
Abstract
Interactions between transmembrane receptors and their ligands play important roles in normal biological processes and pathological conditions. However, the binding partners for many transmembrane-like proteins remain elusive. To identify potential ligands of these orphan receptors, we developed a screening platform using a homogenous nonwash binding assay in live cells. A collection of ~1900 cDNA clones, encoding full-length membrane proteins, was assembled. As a proof of concept, cDNA clones were individually transfected into CHO-K1 cells in a high-throughput format, and soluble PD-L1-Fc fusion protein was used as bait. The interaction between the putative receptor and PD-L1-Fc was then detected by Alexa Fluor 647 conjugated anti-human immunoglobulin G Fc antibody and visualized using the Mirrorball fluorescence plate cytometer. As expected, PDCD1, the gene encoding programmed cell death protein 1 (PD-1), was revealed as the predominant hit. In addition, three genes that encode Fc receptors (FCGR1A, FCGR1B, and FCGR2A) were also identified as screen hits as the result of the Fc-tag fused to PD-L1, which has provided a reliable internal control for the screen. Furthermore, the potential of using a biotinylated ligand was explored and established to expand the versatility of the cDNA platform. This novel screening platform not only provides a powerful tool for the identification of ligands for orphan receptors but also has the potential for small-molecule target deconvolution.
Keywords
Transmembrane receptor proteins are encoded by up to 30% of open reading frames in known genomes.1,2 These proteins and their ligands play important roles in communication between cells in normal biological processes and pathological conditions. Approximately 50% to 60% of marketed drugs target mechanisms on the cell surface, and surface proteins remain the principal targets for new drug discovery.3,4 One large group of transmembrane proteins is the immunoglobulin (Ig) superfamily (IgSF). It consists of a group of cell surface and soluble proteins that share common structural features with immunoglobulins. More than 765 members of this class have been identified with functions in the recognition, binding, or adhesion processes of cells. Many IgSF members are involved in immune checkpoints.
Initiated by ligand-receptor binding, immune checkpoints are stimulatory and inhibitory regulators of the immune system. These regulators are crucial for maintaining self-tolerance and protecting the host from tissue damage. In cancer, immune checkpoint mechanisms are often activated to suppress the nascent antitumor immune response. In the past decade, targeting immune checkpoints has been proven to be a promising anticancer therapy. 5
One group of immune checkpoint proteins is the B7-CD28 superfamily. The B7 family consists of structurally related cell-surface protein ligands, which bind to receptors on lymphocytes that regulate immune responses. Activation of T and B lymphocytes is initiated by engagement of the cell-surface, antigen-specific T-cell or B-cell receptors, but additional signals delivered simultaneously by B7 ligands determine the ultimate immune response. These “co-stimulatory” or “co-inhibitory” signals are delivered by B7 ligands through the CD28 family of receptors on lymphocytes, resulting in the modulation of interleukin production. Interaction of B7-family members with co-stimulatory receptors augments immune responses, while interaction with co-inhibitory receptors attenuates immune responses. 6
Some of the B7 family members and their receptors are well studied. Immune checkpoint co-inhibitor programmed cell death protein 1 (PD-1), encoded by the PDCD1 gene, is present on T cells and can act through binding to either PD-L1 or PD-L2. This event induces T-cell exhaustion and plays a major role in host immune suppression.7,8 CTL-associated antigen 4 (CTLA-4) is a CD-28 homolog present on T cells that binds to B7-1 or B7-2 to downregulate pathways of T-cell regulation. Blocking CTLA-4 or PD-1 and their receptors has been proven to be a promising anticancer therapy. 9 Other members, such as B7-H3, B7-H4, and B7-H5, have been suggested to be co-stimulatory or/and co-inhibitory factors that may have clinical significance in human cancer. However, their receptors have not been identified. Finding the receptors for these new members could shed light on novel immunotherapy mechanisms for cancer.6,10
Recently, several high-throughput membranome platforms have been built to facilitate cell surface receptor deorphanization, early target identification, and monoclonal antibody specificity profiling and target deconvolution. The cell microarray technology developed by Retrogenix provides cell array–based high-throughput screening of cell surface targets.11,12 However, this technology requires fixation of cells, which may change the membrane protein conformation and posttranslational modification; thus, it does not provide physiologically relevant binding information. Other microarray-based live cell screens require specialized equipment for array printing and analysis.13,14 The membrane proteome array technology from Integral Molecular offers a live cell screen using flow cytometry technology. It is optimal for suspension cells and may not be a homogenous assay. 15 An extracellular proteome signal screen has been reported by Five Prime. In their platform, human extracellular domains (ECDs) of membrane proteins carrying an immunoglobulin G (IgG) Fc tag were expressed and purified with a high-throughput expression platform and protein A chromatography. These library proteins were screened for their ability to modulate human T-cell activation. Because the library proteins include only soluble ECDs of single-pass transmembrane classes, their posttranslational modification may be different from the native protein.16,17 The NanoBRET-based proximity binding assay, used as an alternative to a radioligand binding assay, provides a powerful homogenous method to study both protein-protein and protein–small molecule binding in live cells.18–20 However, this technique measures the proximity of test molecules, not real binding. Most NanoBRET studies focused on the binding of a particular membrane protein and a small molecules pair. The small molecule needs to be labeled, and the labeled molecule can be further used as a competition probe. The membrane proteins must possess a Nluc-tag, which will be challenging for the protein library screen.
