
Abstract
Integral membrane proteins (IMPs) play an important role in many cellular events and are involved in numerous pathological processes. Therefore, understanding the structure and function of IMPs is a crucial prerequisite to enable successful targeting of these proteins with low molecular weight (LMW) ligands early on in the discovery process. To optimize IMP purification/crystallization and to identify/characterize LMW ligand-target interactions, robust, reliable, high-throughput, and sensitive biophysical methods are needed. Here, we describe a differential scanning fluorimetry (DSF) screening method using the thiol-reactive BODIPY FL-cystine dye to monitor thermal unfolding of the G-protein-coupled receptor (GPCR), CXCR2. To validate this method, the seven-transmembrane protein CXCR2 was analyzed with a set of well-characterized antagonists. This study showed that the new DSF assay assessed reliably the stability of CXCR2 in a 384-well format. The analysis of 14 ligands with a potency range over 4 log units demonstrated the detection/characterization of LMW ligands binding to the membrane protein target. Furthermore, DSF results cross-validated with the label-free differential static light scattering (DSLS) thermal denaturation method. These results underline the potential of the BODIPY assay format as a general tool to investigate membrane proteins and their interaction partners.
Introduction
Integral membrane proteins (IMPs) represent an important protein class being involved in different cellular processes such as cell-cell interactions, signal transduction, membrane transport, structural integrity of cells, and many more. Approximately 30% of the human genome codes for membrane proteins. Their importance in diseases is reflected by the fact that 44% of all known human pharmaceutical drug targets are membrane receptors (e.g., G-protein-coupled receptors, ligand-gated ion channels, receptor tyrosine kinases). 1 Due to their high complexity and difficulties in handling them biochemically (e.g., low expression levels, tedious purification procedures, and low stability), these targets are extremely challenging to biophysicists and X-ray crystallography experts alike.
The biophysical analysis of interactions between low molecular weight (LMW) ligands and a potential membrane protein drug target is very demanding with regard to sensitivity, specificity, and throughput. Conformational stability is a common property of proteins that can be modulated by the interaction with a LMW ligand. Therefore, thermal shift assays (TSAs) fulfilling some of the aforementioned requirements are valuable tools to characterize proteins and their interactions with ligands. 2 Thermal shift assays are grouped according to their physical detection principle, with these being optical (e.g., UV spectroscopy, circular dichroic, and light scattering) and calorimetric methods of detection (e.g., differential scanning calorimetry and isothermal calorimetry to assess protein stability). The limitations of these labor-intensive methodologies for LMW ligand screening are low throughput and high-protein consumption. Fluorescence-based thermal shift assays such as ThermoFluor, using environmentally sensitive (solvatochromic) fluorophores to monitor protein denaturation, were introduced to enable high-throughput melting temperature (Tm) measurements with reduced protein amounts. Solvatochromic dyes strongly increase their quantum yield of fluorescence upon changing from polar to unpolar environments. 3 This method works very well with soluble proteins but has its limitation with hydrophobic proteins and detergent-solubilized membrane proteins due to strong interference of the dye with hydrophobic components. To overcome this disadvantage, Alexandrov and colleagues 4 used the thiol-reactive dye N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide (CPM) to access the thermal stability of integral membrane proteins.
In this study, we show the use and validation of the thiol-reactive dye, BODIPY FL-L-cystine, as a general tool to monitor thermal unfolding of a detergent-solubilized integral membrane protein to characterize LMW ligands. BODIPY FL-L-cystine offers several advantages due to its spectral properties. As a test system, the C-X-C chemokine receptor (CXCR) 2, a seven-transmembrane G-protein-coupled receptor (GPCR), was used. CXCR2 plays an important role in regulating the migration of different cell types controlling pulmonary functions. Therefore, it is an attractive drug target to treat a variety of lung diseases such as chronic obstructive pulmonary disease (COPD). 5
Materials and Methods
All chemical compounds were of analytical grade and purchased from Sigma-Aldrich (St. Louis, MO). The fluorescence dyes SYPRO Orange and BODIPY FL-L-cystine were purchased from ThermoFisher Scientific (Waltham, MA). The following microtiter well plates were used: 384-well, black, clear-bottom plates (Nunc International, St. Louis, MO) and hard-shell, thin-wall 384-well PCR plates (Bio-Rad, Hercules, CA). Lyophilized bovine β-lactoglobulin (β-LG) and bovine carbonic anhydrase II were purchased from Sigma-Aldrich (St. Louis, MO).
Cloning, Expression, and Purification of CXCR2
The wild-type human CXCR2 gene (encoded by CXCR2; UniProt accession
Insect cell membranes were disrupted by two passes through a cell breaker (EmulsiFlex-C3; Avestin) in low-salt buffer containing 50 mM sodium phosphate (pH 7.5), 10% (w/v) glycerol, 1 mM EDTA, and protease inhibitor cocktail tablets (Roche, Mannheim, Germany). Cell membranes were isolated by high-speed centrifugation at 180,000 g for 1 h. The pelleted membranes were resuspended in 30 mL wash buffer (25 mM sodium phosphate [pH 7.5], 10% glycerol, 150 mM NaCl, 0.1 mM EDTA, protease inhibitor cocktail tablets) per liter culture volume using a Dounce homogenizer and centrifuged again at 180,000 g for 30 min. The washed pellet was resuspended in a minimal volume of wash buffer, flash-frozen with dry ice, and stored at −80 °C until further use. Total membrane protein concentration was quantified by using the bicinchoninic acid method (Pierce, Waltham, MA).
