
Abstract
Kidney fibrosis presents a hallmark of chronic kidney disease. With ever-increasing patient numbers and limited treatment options available, novel strategies for therapeutic intervention in kidney disease are warranted. Fibrosis commonly results from a wound healing response to repeated or chronic tissue damage, irrespective of the underlying etiology, and can occur in virtually any solid organ or tissue. In order to identify targets relevant for kidney fibrosis, we aimed to employ CRISPR screening in primary human kidney fibroblasts. We demonstrate that CRISPR technology can be applied in primary kidney fibroblasts and can furthermore be used to conduct arrayed CRISPR screening using a high-content imaging readout in a whole genome-wide manner. Hits coming out of this screen were validated using orthogonal approaches and present starting points for validation of novel targets relevant to kidney disease.
Introduction
Genetic screens present an unbiased approach to identify factors contributing to biological phenotypes and, with this, the opportunity to identify novel targets for therapeutic intervention.1–3 CRISPR technology especially holds promise in this context compared with RNAi technology since CRISPR has the power to generate real knockouts as compared to variable knockdowns, hence overcoming one of the hurdles of RNAi technology.4–7 Especially the combination of CRISPR with lentiviral delivery has proven to be a powerful screening tool.2,8–11 Delivery of CRISPR components using lentivirus allows the targeting of proliferating and nonproliferating cells, selection for transfected cells, and long-lasting activity of the CRISPR system, thereby maximizing the chance of target editing. Additionally, the integrated lentivirus genome allows the use of pooled screening setups. In pooled screens, a population of cells is infected with a pooled library and the integrated lentivirus genome allows pool deconvolution using next-generation sequencing (NGS) after the experiment. 1 However, while beneficial in terms of library generation and experimental setup, pooled screening approaches are limited in terms of assay readout. Arrayed libraries, on the other hand, allow the combination of CRISPR screening with a vast array of readout options, such as high-content imaging, time-resolved fluorescence energy transfer (TR-FRET), or quantitative PCR (qPCR). 12 In addition, with respect to their basic setup, arrayed CRISPR screens are comparable to phenotypic screens and assays developed for phenotypic screening can be adapted for genetic screens and vice versa. A combination of both screening approaches could therefore maximize the use of a disease-relevant assay for drug discovery.
Here we aimed to evaluate arrayed CRISPR screening in a high-content-based assay developed to identify novel regulators of kidney fibrosis. Organ fibrosis represents a multifactorial condition that can affect virtually every solid organ.13,14 Independent of the source of damage, activation of the resident tissue fibroblasts and their transition into myofibroblasts (pericyte-to-myoblast transition [PMT]) resulting in excessive generation of extracellular matrix is a common feature of organ fibrosis. 15 Targeting the activation of tissue resident fibroblasts and their progression to a myofibroblast phenotype has thus been an opportunity for therapeutic intervention. Primary cells, challenged by an agent relevant for the disease, in combination with a readout reminiscent of a disease manifestation present an attractive system for pharmacological and genetic target identification.16,17 Here we describe the development of a fibrosis-relevant high-content assay in primary human kidney fibroblasts (HKFs) that we combined with CRISPR technology for a whole genome-wide genetic hit identification campaign for novel targets in kidney fibrosis.
Material and Methods
Reagents
Blasticidin (R21001), puromycin (A11138-03), 4× LDS sample buffer (NP0008/7), 10× sample reducing agent (NP0004/9), NuPAGE 4%–12% Bis-Tris Plus gels 10well (NW04120BOX), NuPAGE antioxidant (NP0005), 20× NuPAGE transfer buffer (NP0006), Alexa Fluor 488 goat anti-mouse IgG2a (A21131), and Alexa Fluor 488 goat anti-mouse IgG (H+L) (A11029) were purchased from Thermo Fisher Scientific. (Fisher Scientific GmbH, Schwerte, Germany) Triton-X100, mouse monoclonal anti-alpha-smooth muscle actin (anti-aSMA; clone 1A4), mouse monoclonal anti-collagen type I (C2456), saponin from Quillaja bark (S4521), albumin human (A9731), albumin fraction V (1120180100), and transforming growth factor-β1 (TGF-1) human (A7481) were purchased from Sigma. Design rhodamine phalloidin (PHDR1) was obtained from Cytoskeleton (tebu-bio, Offenbach, Germany). DRAQ5 (DR51000) was obtained from Biostatus (Shepshed, UK). Fasudil hydrochloride (0541/10), GSK 269962 (4009/10), imatinib mesylate (5906/100), and SB431542 (1614/10) were purchased from Bio-Techne GmbH (Wiesbaden-Nordenstadt, Germany). Dasatinib (S1021), nintedanib (S1010), bortezomib (S1013), and carfilzomib (S2853) were purchased from Selleckchem (München, Germany). Polybrene (TR-1003-G) was obtained from Merck (Darmstadt, Germany). Normal goat serum (S-1000) was obtained from Vector Labs (Biozol Diagnostica, Eching, Germany). IRDye 680RD conjugated goat anti-mouse IgG (926-68070) and IRDye 800CW conjugated goat anti-rabbit IgG (926-32211) was obtained from LI-COR (Bad Homburg, Germany). GAPDH (14C10) rabbit monoclonal antibody (mAb; 2118) and Cas9 (7A9-3A3) mouse mAb was obtained from Cell Signaling Technology (Leiden, Netherlands). BioCoat Collagen I 384-well microplates and purified mouse anti-Smad2/3 clone 18 (610842) were from Becton Dickinson (Heidelberg, Germany). MKL1/MRTF-A mouse mAb (sc-390324) was from Santa-Cruz.