In this proof-of-concept study, we used the well-studied PD-1 and PD-L1 interaction and established a live cell homogenous screen platform in CHO-K1 cells transiently expressing membrane proteins from a cDNA library.
The Mirrorball Imaging Cytometer from TTP Labtech is a plate-format high-sensitivity microplate cytometer originally developed for antibody screening. The no-wash, mix-and-read assay protocol has facilitated membrane cDNA library screening in a high-throughput format. Our platform provides a simple, relatively fast, homogenous method to study transmembrane protein-ligand interaction in live cells. It can be applied to early target identification, cell surface receptor deorphanization, monoclonal antibody specificity profiling, and target deconvolution.
Materials and Methods
Membranome cDNA Library
The cDNA library used in screening was obtained from Invitrogen (now ThermoFisher Scientific). It is a part of the ULTIMATE cDNA clone collection, consisting of ~1900 ORF encoding full-length human membrane proteins in the Gateway pCDNA-DEST40 Vector (a complete list of all genes is in
Cell Culture and Transfection
CHO-K1 cells (American Type Culture Collection, Manassas, VA) were grown at 37 °C and 5% CO2 in Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (10565, ThermoFisher/Gibco, Waltham, MA), supplemented with 10% heat-inactivated fetal bovine serum (10082, ThermoFisher Scientific) and 0.5% penicillin/streptomycin (15070063, ThermoFisher Scientific). For reverse transfection, ViaFect transfection reagent (E4981, Promega, Madison, WI) was mixed with Opti-MEM I reduced serum medium (31985, Gibco) at ratio of 1:50. Ten microliters of transfection reagent mixture was added to each well of the 384-well cell assay plate (781091, Greiner Bio-One, Kremsmünster, Austria). A total of 0.5 µL of plasmid DNA at 100 ng/µL (final amount 50 ng DNA/well) was added to each well and mixed three times by a Bravo automated liquid handler (Agilent Technologies, Santa Clara, CA). The cell assay plate was incubated at room temperature for 20 min prior to the addition of CHO-K1 cells at a density of 3000/well in 25 µL of culture medium. The cell plate was then centrifuged at 1000 rpm for 1 min and incubated at 37 °C and 5% CO2 for 48 h. For “forward” transfection, the identical procedure was followed with the noted exception of adding the CHO-K1 cells to the assay plates first, then incubating for 4 h prior to the addition of 10 µL of transfection reagent/DNA mixtures.
Live Cell Screen
At 48 h after DNA transfection, 10 µL of PD-L1-Fc protein (156-B7, R&D Systems, Minneapolis, MN) or 10 µL of Biotin PD-L1 (BAF156, R&D Systems) in culture medium was added to each well in various concentrations. After 30 min of incubation at room temperature, 5 µL of Alexa Fluor 647 conjugated anti-human IgG Fc antibody (409320, BioLegend, San Diego, CA; final concentration 0.4 µg/mL) or 5 µL of Alexa Fluor 647 streptavidin (S32357, ThermoFisher Scientific; final concentration 0.5 µg/mL) in culture medium was added per well. The cell plate was incubated overnight in a cell culture incubator with 5% CO2. Then, 10 µL of Hoechst 33342 (62249, ThermoFisher Scientific) in culture medium (final concentration 100 nM) was added to each well. The plates continued incubating at room temperature protected from light for 1 h prior to detection of fluorescence by the Mirrorball instrument.
Mirrorball Scan and Data Analysis
A Mirrorball system (TTP Labtech, Melbourne, UK) equipped with 405 nm and 640 nm excitation lasers was used. Two channels were used for detection of emitted fluorescence: FL1 (488–540 nm) for Hoechst 33342 and FL3 (650–690 nm) for Alexa Fluor 647. Whole-well scanning was performed, and data were processed using Mirrorball’s Cellista software. The results were exported as the sum of total fluorescence intensity in FL3 normalized over the intensity of FL1. The plate data were analyzed and visualized using Spotfire software (TIBCO Software, Palo Alto, CA).