The frozen membranes were thawed, diluted to a protein concentration of 10 mg/mL with fresh wash buffer, and solubilized with 1.0% lauryl maltose neopentyl glycol 3 (LMNG-3; Anatrace, Maumee, OH) for 2 h at 4 °C. The supernatant was isolated by centrifugation at 180,000 g for 1 h, supplemented with 350 mM NaCl and 5 mM imidazole (pH 7.5), and incubated with TALON metal ion affinity chromatography resin (Clontech) overnight at 4 °C. Typically, 2 mL of resin (slurry) per liter of original culture volume was used. After binding, the resin was washed in batch with 100-g spins for 5 min each with a washing buffer of 25 mM sodium phosphate (pH 7.5), 10% glycerol, 300 mM NaCl, and 0.1% LMNG-3. The resin was poured into a glass column, and bound receptor was eluted in washing buffer supplemented with 200 mM imidazole and with the concentration in NaCl reduced by half. Cobalt-resin eluate was loaded onto anti-Flag M2 resin and washed extensively with buffer composed of 25 mM sodium phosphate (pH 7.5), 10% glycerol, 150 mM NaCl, and 0.1% LMNG-3. Receptor was eluted from the anti-Flag M2 affinity resin with 0.2 mg/mL Flag peptide and further purified by size-exclusion chromatography (Sephadex S200; GE Healthcare, Glattbrugg, CH) in a buffer of 25 mM sodium phosphate (pH 7.5), 10% glycerol, 100 mM NaCl, and 0.01% LMNG-3. The final monodisperse receptor preparation was concentrated to approximately 4 to 5 mg/mL using a 100-kDa molecular weight cutoff Vivaspin concentrator (Vivascience, Göttingen, Germany). Receptor purity and functionality were followed using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). A typical yield of final, purified CXCR2 from 4 liters of cell culture volume was around 2 to 3 mg.
[3H]-Pteridone Saturation Binding
Binding was performed with a range of concentrations of [3H]-Pteridone to construct saturation binding curves, as described by Salchow et al. 6 Experiments were conducted in 96-deep-well plates in assay binding buffer 20 mM HEPES, 10 mM MgCl2, 100 mM NaCl, 1 mM EDTA, and 0.01% (w/v) HSA ± LMNG-3 0.03% (pH 7.4) at room temperature. CHO-CXCR2 cell membranes (10 µg/well, respectively) or detergent purified receptors (0.005 µg/well, respectively) were incubated in 96-deep-well plates at room temperature in assay binding buffer (final volume 0.5 mL) with gentle agitation for 2 h to ensure equilibrium was reached. Following this incubation period, bound and free radioligands were separated by rapid vacuum filtration using a FilterMate Cell Harvester (PerkinElmer, Beaconsfield, UK) onto 96-well GF/B filter plates pretreated with assay buffer and rapidly washed three times with ice-cold 20 mM HEPES containing 1 mM MgCl2 (pH 7.4). After drying (>4 h), 40 µL Microscint 20 (PerkinElmer) was added to each well and radioactivity quantified using single-photon counting on a TopCount microplate scintillation counter (PerkinElmer). Aliquots of [3H]-Pteridone were also quantified accurately to determine how much radioactivity was added to each well using liquid scintillation spectrometry on a Hidex 300SL scintillation counter (LabLogic, Sheffield, UK). In all experiments, total binding never exceeded more than 10% of that added and therefore limiting significant depletion of the free radioligand concentration. 7
Data Analysis and Statistical Procedures
Saturation experiments were analyzed with nonlinear regression using Prism 6.0 (GraphPad Software, San Diego, CA). Specific binding, obtained by subtracting nonspecific binding from total binding, was fitted to a one-site binding model that describes a rectangular hyperbola or binding isotherm. Where comparisons of equilibrium saturation binding parameters were made, statistical significance was determined by unpaired t test.
Differential Scanning Fluorimetry
Differential scanning fluorimetry (DSF) experiments were carried out in 384-well PCR plates and measured with a CFX384 real-time PCR instrument (Bio-Rad). Protein unfolding was monitored by measuring fluorescence of thiol-reactive dye BODIPY FL-L-cystine. The standard assay conditions were 50 mM HEPES (pH 7.4), 100 mM NaCl, 0.001% LMNG-3, and 1% DMSO with protein concentrations 0.025 to 0.2 mg/mL and BODIPY FL-L-cystine concentrations 0.675 to 20 µM. All assays were performed in 10 µL. The plates were sealed with a clear, optical foil (Bio-Rad) before starting the heating process in the PCR instrument. For all experiments, a heating rate of 1 °C/min was used. The fluorescence was detected by using the FAM filter set (450–490 nm excitation, 515–530 nm emission). The fluorescence was measured every 0.2 or 0.5 °C temperature interval. To determine the stabilization of CXCR2 by ligands, control wells containing the protein without ligand and 1% DMSO were included in each experiment. Melting temperature shifts (ΔTm) more than 3-fold the standard deviation of Tm controls were classified as significant stabilization. The standard deviation of Tm was in the range of 0.1 to 0.3 °C for all experiments. The Tm was determined by plotting the fluorescence values against temperature and fitting the data to the Boltzmann equation using an in-house software program. The inflection point of the first derivative of the melting curve represented the protein Tm.