Lentivirus Constructs and Screening Library
Cas9 lentivirus (LentiArray Cas9 Lentivirus [A32064]) was from Thermo Fisher Scientific. gRNA lentivirus was ordered from Sigma as QuickPick KO gRNAs with two different gRNAs/means per target gene, and for hit follow-up activities two additional gRNAs were chosen: The “Guaranteed Predesigned gRNA” panel from Sigma is based on the predicted efficiency score. Designs for QuickPick clones were taken from the Sanger Arrayed CRISPR library (Sigma), an arrayed lentivirus gRNA library encompassing the whole genome with two different gRNAs/means per target provided in two separate wells of a 384-well plate. 12 The Sanger Arrayed library was also used for genome-wide screening. Titers of the used lentivirus were determined using a p24-based enzyme-linked immunosorbent assay (ELISA) at the vendor and multiplicities of infection (MOIs) in assays were calculated as the fraction of added transduction units (TU) per cell in the well during infection. For the Sanger library, p24 titer information is given for 20 representative wells/384-well plate; in total, 5.5% (n = 2040) of the lentivirus contained in the library p24 titer is provided.
Cell Culture
HKFs (ASE-5213) were purchased from CellSystems (Troisdorf, Germany). Cells were maintained at 37 °C in a humidified 5% CO2 atmosphere and cultured in FibroLife S2 Medium Complete Medium (LL-0011) from CellSystems (Troisdorf, Germany) containing 100 IU/mL penicillin and 100 µg/mL streptomycin (15140-122) from Thermo Fisher Scientific.
Human pulmonary fibroblasts (HPFs; C12360) were purchased from PromoCell (Heidelberg, Germany). Cells were maintained at 37 °C in a humidified 5% CO2 atmosphere in Dulbecco’s modified Eagle’s medium (DMEM)/Ham’s F-12 (1:1) with 2.5 mM
Normal rat kidney (NRK-49f) cells (CRL 1570) were purchased from ATCC (Wesel, Germany). Cells were maintained at 37 °C in a humidified 5% CO2 atmosphere in DMEM (30-2002) from ATCC, 10% FCS, and 100 IU/mL penicillin and 100 µg/mL streptomycin.
PMT Assays
For PMT in HKFs, cells were seeded into BioCoat Collagen I 384-well microplates at a density of 4000 cells/well in FibroLife S2 medium complete medium and left overnight at 37 °C in a humidified 5% CO2 atmosphere. The following day, the media was removed and the cells were washed two times in an assay media consisting of DMEM/Ham’s F-12 (1:1), with 2.5 mM
To assess PMT in HPFs, these were seeded into BioCoat Collagen I 384-well microplates at a density of 2500 cells/well in an assay medium (DMEM/Ham’s F-12 (1:1), with 2.5 mM
For experiments with NRK-49f cells, the cells were seeded into BioCoat Collagen I 384-well microplates at a density of 4500 cells/well in an assay medium (DMEM, 0.3% FCS, and 100 IU/mL penicillin and 100 µg/mL streptomycin). Plates were incubated overnight at 37 °C in a humidified 5% CO2 atmosphere. The following day, compound treatment was done as described above and cells were stimulated by the addition of TGF-β at a final assay concentration of 3 ng/mL (EC80), and the plates were incubated for 48 h at 37 °C in a humidified 5% CO2 atmosphere prior to fixation and staining.
Immunofluorescence Staining
To stain for aSMA and Cas9, the media on the cells was discarded by inversion of the plate and cells were fixed for 20 min with 4% paraformaldehyde (PFA) solution in phosphate-buffered saline (PBS). After fixation, plates were washed three times with PBS and blocked by the addition of a blocking buffer (3% v/v normal goat serum, 2% w/v albumin fraction V, and 0.1% saponin in PBS) for 30 min at room temperature. The blocking buffer was discarded by inversion of the plate and the plates were incubated with anti-aSMA antibody (4 µg/mL) or anti-Cas9 antibody (5 µg/mL) in blocking buffer for 1 h at 37 °C. Plates were washed three times in PBS and incubated for a further 1 h at room temperature with Alexa Fluor 488 goat anti-mouse IgG2a (1 µg/mL) and rhodamine phalloidin (28 nM) in blocking buffer. Plates were washed three times in PBS before the addition of Draq5 (10 µM in PBS). Plates were sealed and images were acquired on a PerkinElmer Opera QEHS imaging device using a 10× air objective for aSMA and 20× air objective for Cas9. Per sample, 5 fields of view were acquired with 2 × 2 binning.
To stain for SMAD2/3, plates were fixed with 4% PFA solution in PBS and then blocked in a blocking buffer (5% v/v normal goat serum and 0.2% v/v Triton X-100 in PBS) for 1 h at room temperature. Plates were washed three times with PBS and anti-SMAD2/3 mAb was added (1.25 µg/mL) in antibody dilution buffer (5% v/v normal goat serum in PBS), and the plates were incubated for 2 h at room temperature. Plates were washed three times in PBS and Alexa Fluor 488 goat anti-mouse IgG (H+L) in antibody dilution buffer was added (1 µg/mL), and the plates were incubated for 1 h at room temperature. Plates were washed three times in PBS before the addition of Draq5 (10 µM in PBS). Plates were sealed and images were acquired on a PerkinElmer Opera QEHS imaging device as described above.
To assess collagen I staining, the media was discarded by inversion of the plate and cells were fixed for 10 min with methanol at 4 °C. After fixation, plates were washed three times with PBS and blocked by the addition of a blocking buffer (6.25% v/v FCS in PBS) for 30 min at room temperature. The blocking buffer was discarded by inversion of the plate and the plates were incubated with anti-collagen type I antibody (4 µg/mL) in blocking buffer for 2 h at room temperature. Plates were washed three times in PBS and incubated for a further 1 h at room temperature with Alexa Fluor 568 goat anti-mouse IgG1 (y1) (1 µg/mL) in blocking buffer. Plates were washed three times in PBS before the addition of Draq5 (10 µM in PBS). Plates were sealed and images were acquired on a PerkinElmer Opera QEHS imaging device as described above.