Membranome cDNA Library Protein Functional Classification
The genes in the membranome were mapped to Ingenuity Pathway Analysis (IPA) using gene symbol (Qiagen, Hilden, Germany; https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis), and the functional classes of the 1761 genes (of the 1847 genes that were mapped to IPA) were obtained from IPA. The molecule family is based on evidence within the Ingenuity Knowledge Base, and each molecule is assigned a single molecule type based on its primary role. The frequency of each functional class is plotted in
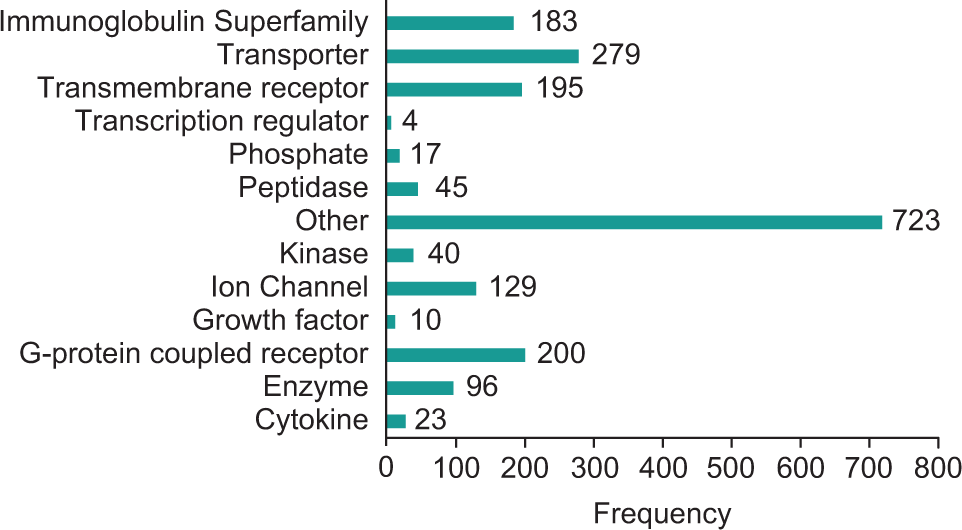
Membranome cDNA library protein functional classes.
Results
Membranome cDNA Library Selection
The list of 1900 genes was generated from Invitrogen’s (now ThemoFisher’s) ULTIMATE cDNA clone collection by selecting annotated genes in the human genome that are predicted to code for plasma or membrane proteins. The candidate clones were further filtered to include those with predicted transmembrane regions and excluded the olfactory receptors. A comprehensive list of library cDNAs is provided in
Design of Live Cell Homogenous Membranome cDNA Library Screen Assay
The library is a collection of ~1900 cDNA clones, encoding full-length membrane proteins (