Differential Static Light Scattering
The thermal stability of membrane proteins was determined by differential static light scattering (DSLS) using a StarGazer-384 (Harbinger Biotechnology and Engineering Corporation, Markham, Ontario, Canada). This method follows protein stability by measuring the intensity increase of scattered light during protein denaturation using a CCD camera-based system. 8 The assay conditions were 50 mM HEPES (pH 7.4), 100 mM NaCl, 0.001% LMNG-3, and 1% DMSO. In total, 25 µL/well CXCR2 at 0.4 mg/mL was dispensed in a black clear-bottom 384-well plate (Nunc International) and covered with 25 µL mineral oil to block evaporation. The 384-well plate was placed on the heating block of a StarGazer-384 instrument and heated at 1 °C/min from 25 to 80 °C. Every 0.5 °C, the change of scattered light was monitored by taking an image of the entire microtiter well plate. To convert the pixel intensity into scattered light values, a preselected well area was analyzed by the manufacturer’s software, StarGazer_Intensity (Harbinger Biotechnology and Engineering Corporation). The obtained intensity values were plotted against the temperature. The curves were fitted using the Boltzman equation to determine the aggregation temperature (Tagg).
Results and Discussion
Purification and Characterization of Recombinant Detergent Solubilized CXCR2
Human Flag-His10 tagged CXCR2 was expressed in Sf21 insect cells. To identify the most suitable detergent for solubilization of CXCR2 from insect cell membranes, different detergents (zwitterionic, polar, and nonionic detergents such as neopentyl glucosides) were tested by anti-Flag Western blot analysis. The extraction of CXCR2 with LMNG-3 proved to be most efficient to obtain receptor (data not shown). LMNG-3–extracted CXCR2 was purified through a series of chromatographic steps, including metal chelate, anti-Flag affinity, and size-exclusion chromatography columns. Peak fractions displaying appropriate molecular weight were pooled and subjected to the next chromatographic step. This purification scheme yielded typically 0.5 to 1 mg of >95% pure receptor per liter of insect cell culture ( Fig. 1A ). To check the pharmacological binding properties of the detergent-solubilized CXCR2, a radioligand binding assay was established and a comparison was made of the binding properties of [3H]-pteridone in a preparation of purified recombinant CXCR2 receptors and membranes from CXCR2-expressing Chinese hamster ovary (CHO) cells. The observed saturation binding curves generated the same Kd values for pteridone ( Fig. 1B ), whereas the receptor expression levels were strongly increased for the purified recombinant CXCR2 from Sf21 cells. These data demonstrate that the detergent-solubilized CXCR2 is functional after purification in LMNG-3, and hence this material should serve as a good model to study ligand interaction by TSA.
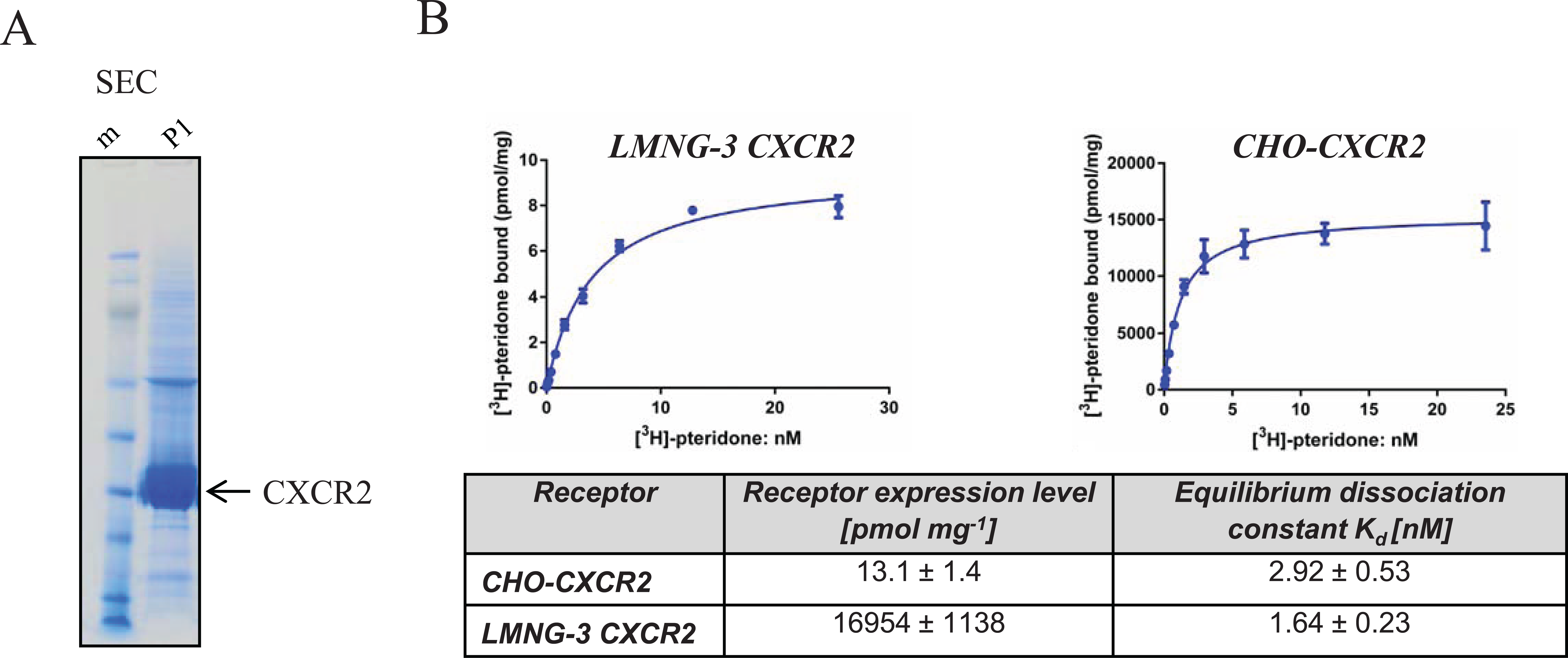