Image Analysis
Once the images were acquired on a PerkinElmer Opera QEHS imaging device, the associated ACapella scripting environment was used to analyze the data. To quantify aSMA expression levels, DRAQ5 was used for cell nuclei detection and rhodamine-labeled phalloidin as an actin marker was used to estimate the cell area. Within the cytoplasmic area, which was calculated as the cell region minus the nucleus region, the aSMA intensity was determined per cell (
To detect collagen I positive cells, a similar approach to that for aSMA was used. For the collagen assay nuclei detection was based on the DRAQ5 signal, which was also used for cell outline detection. In cases where cytoplasmic DRAQ5 staining was not above background, a ring-like region around the nucleus was used as a substitution to determine the cytoplasmitic region. Similar to the background normalization done for the aSMA readout, the collagen I intensity estimated within the cytoplasm was normalized against the mean background intensity. Per cell, an intensity ratio was calculated by dividing the cytoplasmic collagen I intensity by the average collagen I intensity of the background. Cells were selected as positive if the intensity ratio was ≥3 (
To assess SMAD2/3 translocation, DRAQ5 was used for nuclei detection and cell outline estimation. SMAD2/3 intensity was determined within the nucleus and cytoplasm region separately and used to calculate a SMAD2/3 intensity ratio. Here, SMAD2/3 intensity within the nucleus was divided by SMAD2/3 intensity within the cytoplasm region. The ratio was calculated per cell and then averaged per field and per well. If the ratio was above a threshold of 3, a cell would be counted as positive for SMAD2/3 translocation to the nucleus (
Western Blotting
Samples were combined with LDS sample buffer and reducing agent and heated for 10 min at 95 °C. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) was done using NuPAGE 4%–12% Bis-Tris gels and transferred to nitrocellulose using the XCell II Blot Module (Thermo Fisher Scientific). Membranes were blocked in LI-COR blocking buffer and then incubated with the primary antibodies overnight. Primary antibodies were used at 1:250 (anti-Cas9), 1:500 (anti-MKL1), and 1:1000 (anti-GAPDH) in Tris-buffered saline with Tween 20 (TBST). After washing with TBST (three times for 10 min), blots were incubated with the appropriate IRDye-labeled secondary antibodies for 2 h and washed again as above before visualization using Odyssey infrared imaging (LI-COR Biosciences).
Transcriptomics Analysis
HKFs (64,000 cells/well) and HPFs (40,000 cells/well) were seeded into 24-well tissue culture plates in their appropriate cell culture media and overnight at 37 °C in a humidified 5% CO2 atmosphere. The following day, the cells were washed two times in assay media and a final volume of assay media was added and the plates were left overnight at 37 °C in a humidified 5% CO2 atmosphere. The following day, cells were stimulated by the addition of TGF-β at EC80, and the plates were incubated for 4 h at 37 °C in a humidified 5% CO2 atmosphere prior to RNA isolation using the Qiagen Rneasy Mini Kit. RNA sequencing libraries were generated using the Illumina TruSeq Stranded mRNA Library Preparation kit and run on an Illumina Hiseq2500 device using 1 × 50 bp single-end reads. RNAseq reads were aligned to the human reference genome (GRCh38) using STAR (v2.5.3a).18,19 Reads per gene were counted using the “—quantMode GeneCounts” option of STAR aligner, using Ensembl 91 genome annotation as a reference. Counts were log normalized in R (v3.4.1) and a principal component analysis (PCA) was constructed based on 500 genes with the highest variance of expression level using the R function prcomp.
HKF Cas9 Cell Line Generation
HKFs at early passage were seeded into 12-well plates at a density of 100,000 cells/well in FibroLife S2 Medium Complete Medium and left overnight at 37 °C in a humidified 5% CO2 atmosphere. LentiArray Cas9 Lentivirus was added in the presence of 8 µg/mL polybrene at a final MOI of 5. The plate was spun at 930g, 30 °C for 1 h and then incubated overnight at 37 °C in a humidified 5% CO2 atmosphere. Selection was done using blasticidin at 5 µg/mL until noninfected control well cells were dead. Cells were expanded for characterization and storage.
CRISPR PMT Assay
HKF-Cas9 cells were seeded into BioCoat Collagen I 384-well microplates at a density of 750 cells/well in FibroLife S2 Medium Complete Medium and left overnight at 37 °C in a humidified 5% CO2 atmosphere. The following day, the cells were spin infected at 930g/30 °C with gRNA lentivirus at different MOIs in the presence of polybrene (8 µg/mL) for 1 h. Plates were incubated overnight at 37 °C in a humidified 5% CO2 atmosphere. The following day, the media was removed and fresh media was added containing puromycin (0.6 µg/mL), and the cells were incubated for another 5 days at 37 °C in a humidified 5% CO2 atmosphere. The media was removed and the cells were washed two times in assay media and incubated overnight at 37 °C in a humidified 5% CO2 atmosphere with a final addition of assay media. The following day, cells were stimulated with TGF-β at a final assay concentration of 1 ng/mL for 48 h at 37 °C in a humidified 5% CO2 atmosphere and further processed as described above. To assess for differences in SMAD2/3 translocation, cells were treated as described above; however, treatment following starvation was only for 6 h.
Whole Genome-Wide Screening Assay
Due to the liability of lentivirus to freeze and thaw cycles, two screening-ready intermediate plates were generated from each stock plate (containing 20 µL of 10,000 TU/µL average per construct) prior to screening. To this end, 15 µL of cell culture media was added in an intermediate plate (Multidrop) and 2 µL of lentivirus from each stock plate was added using a CyBi Selma with a 384-well head (Analytik Jena). Plates were stored at −80 °C until needed. One day prior to screening, HKF-Cas9 cells were seeded into BioCoat Collagen I 384-well microplates at a density of 750 cells/well in 30 µL of FibroLife S2 Medium Complete Medium (Multidrop) and left overnight at 37 °C in a humidified 5% CO2 atmosphere. The following day, 5 µL of 8× polybrene medium (8 µg/mL final) was added onto the cells (Multidrop), followed by transfer of 6 µL of virus supernatant from intermediate plates using a CyBi Selma with a 384-well head (Analytik Jena). With the titer information available for the library we calculated the resulting MOI during infection.
