effective gene editing ( Suppl. Fig. S3 ).
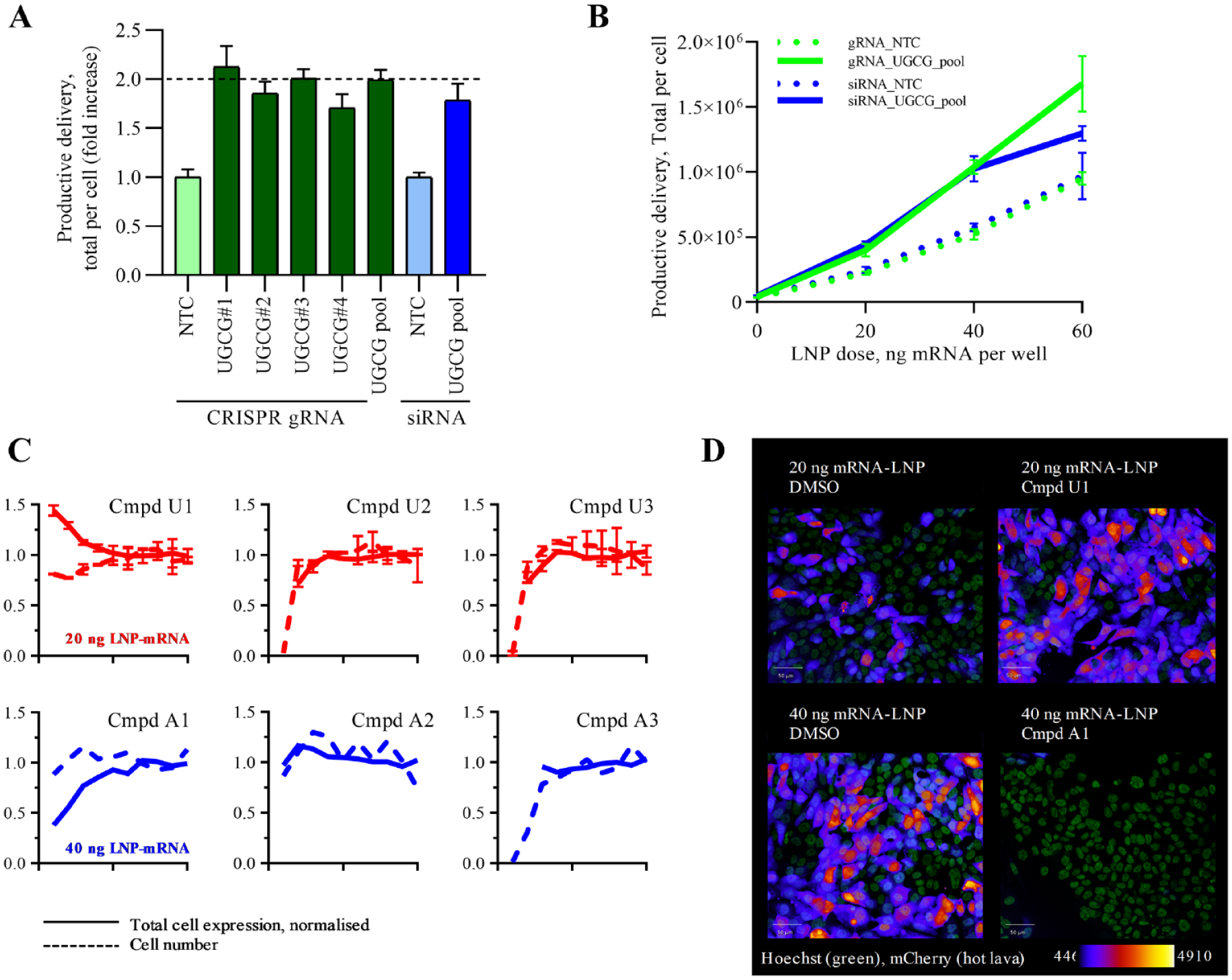
UDP-glucose ceramide glucosyltransferase (UGCG) and V-type proton ATPase validation (ATP6V). (A) Reverse transfection of individual cr:tracrRNA and small interfering RNA into NCI-H358 (wild-type and Cas9) cells and at 96 h addition of 20 ng lipid nanoparticle–encapsulated messenger RNA (LNP-mRNA) and productive delivery quantified at 120 h. (
We have shown that at between 10 and 60 ng LNP-mRNA per 384-well, there is a linear relationship with the measured total protein expression ( Fig. 2H ). To understand whether cells retained this linear relationship following the loss of UGCG, we performed dose-response experiments in cells in which the UGCG gene was knocked out using CRISPR or knocked down using siRNA. These experiments show that at each dose tested, an approximately twofold increase greater than control was observed for the measured total protein expression per cell ( Fig. 6B ).