Purification and characterization of recombinant human CXCR2: (
Use of BODIPY FL-L-Cystine Dye as a Cysteine Sensor in Thermal Shift Assay to Follow Membrane Protein Unfolding
Several groups proved that the thiol-reactive CPM dye is a valuable tool to follow unfolding of integral membrane proteins. In membrane proteins, cysteine residues are often buried in the transmembrane domains, limiting their exposure to aqueous environment. Therefore, these residues can serve as sensors to monitor thermal unfolding of proteins. During the unfolding process, sulfhydryl groups of cysteine residues get accessible for a thiol-reactive dye, forming a covalent bond and releasing the fluorescence quench. 4 This method can be applied to optimized buffer conditions for protein expression, purification, and crystallization studies. 9 A major drawback of CPM dye, a coumarin derivative, is its short-wavelength excitation/emission maximum of 384/470 nm, which is prone to compound autofluorescence artifacts in fluorescence intensity assay formats. 10 In addition, the spectral properties of CPM dye are compatible with only a limited number of commercially available 96-well microtiter plate real-time PCR machines, strongly reducing its use as a high-throughput screening (HTS) method in the 384- or 1536-well plate format. Therefore, thiol-reactive dyes with longer wavelength excitation spectra could minimize potential compound autofluorescence effects and enable the use of standard real-time PCR equipment with 384/1536-well microtiter plate capabilities, enabling HTS. The commercially available BODIPY FL-L-cystine (excitation, 505 nm; emission, 513 nm) fulfills all criteria as a potential dye for the application in thermal shift assays with membrane proteins. During the thermal heating process, BODIPY FL-L-cystine showed very low background fluorescence ( Fig. 2B ), since the disulfide bridge between BODIPY FL-L-cystine molecules is very stable and protects spontaneous release of fluorescence quench. This dye was used for a soluble protein α-tocopherol transfer protein in DSF. 11 BODIPY FL-L-cystine consists of two BODIPY FL molecules that are connected via a disulfide bridge between two cysteine residues ( Fig. 2A ). In the dimeric disulfide state, the fluorescence is quenched, whereas the reduction of the disulfide bridge leads to the release of a monomeric dye molecule, unquenching the fluorescence. To test the BODIPY FL-L-cystine dye for hydrophobic proteins, the thermostability of bovine β-LG, a globular milk protein representing the prototype for a hydrophobic protein, was analyzed. Under physiological conditions, β-LG is a dimer consisting of two monomers spanning 162 amino acid residues. Each monomer contains three cysteine residues. Two cysteines are fixed in an intramolecular disulfide bridge. The third cysteine residue offers a free thiol group, which could serve as a reaction point for the thiol-reactive dye to follow unfolding of β-LG.