Data Analysis
Z′ factors and signal-to-background (S/B) ratios were calculated as described. 20 For pathway analysis, gProfiler 21 was used with the following parameter: Statistical Domain scope=All known genes; Significance Threshold=Benjamini-Hochberg FDR; User Threshold=0.05. Enrichment analysis focused on Wikipathways. Error bars show the standard deviation of technical replicates (≥3). EC50 and IC50 values were determined using the Prism software nonlinear regression and a four-parameter fit (GraphPad Prism Software, La Jolla, CA). RNAseq and image analysis were processed as described above. All experiments except primary screening, hit confirmation, and RNAseq analysis have been independently repeated at least once.
Results
Establishment of a High-Content Assay for Fibroblast Activation in Primary Kidney Fibroblasts
In order to establish a phenotypic cellular PMT assay, we focused our activities on using primary kidney fibroblasts, thus establishing a cellular system that employs the disease-affected cell type from the affected organ, as organ-specific differences in fibroblasts have been described. The induction of aSMA is a widely described marker for PMT, which can robustly be induced by treatment with TGF-β,22,23 a master inducer of fibrosis. As shown in
Figure 1A
, we were able to identify three different primary fibroblast lines that show low baseline levels of aSMA and induction upon TGF-β treatment. Following optimization of assay parameters such as cell density and treatment time, we were able to develop a robust assay for measuring the induction of aSMA following TGF-β treatment with comparable EC50 values for each of the three lines (
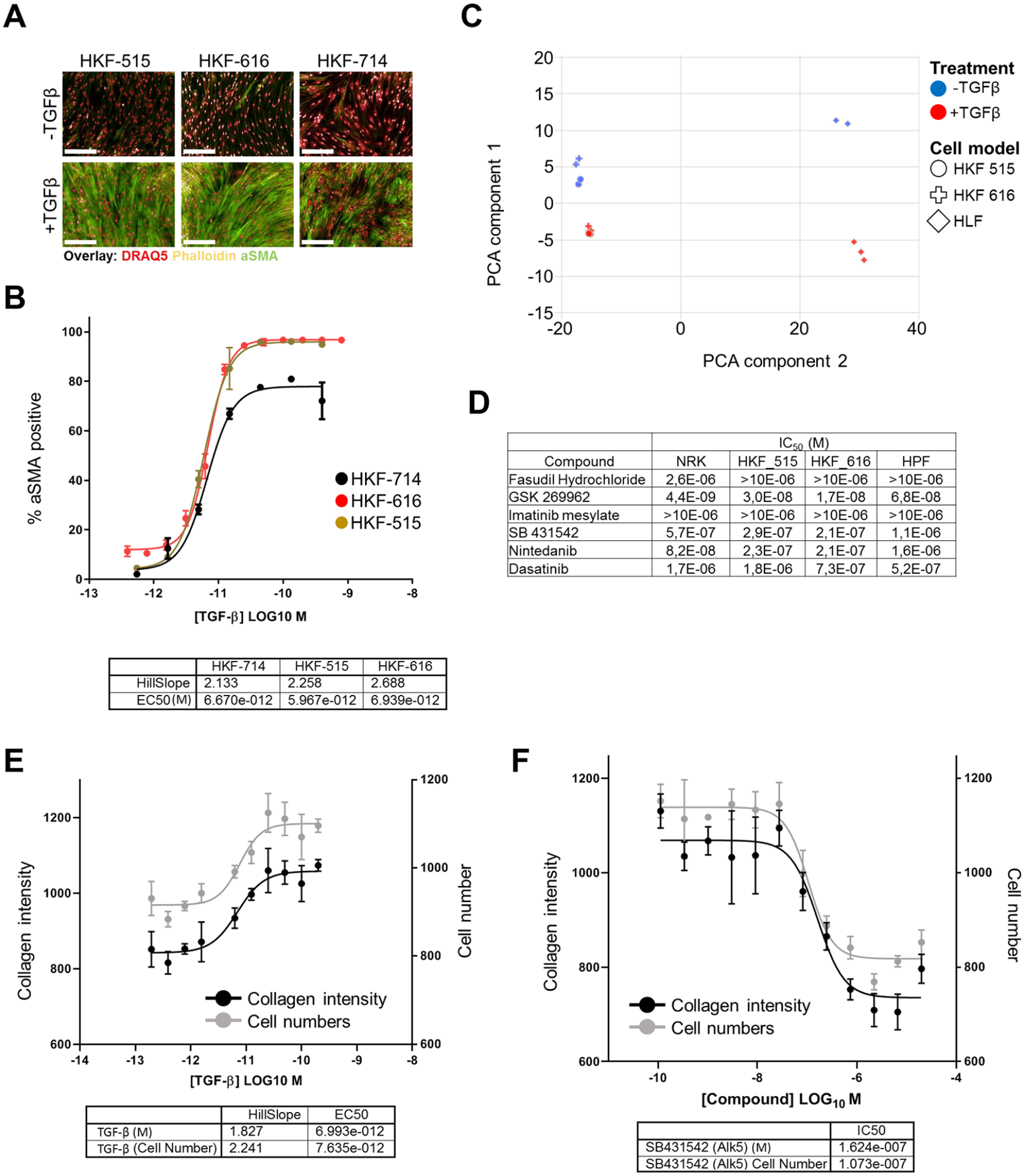
Development of a PMT assay in primary HKFs. (
Application of CRISPR Technology in Primary Kidney Fibroblast Model
Having identified two cell lines with robust responses to TGF-β stimulation, we tested whether these would be amenable to genetic manipulation using CRISPR technology. With the prospect of maximizing editing rates due to constitutive expression of the CRISPR components and the opportunity to select transfected cells, we focused on using a lentivirus-based CRISPR system for these tests. We derived stable Cas9-expressing cell pools from early passage cells and characterized these for Cas9 expression. As shown in