To further assess the validity of our findings, we wanted to confirm whether the enhancement of LNP-mRNA delivery caused by the genetic deletion of the UGCG gene could be recapitulated by small-molecule inhibition. We queried internal compound databases and identified three UGCG small-molecule inhibitors (U1, U2, and U3). In addition, we wanted to understand from a mechanistic perspective whether we could block delivery with chemical inhibition of ATP6V and so also selected three ATP6V inhibitors (A1, A2, and A3). All compounds were profiled as 10-point dose response by co-dosing along with LNP-mRNA in the standard assay format. Compound U1 successfully elicited a 1.5-fold increase in LNP-mRNA delivery without significant effects on cell health ( Fig. 6C ). Two other UGCG compounds, U2 and U3, showed significant cytotoxicity at high concentrations. Because our screening data indicate that cells can tolerate complete genetic ablation of UGCG gene, we argue that U2 and U3 compounds might have off-target effects resulting in toxicity, especially considering the relatively high concentrations at which toxicity was observed.
Compound A1 resulted in a significant block of productive LNP-mRNA delivery, with little effect on the morphology of the cell nuclei ( Fig. 6D ). Compound A2 was inactive and A3 was toxic, likely for reasons explained previously for U2 and U3 compounds.
To assess whether UGCG elicits a pleiotropic effect on core cellular processes rather than a more selective effect on productive LNP-mRNA delivery, we preformed siRNA knockdown of this gene in a series of internal assays looking at endocytosis, protein synthesis, and cell metabolism. We did not observe any significant effect of UGCG knockdown on these cellular processes (data not shown), supporting the hypothesis that UGCG plays a key role specifically in productive LNP-mRNA delivery.
Discussion
We demonstrate here the development, validation, and application of the first druggable genome (7795 genes) scale arrayed CRISPR platform, which we have used to identify genes that increase and decrease the productive delivery of LNP-mRNA. The screen identified 130 putative hits that were subsequently taken into hit confirmation, in which a total of 44 genes were validated as having a significant effect on either increasing (37 genes) or decreasing (17 genes) the productive delivery of the LNP-mRNA. Network analysis (STRING) of these validated hits built further confidence in the robustness of the screen as greater than 60% of confirmed hits mapped to a small number of highly interconnected clusters in a network filtered for high confidence interactions only. Eighty-six targets were unsurprisingly devalidated, as we had initially used inclusive hit thresholds to ensure we did not wrongly exclude false-negatives, which might have elicited only a modest phenotype.
A significant number of genes that increased delivery centered on pathways involved with host cell transcription (e.g., MED and TAF families) and even included RNA polymerase II itself (POLR2C). Whether this effect is due to direct modulation of the expression of genes involved with LNP delivery or to other indirect mechanisms remains to be established. From a therapeutic perspective, these genes would be more challenging to target via small molecules and more likely to represent a safety liability, and so they were not characterized further.
The knockout of genes in the protein ubiquitination pathway improved productive delivery of LNP-mRNA (e.g., CUL1; see Fig. 5A ). This is likely to result from reduced degradation of the mCherry protein produced from the mRNA cargo, rather than a direct effect on enhancing the uptake and release of LNP. However, it remains to be investigated whether these pathways are specific to the production of the mRNA cargo (mCherry mRNA in this case).