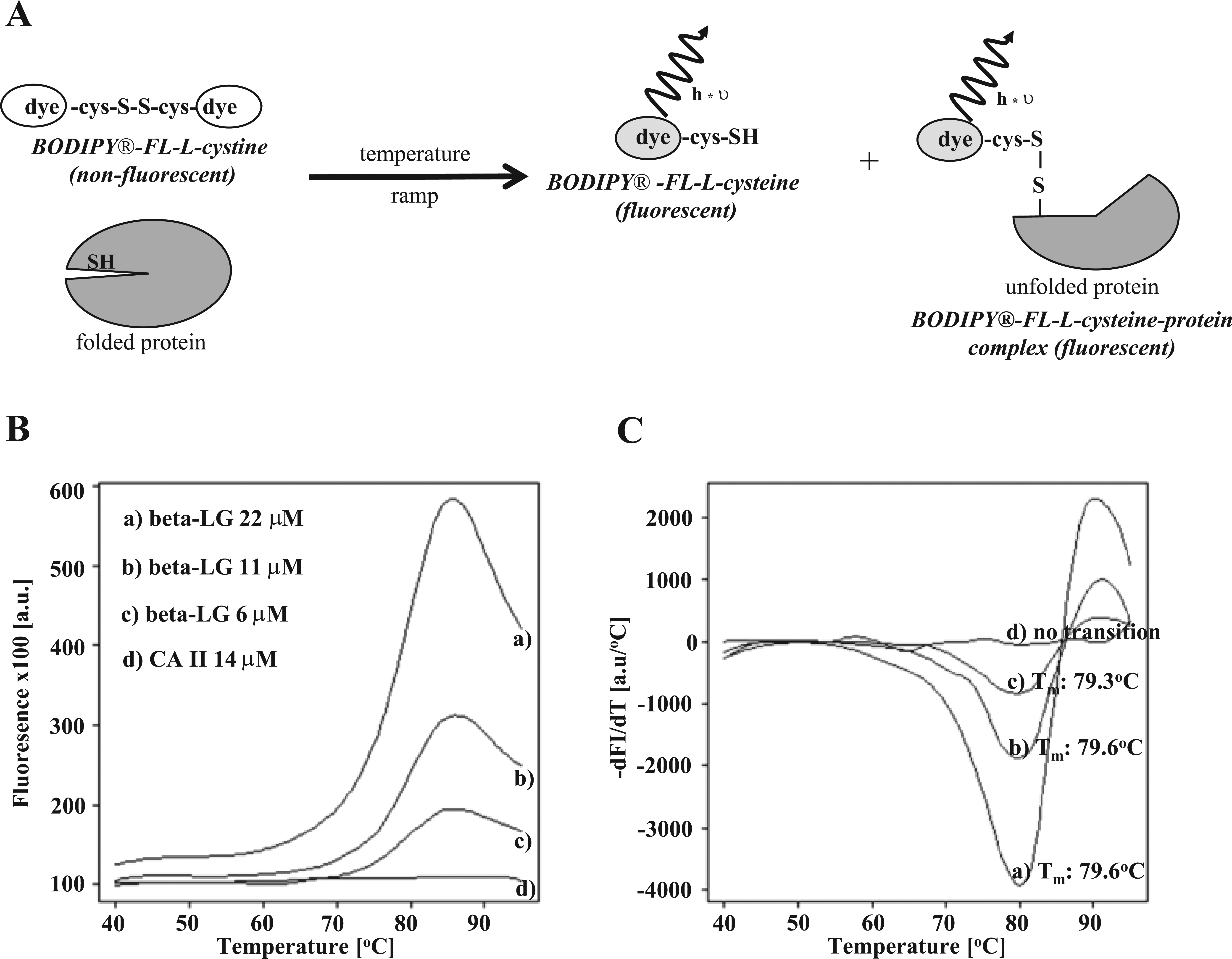
Validation of BODIPY FL-L-cystine as a thiol-sensor in differential scanning fluorimetry (DSF). (
Different β-LG concentrations (6–22 µM) were tested with 20 µM BODIPY FL-L-cystine in a DSF experiment producing reliable melting curves ( Fig. 2B ). The determined melting temperatures for the different β-LG concentrations were very constant, between 79.3 and 79.6 °C ( Fig. 2C ). These values were in good agreement with the published data using the CPM dye. 4 Due to its high hydrophobicity, unfolding of β-LG could not be analyzed with standard solvatochromic dyes such as SYPRO Orange, which produced high background fluorescence in folded protein stage (data not shown). To prove the specificity of fluorescence signal, bovine carbonic anhydrase II containing no cysteine residues in the protein primary structure was used as a negative control. As shown in Figure 2B , C , no significant temperature-dependent increase of the fluorescence signal was observed for carbonic anhydrase II. This clearly indicated that the measured fluorescence signal using β-LG reflected the accessibility of the cysteine residue by the BODIPY dye during the heat-induced unfolding process.
To further investigate the use of thiol-reactive BODIPY dye, the human seven-transmembrane GPCR, chemokine receptor CXCR2, was chosen as a model system to monitor unfolding of an integral membrane protein. The human CXCR2 primary structure contains nine cysteine residues that could serve as potential sensors for the thiol-reactive dye. According to a secondary structure model by Catusse et al., 12 five of the nine cysteine residues are buried inside of transmembrane helices, reducing the accessibility of the SH groups for the thiol-reactive dye in the folded receptor stage.
In an initial experiment, CXCR2 was unfolded in a temperature gradient from 20 to 95 °C, and protein denaturation was monitored by an increase of BODIPY FL fluorescence. Under these conditions, reproducible melting curves were recorded with a Tm of 61.8 ± 0.2 °C ( Fig. 3A ). Further assay development showed that several parameters influenced the measured apparent Tm values: (1) the molar ratio between protein/BODIPY dye and (2) the velocity of temperature increase during the unfolding process. In the case of CXCR2, a molar excess of dye decreased Tm, whereas an excess of protein caused an increase ( Fig. 3B ). CXCR2 protein concentrations >150 mg/mL (3.3 µM) could not be combined with a high dye concentration (20 µM), which caused a saturation of photomultiplier tube in the real-time PCR machine.