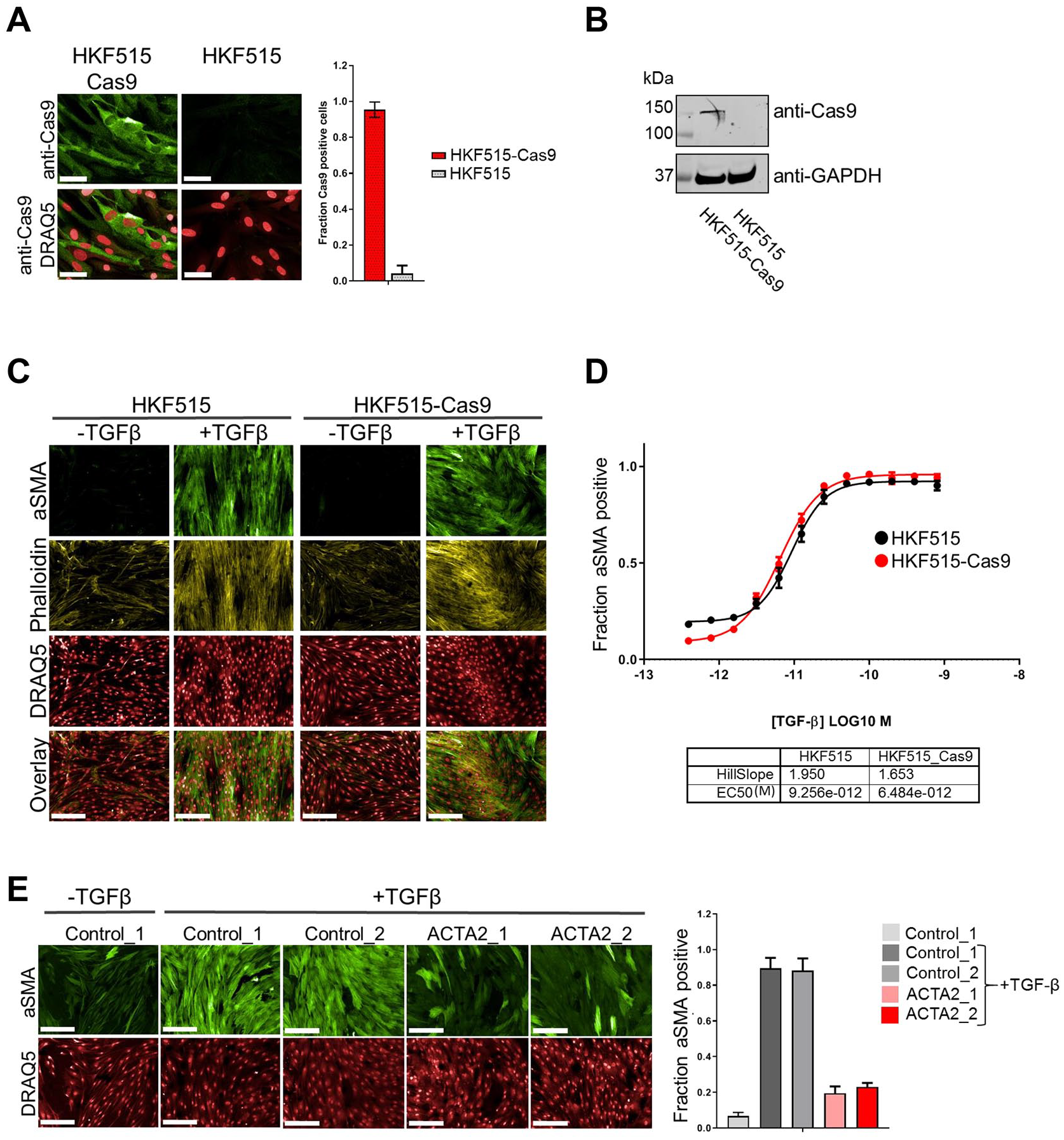
Application of CRISRP technology in primary HKFs. Cells were infected with Cas9 carrying lentivirus at p2, stable pools selected with blasticidin and expanded until p9 (HKF515-Cas9), alongside control cells without Cas9 that were expanded to the same passage (HKF515). (
Next, we tested whether CRISPR technology could be used to manipulate targets upstream of aSMA. We focused on the genes for TGF-β receptors (TGFBR1 and TGFBR2) as well as MKL1. While TGFBR1 and TGFBR2 mediate direct activation of the ACTA2 gene following TGF-β treatment via the SMAD pathway, MKL1 has been shown to function by integrating cytoskeletal cues into aSMA activation.22,23,27 As shown in Figure 3A , gRNAs targeting TGFBR1 and TGFBR2 are able to reduce aSMA induction, albeit to various degrees. Importantly, also targeting MKL1 reduces aSMA induction, thus indicating that more indirect regulatory inputs into the ACTA2 gene can also be identified using CRISPR in the PMT assay. Despite showing an effect in our PMT assay, targeting TGFBR1 and TGBFR2 did not completely block aSMA induction, even if targeting TGFBR1 and TGBR2 in combination (data not shown). TGF-β treatment induces cell proliferation and, as shown in Figure 3B , targeting of TGFBR1 and TGFBR2 effectively blocked TGF-β-induced proliferation, 26 while gRNAs against ACTA2 or MKL1 had no effect on this phenotype. This indicates that unedited cells can mask TGFBR1/2-targeted cells in assays with prolonged incubation time. We therefore also evaluated the effect of targeting TGFBR in a more proximal assay by testing for impact on TGF-β-induced translocation of SMAD2/3 into the nucleus. 28 As shown in Figure 3C , translocation of SMAD2/3 was impaired by ~50% by targeting TGFBRs, thus showing that despite comparably little effect on aSMA induction, TGFBR-induced pathways can still be impaired using CRISPR in this format. Taken together, these data show that CRISPR technology can be applied in primary HKFs and that it has the potential to efficiently manipulate ACTA2 as well as genes upstream of ACTA2, and that effects on aSMA can be measured in an imaging-based PMT assay.