Genes from the Wnt signaling pathway were identified as having roles in both the enhancement and inhibition of productive LNP-mRNA delivery. Here, the knockout of beta-catenin enhanced LNP delivery, suggesting that Wnt signaling is not favorable to delivery. In corroboration of this, the knockout of GNB2L1/RACK1, a known suppressor of the Wnt pathway, was shown to significantly inhibit productive delivery. This is evidence of hit detection in the primary pathway (beta-catenin) driving one phenotype, as well as negative regulators of the same pathway (GNB2L1) driving the inverse phenotype.
One of the most interesting hits identified was UGCG. This enzyme catalyses the first step of glycosphingolipid biosynthesis synthesis, a family of lipids that play a key role in mediating membrane trafficking and signal transduction. Here, most excitingly, we could reproduce this phenotype with a small-molecule inhibitor, U1, co-dosed at the same time as the LNP-mRNA. The confirmation that small-molecule inhibition of UGCG also promotes LNP-mRNA delivery is important. It means this phenotype does not manifest as the result of cellular adaptation in response to chronic depletion of the UGCG protein and subsequent depletion of GSLs; rather, it suggests an acute loss of UGCG catalytic activity driving this phenotype. This is critically important from a therapeutic perspective, as LNP-mRNA would be co-dosed to patients with small-molecule enhancers to improve localized delivery.24,25
We also characterized small-molecule inhibitors of ATP6V and confirmed that these compounds can ablate productive cellular uptake of LNP-mRNA. The endosomal trapping of reagents such as LNP-delivered macromolecules is well known, and so it is likely that blocking the acidification of endosomes and stopping their maturation through the endocytic pathway is not favorable to delivery. 2 Although not relevant for therapeutic intervention, this finding presents an opportunity to screen panels of different LNP chemistry in the presence of compound A1 to identify those that are not dependent on vesicle acidification, thus offering the potential to identify novel LNPs with reduced propensity to be trapped inside endosomes.
To our knowledge, this is the first application of druggable-scale arrayed CRISPR screening to increase our understanding of mechanisms modulating productive LNP-mRNA delivery.
CRISPR screening is not without limitations, and concerns over off-target activity remain and reinforce why a robust target validation cascade with different modalities is an essential part of any target discovery project. Nevertheless, our data show the unprecedented power of functional genomics and CRISPR screening technology to shed new light on new biological pathways. To understand the therapeutic relevance of translating these findings into an in vivo setting is an essential next step.
Supplemental Material
Supplementry_figures – Supplemental material for Arrayed CRISPR Screening Identifies Novel Targets That Enhance the Productive Delivery of mRNA by MC3-Based Lipid Nanoparticles
Supplemental material, Supplementry_figures for Arrayed CRISPR Screening Identifies Novel Targets That Enhance the Productive Delivery of mRNA by MC3-Based Lipid Nanoparticles by Douglas Ross-Thriepland, Aurelie Bornot, Larissa Butler, Arpan Desai, Himjyot Jaiswal, Samantha Peel, Morag Rose Hunter, Uchechukwu Odunze, Beverley Isherwood and Davide Gianni in SLAS Discovery
Supplemental Material
Supplementry_table_and_methods – Supplemental material for Arrayed CRISPR Screening Identifies Novel Targets That Enhance the Productive Delivery of mRNA by MC3-Based Lipid Nanoparticles
Supplemental material, Supplementry_table_and_methods for Arrayed CRISPR Screening Identifies Novel Targets That Enhance the Productive Delivery of mRNA by MC3-Based Lipid Nanoparticles by Douglas Ross-Thriepland, Aurelie Bornot, Larissa Butler, Arpan Desai, Himjyot Jaiswal, Samantha Peel, Morag Rose Hunter, Uchechukwu Odunze, Beverley Isherwood and Davide Gianni in SLAS Discovery
Footnotes
Acknowledgements
We would like to thank Martin Booth for automation support, Marcello Maresca for gene editing advice, and Jenny Bradley and Anne Goeppert for help with cell line generation.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors were employed by AstraZeneca whilst performing the work for this paper.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.