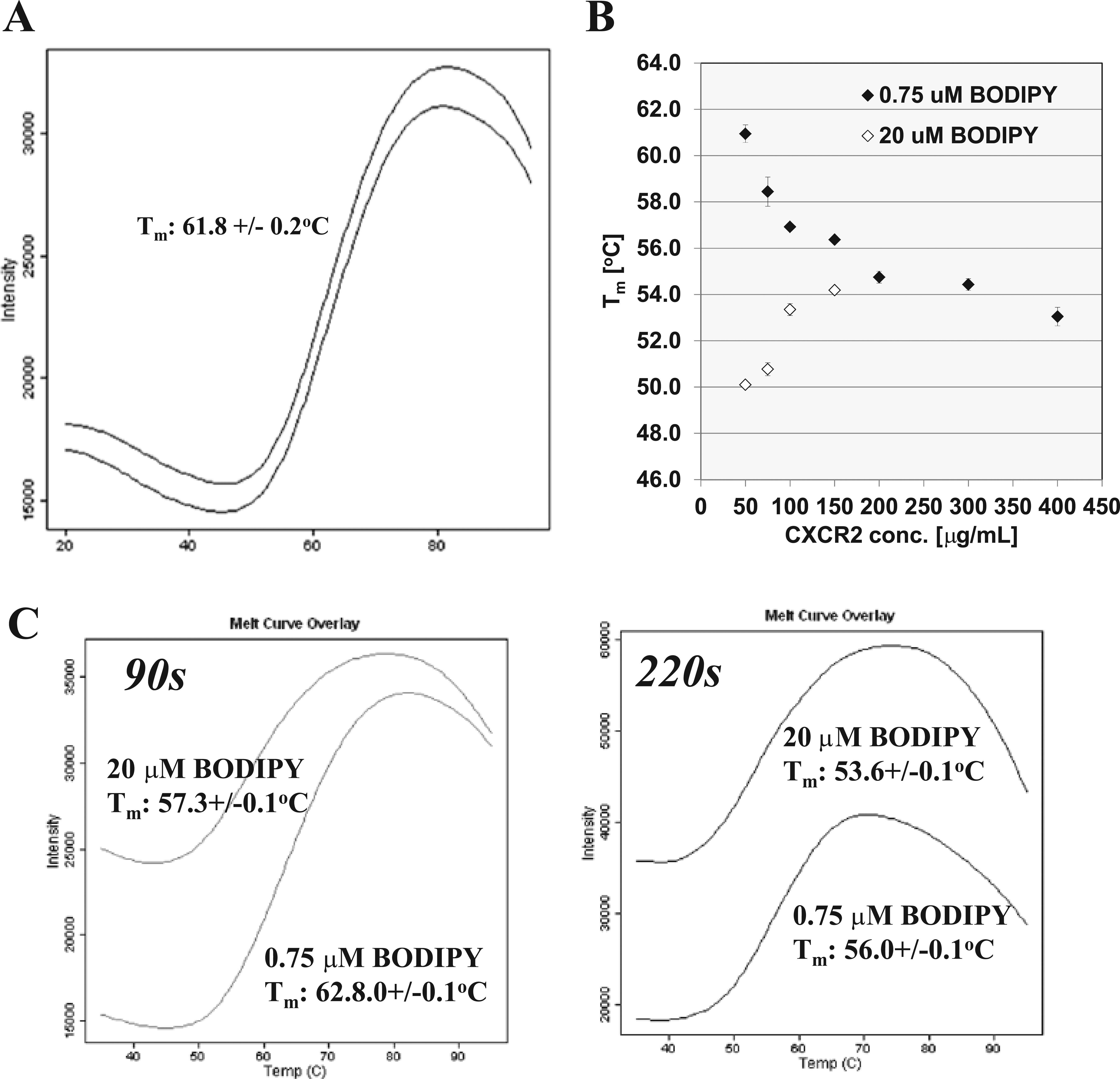
Parameters influencing Tm determination. (
The effect of molar ratio between two reactants in a chemical reaction could be compensated by an increase of the reaction time, as seen in Figure 3C . The reaction time was elongated from 90 s to 220 s per 1 °C, leading to a Tm decrease of 6.8 °C for the low dye concentration. Our experience with other membrane proteins (e.g., transmembrane enzymes, GPCRs) showed that the strength of this effect is very protein specific. Therefore, these parameters have to be evaluated during assay development to establish a robust, reliable assay. For all following experiments, CXCR2 protein/dye ratios of 0.1 and 2.9, respectively, were used, displaying constant Tm values.
Good Correlation of CXCR2 Tm and Tagg Values Determined in DSF and DSLS
DSLS has been shown as a valuable method to follow thermal denaturation of membrane proteins reconstituted in detergent micelles. 13 This label-free technique measures the intensity increase of scattered light during the thermal unfolding and aggregation of proteins, enabling the determination of an aggregation temperature (Tagg). In many cases, Tagg correlates very well with the corresponding Tm being measured by DSF. 14 Therefore, Tagg of CXCR2 was analyzed to confirm the Tm, which was measured by an excess of thiol-reactive BODIPY dye. The thermal profiles of CXCR2 in DSLS and DSF ( Fig. 4A , B ) demonstrated a similar protein stability reflected by a Tagg of 55.1 ± 0.6 °C and a Tm of 53.5 ± 0.3 °C. The comparability of the results underlines the applicability of the BODIPY dye to determine the Tm of an integral membrane protein.