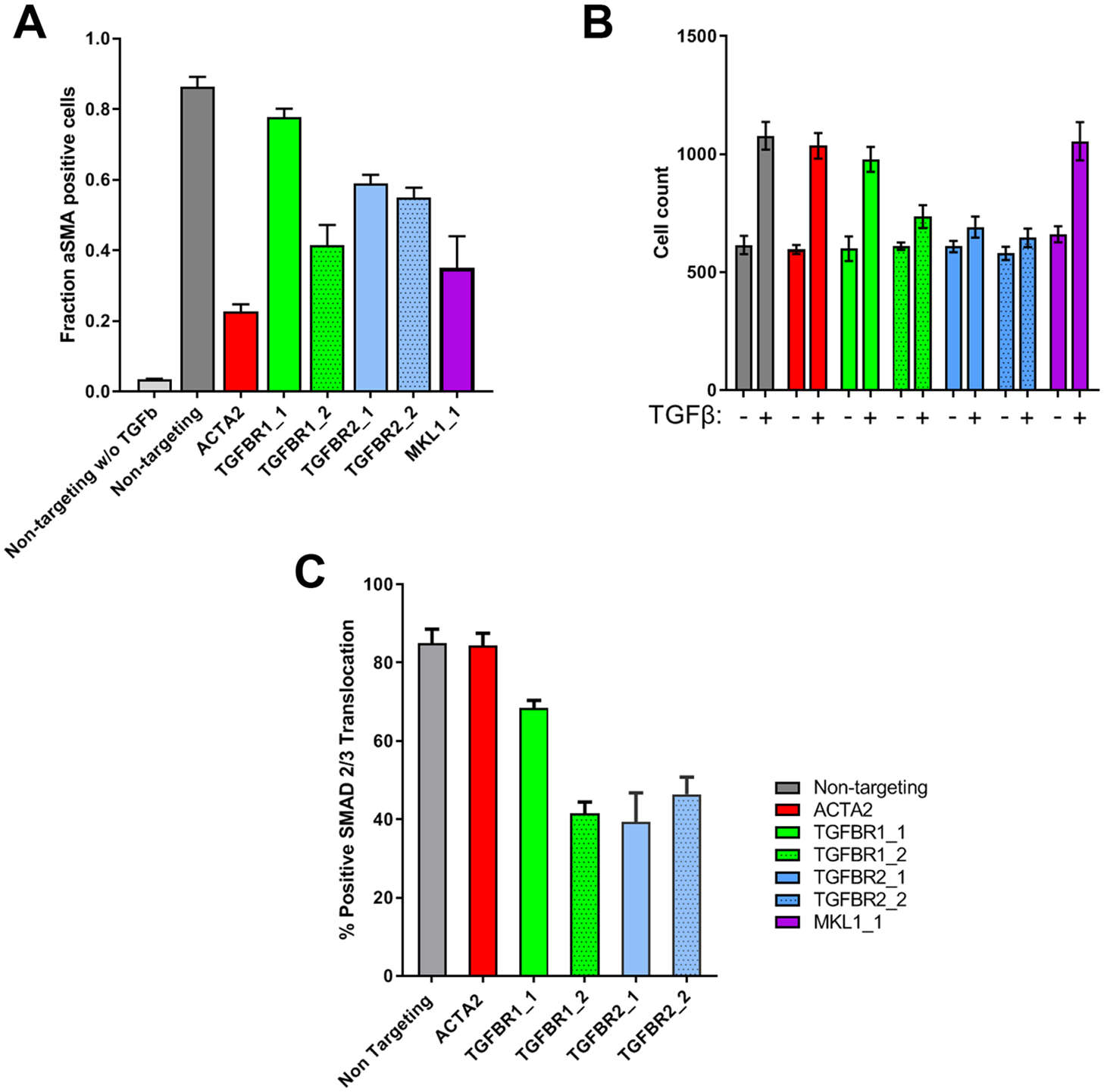
Characterization of CRISPR effectivity on PMT response in kidney fibroblasts. (
Setup of Large-Scale Screen and Primary Screen
Having confirmed that CRISPR technology can efficiently be applied in primary fibroblasts, we aimed to extend the assay to screening an arrayed library encompassing the whole genome.
12
While the general setup of an arrayed lentivirus-based CRISPR library is similar to other formats such as siRNA or small-molecule libraries, due to the production process the amount of active virus provided differs between wells.
12
Differing titers could pose a challenge for large-scale screening if the assay is affected by high amounts of virus. As shown in
Figure 4A
, we do observe an effect of high MOIs (>20) on the induction of aSMA following TGF-β treatment in primary kidney fibroblasts. This indicates that, if possible, the added amount of lentivirus should fall between MOIs of 2 and 20. As shown in
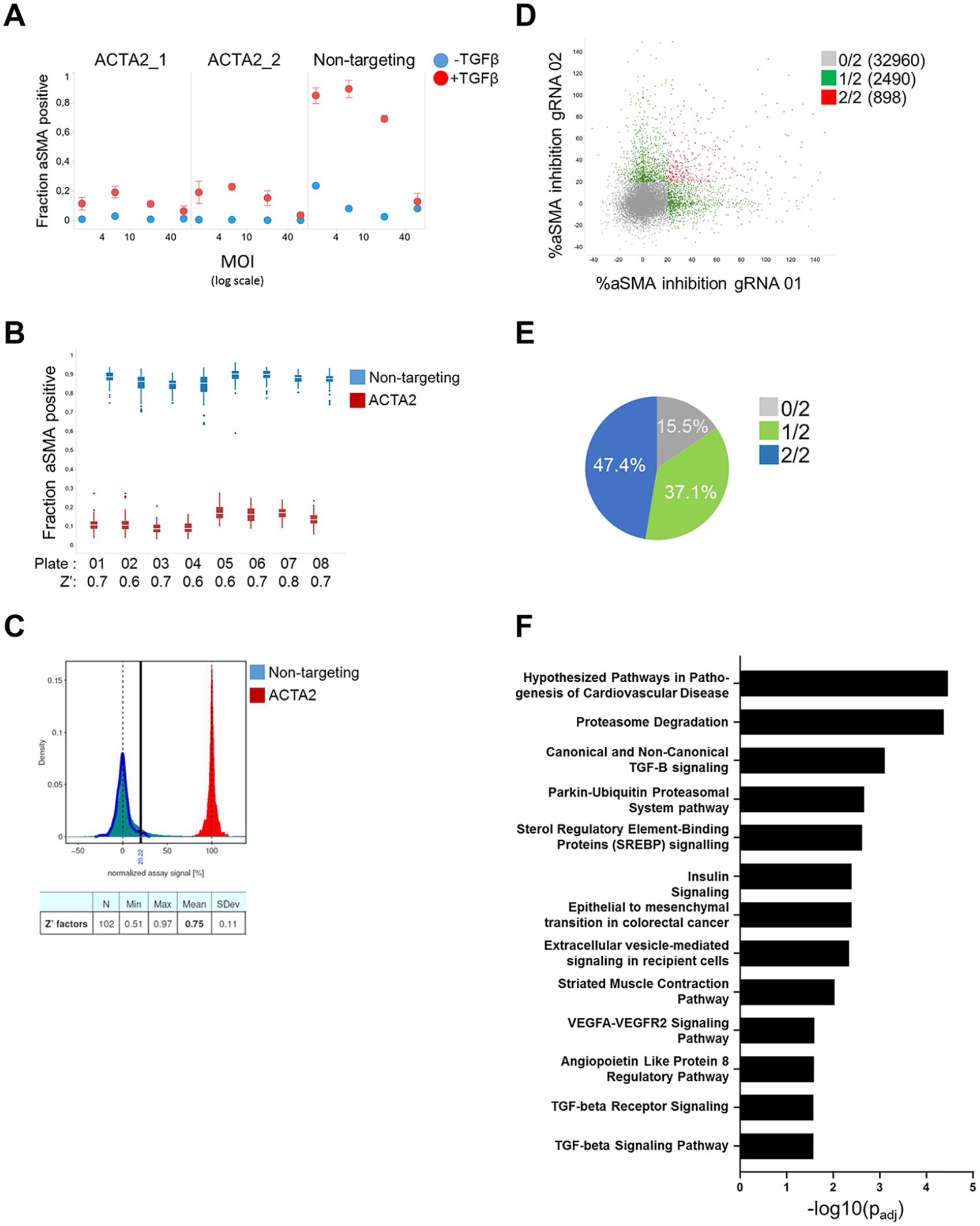
Whole genome arrayed CRISRP screening in primary HKFs. (
Orthogonal Confirmation of Proteasome Inhibition as Effective in Blocking Kidney Fibroblast Activation
We have observed a strong enrichment of confirmed hits around proteasomal degradation. As a proof of concept whether our screen was able to identify relevant hits in the area of kidney fibrosis, we tested the proteasome inhibitors bortezumib and carlizomib in our PMT assay. As shown in
Figure 5A