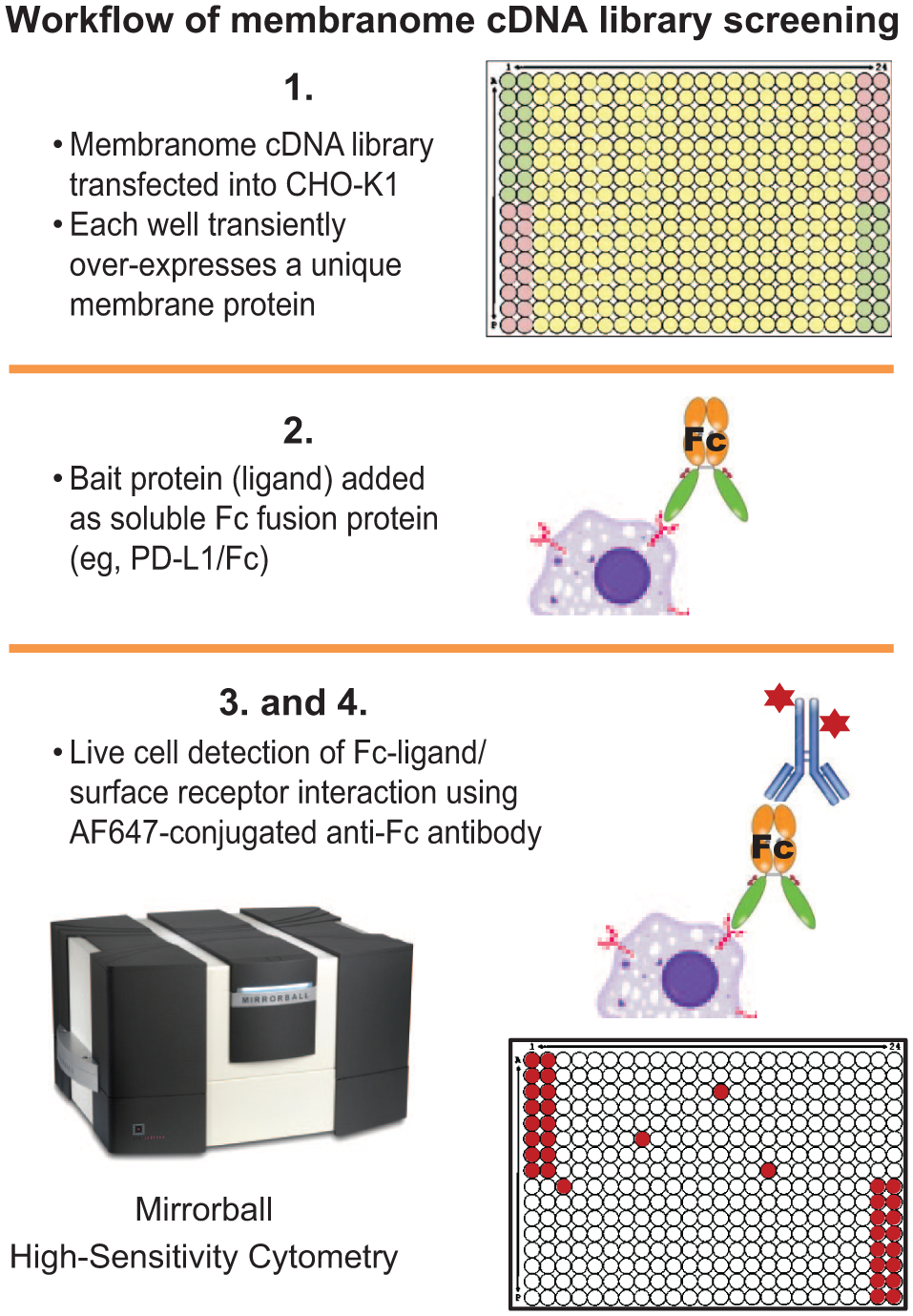
Workflow of membranome cDNA library screening. Each cDNA was individually transfected into CHO-K1 cells in a 384-well format. At 48 h after transfection, transiently expressed membrane protein was probed by a soluble Fc-fusion bait protein (PD-L1-Fc fusion protein in our case). The membrane protein-ligand binding was measured using Alexa Fluor 647 Fc conjugated antibody, detected by the Mirrorball instrument.
Recombinant Human PD-L1-Fc Fusion Protein Binds to Overexpressed PD-1 in a Concentration-Dependent Manner
To determine whether recombinant human PD-L1-Fc fusion protein can be used as a bait protein, we conducted a concentration titration of PD-L1-Fc using CHO-K1 cells overexpressing PD-1 (
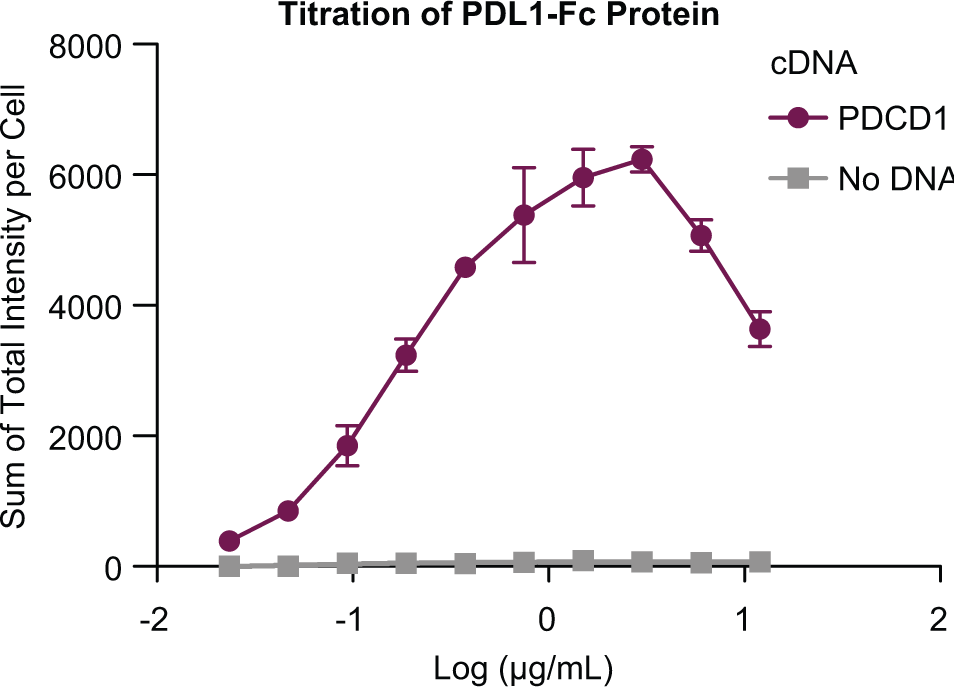
Recombinant human PD-L1- Fc fusion protein binds to overexpressed PD-1. CHO-K1 cells overexpressing PD-1 were treated with increasing amounts of recombinant PD-L1-Fc fusion protein and 0.4 µg/mL AF647-antibody. Nuclei were counterstained with Hoechst 33342. The Mirrorball instrument was used to detect fluorescence. The AF647 fluorescence intensity of each well is normalized by the cell number (nuclei) in the well.