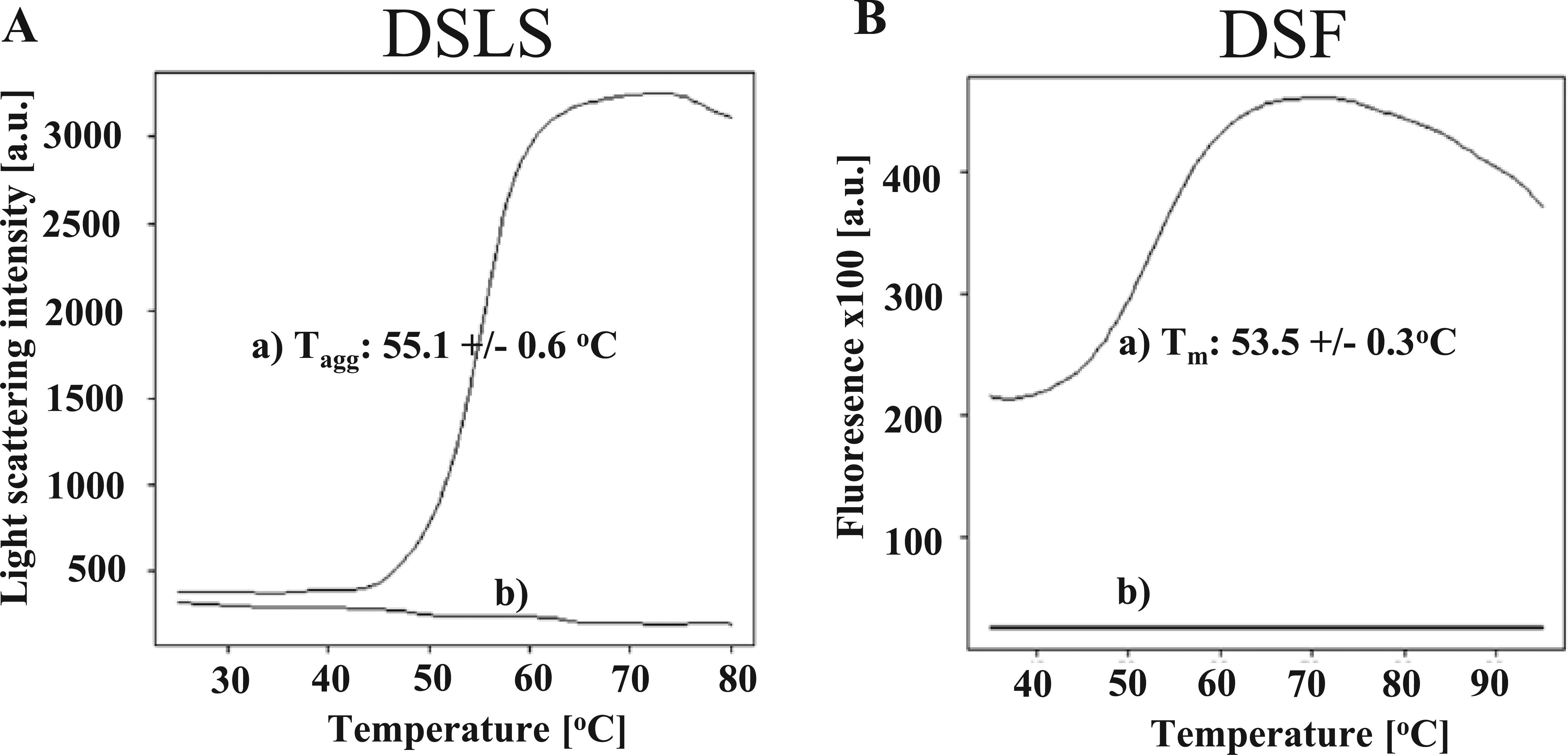
Comparison of differential scanning fluorimetry (DSF) and differential static light scattering (DSLS): CXCR2 melting curve profiles with determined Tagg and Tm. (
Stabilization of CXCR2 by Low Molecular Weight Antagonists
A major application of thermal shift assays is the identification and analysis of low molecular weight ligands that stabilize a target protein of interest. 3 The observed stabilization is a good indicator for a direct physical interaction between both components. To test this application, CXCR2 was analyzed with four well-characterized CXCR2 antagonists at concentrations of 1 to 10 µM using the BODIPY DSF assay ( Fig. 5 ): (a) Sch527123 2-hydroxy-N,N-dimethyl-3-{2-[[(R)-1-(5-methyl-furan-2-yl)-propyl]amino]-3,4-dioxo-cyclobut-1enylamino}-benzamide, (b) pteridone-1, (c) SB265610 N-(2-Bromophenyl)-N′-(7-cyano-1H-benzotriazol-4-yl)urea, and (d) SB225002 N-(2-Bromophenyl)-N′-(2-hydroxy-4-nitrophenyl)urea) ( Fig. 5D ).6,12 As negative controls, cyanopindolol hemifumarate, a β2-adrenergic receptor antagonist, and DMSO were used. 15 All tested antagonists significantly stabilized CXCR2 up to 18.7 °C in comparison to the negative controls ( Fig. 5A , B ). The observed melting point shift (ΔTm) of CXCR2 was dependent on the antagonist concentration, as shown in Figure 5A for pteridone-1 (ΔTm(1µM): 7.3 ± 1.1 °C to ΔTm(10µM): 11.6 ± 0.4 °C).