, both are able to block induction of smooth muscle actin following TGF-β treatment in kidney fibroblasts. Although both inhibitors are cytotoxic at high concentrations, especially for bortezumib there is a clear difference between inhibition of aSMA induction and reduction of cell numbers below the numbers observed for SB4311542. Importantly, these finds could be reproduced in a second donor (HKF_616) (
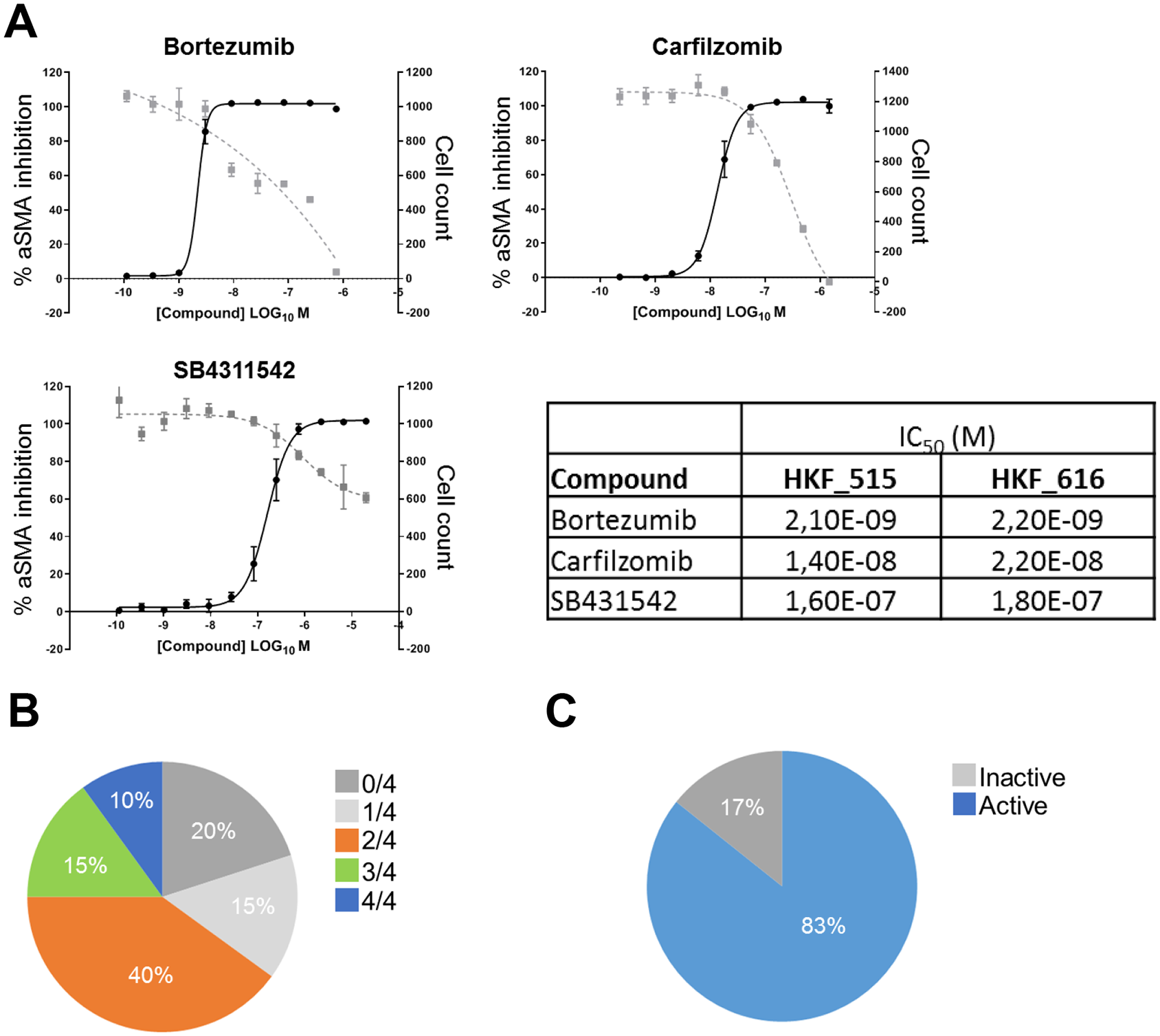
Overview of hit follow-up activities. (
Discussion
Phenotypic drug discovery approaches have the prospect of higher translatability of preclinical findings to the clinic,17,34 especially if phenotypic approaches are developed combining an assay system with a clear link to the disease by using primary or induced pluripotent (iPS)-derived human cells, a readout close to disease pathology, and a disease-relevant trigger preferentially without exogenous stimulation. 16 However, while phenotypic drug discovery has the potential to discover first-in-class small-molecule drugs, identifying the relevant targets responsible for this activity can pose a significant roadblock in the development of a compound. 34 Unbiased genetic screening approaches such as RNAi or CRISPR offer to bridge phenotypic and target-based drug discovery. 35 Ideally, both screening approaches utilize the same disease-relevant assay and thereby have the prospect of complementing each other: on the one hand providing interesting chemical matter and on the other hand identifying targets modulating the disease-relevant phenotype.