Reverse Transfection Enables High-Throughput Automation
The conventional forward transfection method requires that cells are plated a day or hours first to allow cells to be fully attached prior to the addition of the transfection lipid/DNA mixture. This workflow is not amenable to automation. Conversely, in the method of reverse transfection, the transfection lipid/DNA mixture is added to the plate wells just prior to adding the cell suspension, and all procedures can be performed in the same well of a multiwell assay plate. To design the assay workflow to be more easily adapted to automation processes, we optimized the reverse transfection method based on the PD-L1-Fc concentration titration and eGFP expression efficiency at 72 h posttransfection (
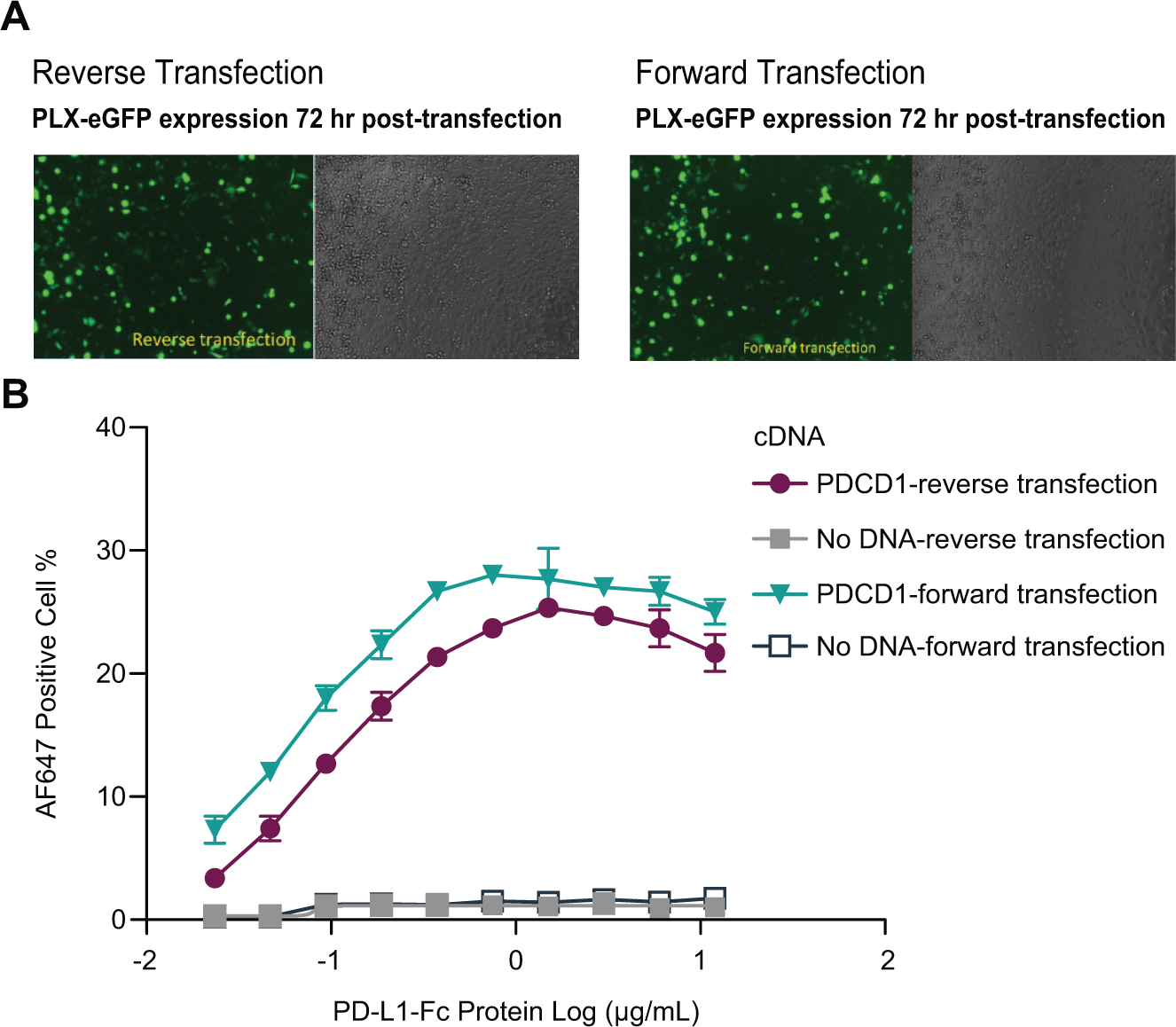
Comparison of transfection methods. eGFP plasmid was transfected in CHO-K1 cells. Seventy-two hours after transfection, eGFP fluorescence and phase contrast images were captured using fluorescent microscopy (
Proof-of-Concept Screen Using PD-L1-Fc Fusion Protein as Bait
To perform a proof-of-concept screen, we used PD-L1-Fc fusion protein as the bait to screen a cDNA library, which consisted of ~1900 plasmid cDNAs of full-length membrane proteins in plasmid vector PCDNA3. Each plasmid was purified and the sequence verified. Each plasmid cDNA was diluted to 0.1 µg/mL in TE buffer and individually dispensed into separate wells of a Labcyte 384-well polypropylene microplate. The screen was conducted in high-throughput 384-well format (
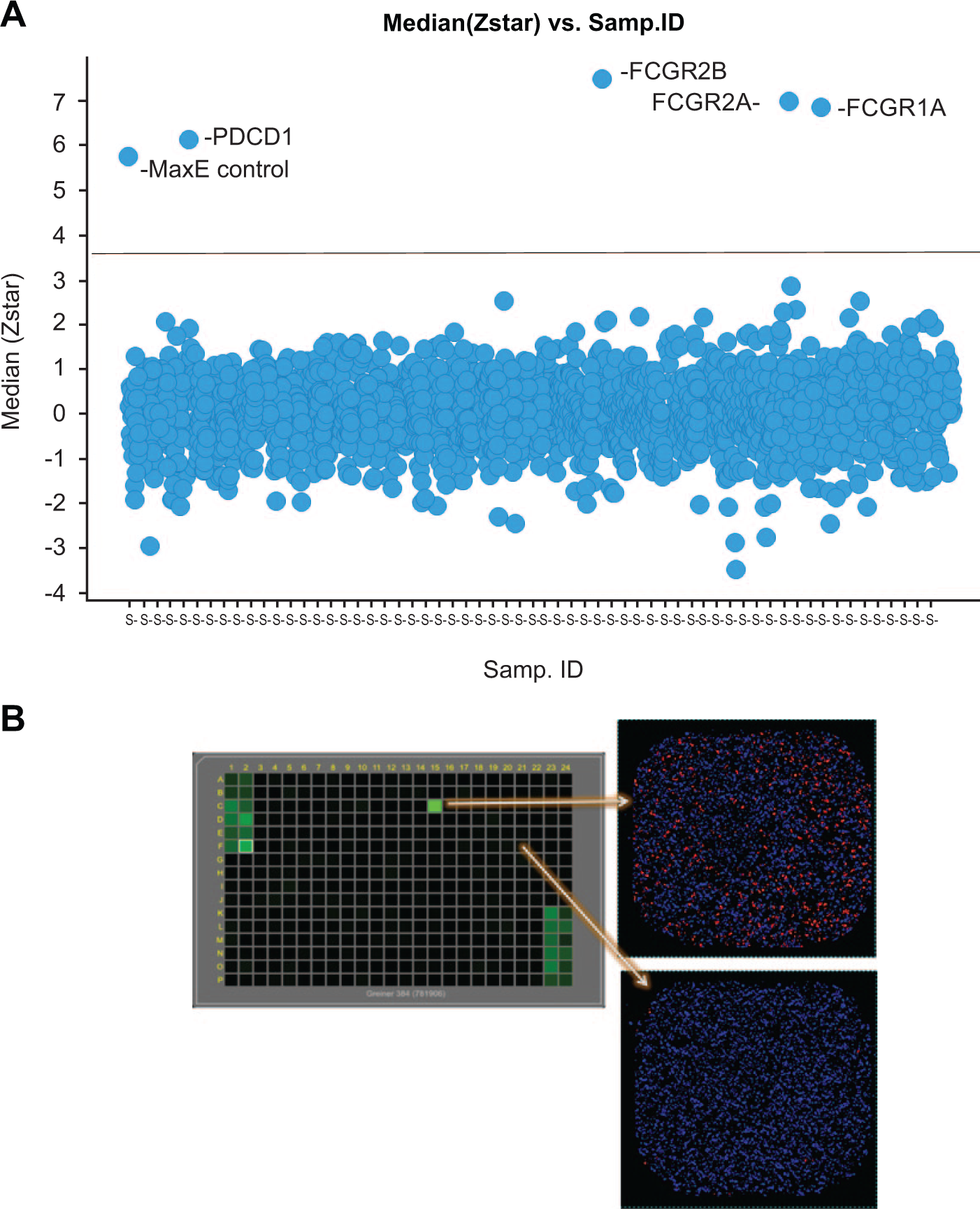
PDCD1 and FCGR1A, FCGR1B, and FCGR2A receptor-ligand interactions were identified by the library screen. A library of ~1900 cDNA clones, encoding full-length membrane proteins, was transiently expressed in CHO-K1 cells in 384-well plate format. The screen was conducted twice (
The screen was performed in duplicate. The raw data of each well were log-transformed. The following normalization was done plate by plate: data in each well were normalized by subtracting the median of the sample wells and then dividing by the median absolute deviation from the median). These are called Z*-scores. The medians of the Z*-scores of the same sample were computed. The Z*-score cutoff was set to 3.5, so that the fraction of negative control wells that exceed it was less than 0.5%, to keep the false-positive rate to a minimum. The projected false-positive rate based on the min-effect wells for the screen was ~0.3%. Four cDNAs met this Z*-score cutoff. They were PDCD1, FCGR1A, FCGR1B, and FCGR2A. The latter three encode high-affinity immunoglobulin gamma Fc receptor I, high-affinity immunoglobulin gamma Fc receptor IB, and low-affinity immunoglobulin gamma Fc region receptor II-a, respectively. Since the bait protein was an Fc fusion protein, it was expected that these three Fc-receptor cDNAs would be identified.