Here we describe the development of a phenotypic screening assay in the area of kidney fibrosis employing primary disease-relevant cells from the affected organ combined with a disease-relevant readout and relevant trigger. Although treatment with TGF-β presents an exogenous trigger and has potential to introduce bias into the results by identifying stimulus-related mechanisms, TGF-β has been shown to be increased in kidneys from chronic kidney disease patients and shown in mouse models to be responsible for increased matrix deposition and fibroblast proliferation.22,36 Our data show that a robust PMT assay can be developed using primary kidney fibroblast, showing increased proliferation, accumulation of collagen, and induction of aSMA. Importantly, our pharmacological as well as transcriptional characterization shows that primary kidney fibroblasts are different than an established fibroblast cell line from rat kidney and normal lung fibroblasts, thus suggesting that this model can identify factors specific to human kidney fibrosis in addition to more general fibrotic regulators.25,26
Our main interest in this work was to evaluate whether CRISPR technology can be applied to such a primary cell system and whether this approach could be used to screen a whole genome library to identify novel targets in kidney fibrosis. Our data shows that a lentivirus-based CRISPR can not only be applied in primary kidney fibroblasts, but also be expanded to screen a whole genome-wide library. While several reports have shown successful utilization of CRISPR technology in primary fibroblasts from different origin,26,37,38 to our knowledge this work describes the first whole genome-wide screen in such a model. Our data shows that in this setting, our highly robust CRISPR PMT assay can identify both TGF-β-dependent (TGFBR1/2, SMAD4) and independent factors (MKL1). 23 With the example of proteasomal inhibition we also identified modalities of fibrosis attenuation that potentially involve both of these modalities. 29
Lentivirus-based systems offer the possibility to apply CRISPR technology to a vast number of proliferating and nonproliferating cell types and have shown its potential in numerous pooled screens published in different biological settings.1,5 Here we show that this can also be achieved with an arrayed lentivirus-based library. Arrayed libraries significantly broaden the assay readouts that can be screened using such a library, and also do not involve NGS-based screening deconvolution.12,39 On the other hand, arrayed lentivirus-based libraries also present certain challenges that need to be taken into account when attempting to perform an array-based CRISPR screen. As lentiviruses are intrinsically liable to freeze and thaw cycles, which result in reduction of titer, 40 these need to be carefully tracked during a screening campaign and minimized, for example, by generating replicate screening plates from the original plate as backups in case of plate failure or as a source for hit confirmation activities. Furthermore, titers for each lentivirus in the library differ, which results in a spread of MOI in the assay that can impact the cells due to infection stress. In our example, we estimated that up to 6.6% of hits could be false positives due to high lentivirus titer. The four targets, which we were not able to confirm with any gRNA during hit follow-up activities, could therefore present examples of false positives stemming from high virus titer. Chemically synthesized sgRNA libraries would offer an alternative without these liabilities, but potentially at the expense of a more limited applicability across cell types and lower efficiency. In both cases, hits should be followed up by employing orthogonal approaches such as RNAi, RNP-based CRISPR, or tool compounds to eliminate false positives stemming from using a certain screening approach. Independently of these challenges, we were able to identify high-confidence hits in our screen and confirm these in hit follow-up activities. These hits present potentially new exciting targets in the context of kidney fibrosis.
Supplemental Material
Supplemental_Material_for_Tuner_et_al – Supplemental material for A Whole Genome-Wide Arrayed CRISPR Screen in Primary Organ Fibroblasts to Identify Regulators of Kidney Fibrosis
Supplemental material, Supplemental_Material_for_Tuner_et_al for A Whole Genome-Wide Arrayed CRISPR Screen in Primary Organ Fibroblasts to Identify Regulators of Kidney Fibrosis by Robert J. Turner, Stefan Golz, Carina Wollnik, Nils Burkhardt, Ina Sternberger, Uwe Andag and Hauke Cornils in SLAS Discovery
Footnotes
Acknowledgements
We would like to thank all members of the Evotec/Bayer Kidney collaboration for helpful discussions and insight during this project.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Robert J. Turner, Carina Wollnik, Ina Sternberger, Uwe Andag, and Hauke Cornils are employed by Evotec SE and Evotec International GmbH, and their research and authorship of this article was completed within the scope of their employment with Evotec SE and Evotec International GmbH. Stefan Golz and Nils Burkhardt are employed by Bayer AG, and their research and authorship of this article was completed within the scope of their employment with Bayer AG.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.