PDCD1 Can Be Identified Using Biotin-PD-L1 as Bait
The large size of the Fc may be expected in some instances to interfere in the binding of the soluble bait protein to its cell surface binding partner. Alternative tags with a smaller size might be better suited for these bait proteins. To expand the scope of the screening platform, we explored whether a biotinylated protein would serve as a bait using biotinylated PD-L1 as a proof-of-concept tool protein (
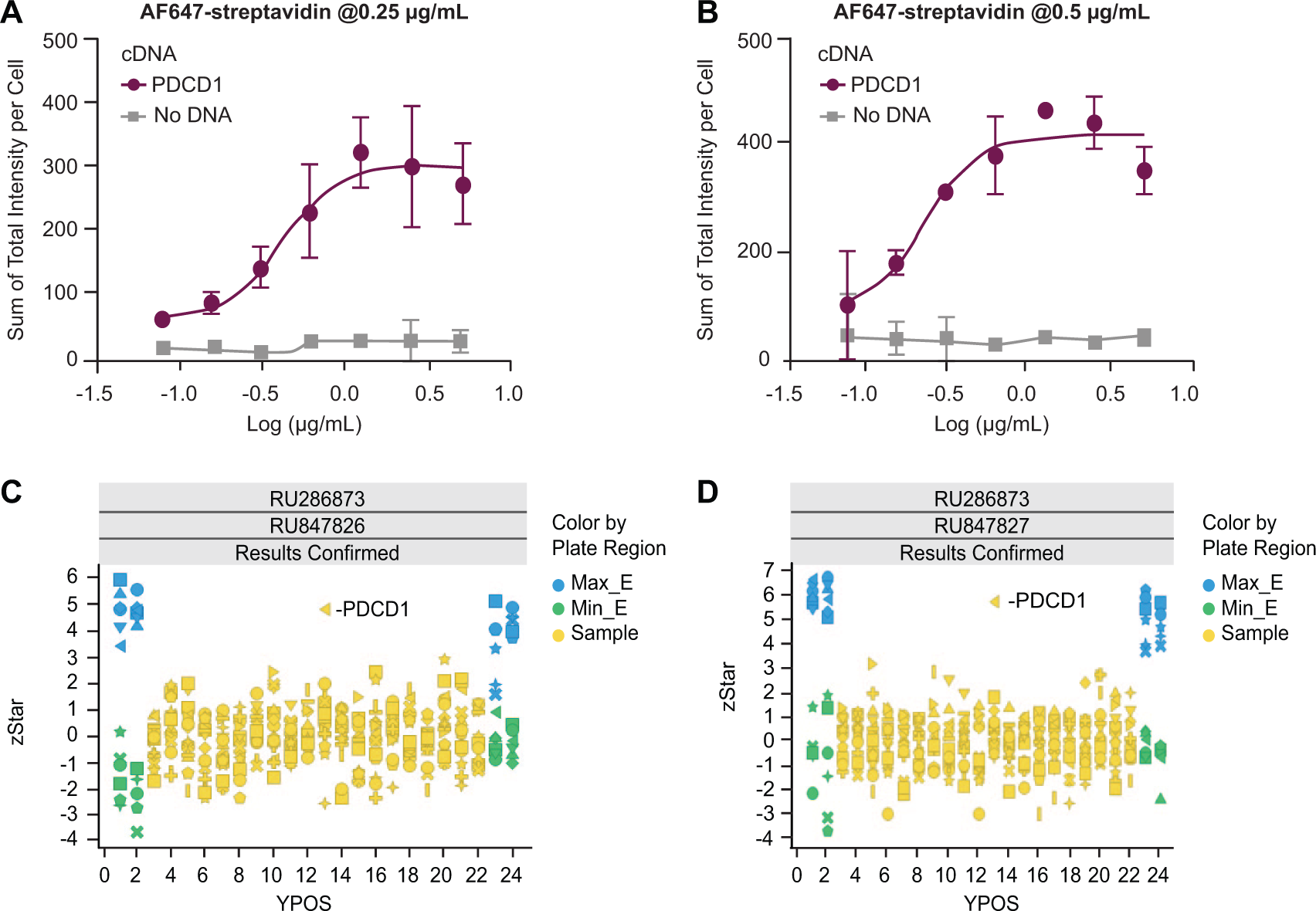
PDCD1 was identified from the library plate using biotinylated PD-L1 as bait ligand. (
Discussion
In this study, we designed and successfully demonstrated a reliable platform to screen a membrane protein cDNA library using a PD-1/PD-L1-Fc pair as proof of concept. We were able to identify PDCD1 as one of the positive hits from ~1900 membrane protein cDNAs. The Fc receptors encoding cDNAs, FCGR1A, FCGR1B, and FCGR2A were also identified from the library, which further validates the success of the screen design. These Fc-receptors also serve as reliable internal controls. Compared with the NanoBRET-based proximity binding assay using stable transfected cells, our platform does not require an N-terminal Nluc tag in library proteins, and the transient transfection makes screening more time efficient and cost-effective. Another key advantage of this platform is the ability to detect protein-ligand interactions in live cells. This enables the screen to be performed with the membrane proteins in their native conformations. In addition, the homogenous nature of the experimental design with no wash or media changes makes the screen ideal for high-throughput automated operations and for producing robust assay statistics. Our platform enables the study of the binding of a bait protein to thousands of library proteins simultaneously and can be used to deorphanize membrane receptors. Although our screen was conducted in CHO-K1 cells, the method can be adapted to other easy-to-transfect cell lines.
We also showed that a biotinylated bait protein provides an advantage over the use of an Fc-tag, which may sterically interfere with protein binding of some receptors. Conjugation of biotin to a molecule can be achieved not only through a bioengineered Avi tag but also by direct labeling. Therefore, genetically engineering a tag into a protein construct at an early stage is not necessary. Furthermore, it is expected to broaden the scope of the platform to include small molecules as bait, which could be valuable for target deconvolution purposes.
During our assay validation, besides PD-1 and PD-L1, other Fc-tagged bait proteins (i.e., PD-L2, CD28, and Notch1) were tested, and specific binding to their reported ligands PD-1, CD80, and DDL4 were observed (data not shown). The platform has already been successfully used internally to deorphan membrane receptors, such as B7-H3, B7-H4, ILT3, and so on. Novel interactions have been identified, and results will be reported in future publications.
The platform also has its limitations. First, there were more than 5000 nonolfactory membrane proteins identified. We have expanded our library from 1900 to 2900 cDNAs, which covers only ~60% of the expected total. A membranome cDNA library covering a higher percentage of known nonolfactory membrane proteins would be desirable.
Second, protein expression levels may not be equal in all transfections. For example, it is well known that ion channels are usually more difficult to transfect and thus may express at much lower levels. The low expression level of certain library proteins may lead to false-negative results. In that sense, the screen is an inclusion but not an exclusion screen. One option would be to categorize the cDNAs into different groups based on efficiency of transfection and to apply optimized transfection strategies to different groups. All constructs in our cDNA library carry a V5 tag, which could facilitate the assessment of target protein expression level by using immunostaining or enzyme-linked immunosorbent assay.
Third, the quality of bait protein could play a crucial role in the success of the screen. The bait protein used in our screen is the ECD of human PD-L1 and human IgG1 Fc region (C-terminal) fusion protein. It forms a homodimer in the solution, which may help amplify the signal. The Fc-fusion protein strategy may not be applicable in cases in which the parent protein is small, as the Fc tag may hinder its binding to the surface protein. Validation of biotinylated PD-L1 as bait protein not only provides an alternative option for the protein tag but also should expand the application to small molecules.
The reported equilibrium dissociation constant (Kd) for PD-L1 to PD-1 is between 11 nM and 770 nM in vitro depending on the method of measurement.21–23. Protein interaction affinities beyond this range may not be detected with the current platform. This method may also not be suitable for identifying transient protein interactions. It is interesting to note that we could not identify B7-1 (CD80), which is known to bind to PD-L1 with a weaker affinity than PD-1 (the reported Kd for PD-L1 is 1.4 uM in vitro 21 ). A recent study suggests that PD-L1 binds to B7-1 only in cis on the same cell surface, 24 which may explain the lack of an observed interaction in this system.
Lastly, even though CHO-K1 cells provided a simple experimental cellular background for screening, it is possible that some protein-ligand interactions may need other factors such as the presence of multiple surface proteins. In such a case, a physiologically relevant cell system may need to be considered.
To our knowledge, this is the first homogenous live cell binding assay to study membrane protein-ligand interactions. This approach provides a powerful screening tool for discovery of ligand-receptor interactions in live cells and may be applied to early target identification, cell surface receptor deorphanization, monoclonal antibody specificity profiling, and target deconvolution.
Supplemental Material
DS_DISC873069 – Supplemental material for Live Cell Membranome cDNA Screen: A Novel Homogenous Live Cell Binding Assay to Study Membrane Protein-Ligand Interaction
Supplemental material, DS_DISC873069 for Live Cell Membranome cDNA Screen: A Novel Homogenous Live Cell Binding Assay to Study Membrane Protein-Ligand Interaction by Xun Shen, Elizabeth Smith, Xi Ai, William T. McElroy, Andy Liaw, Tony Kreamer, Meiping Chang, Kristine Devito, Edward Hudak, Serena Xu, Yi Pei, Sylvie Sur, Andrea Peier and Jing Li in SLAS Discovery
Footnotes
Acknowledgements
We thank Adam Weinglass, Mary Jo Wildey, Maria Webb, and Kun Liu for their support and valuable comments on the article.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.