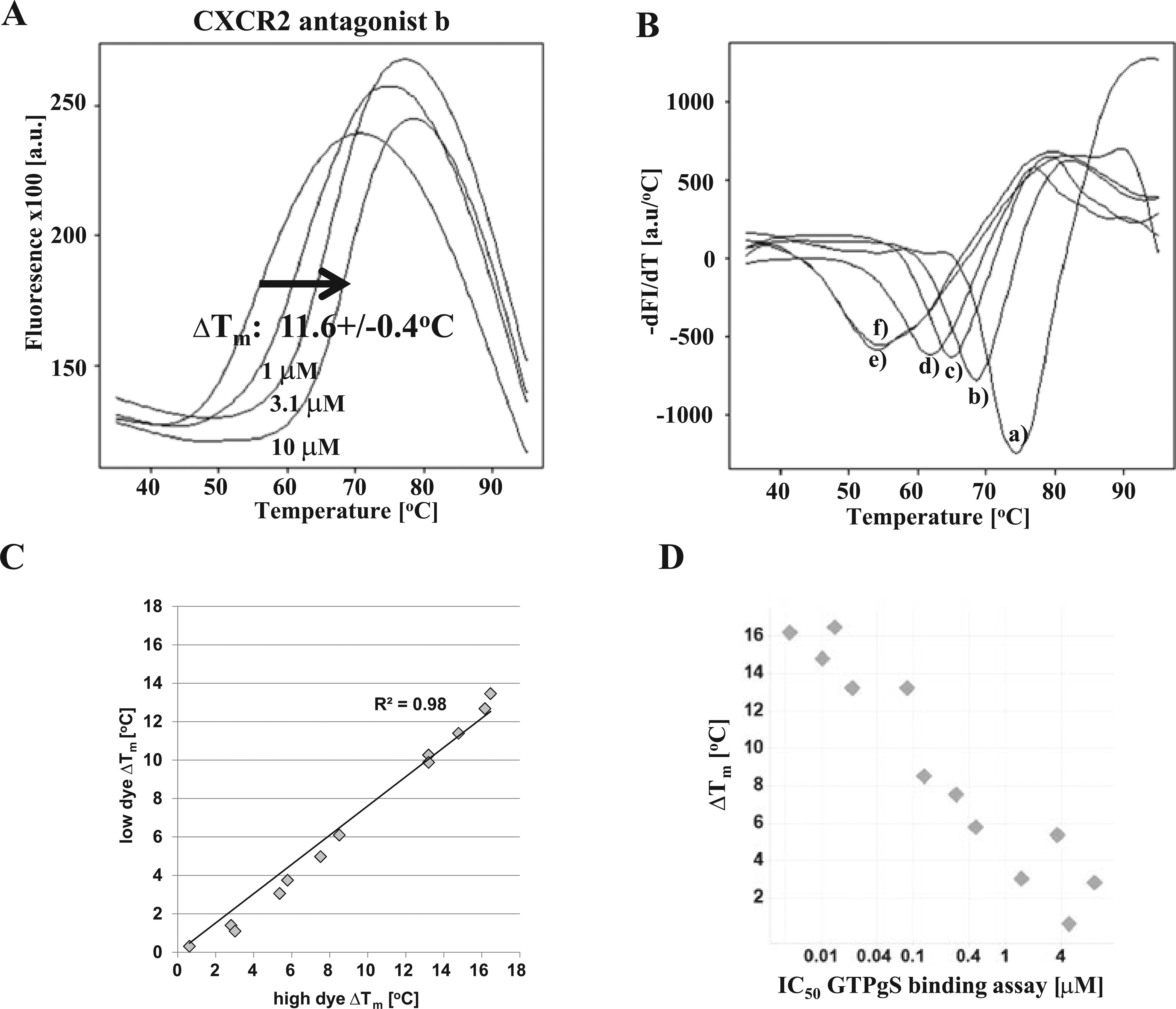
Stabilization of CXCR2 by antagonists and their correlation with activity. (
As shown before in Figure 3 , the molar ratio between protein and dye influenced the measured apparent Tm values of CXCR2. To further investigate whether the protein/dye ratio affects the determined ΔTm values and ranking of potential ligands, 14 LMW antagonists of CXCR2 with IC50 values between 1 nM and 10 µM in a GTPγS binding assay were analyzed at dye deficit (0.3-fold) and excess (10-fold). Under both assay conditions, the rank order of compounds was preserved, which was reflected by a high correlation (R2 = 0.98) between the measured ΔTm values in both formats ( Fig. 5C ). Furthermore, the strength of observed ΔTm correlated highly with the IC50 values, which were determined in the GTPγS binding assay ( Fig. 5D ). These results underline the stability, reproducibility, and validity of the new DSF assay format. Finally, stabilization of CXCR2 upon ligand binding was compared in DSLS and DSF. Therefore, four antagonists were tested in both assay formats. This experiment demonstrated a high correlation of the observed stabilization in the label-free DSLS and fluorescence-based DSF assay format ( Fig. 6 ). Between both assay formats, the rank order of ligands was preserved and the determined ΔTagg correlated with the corresponding ΔTm values.
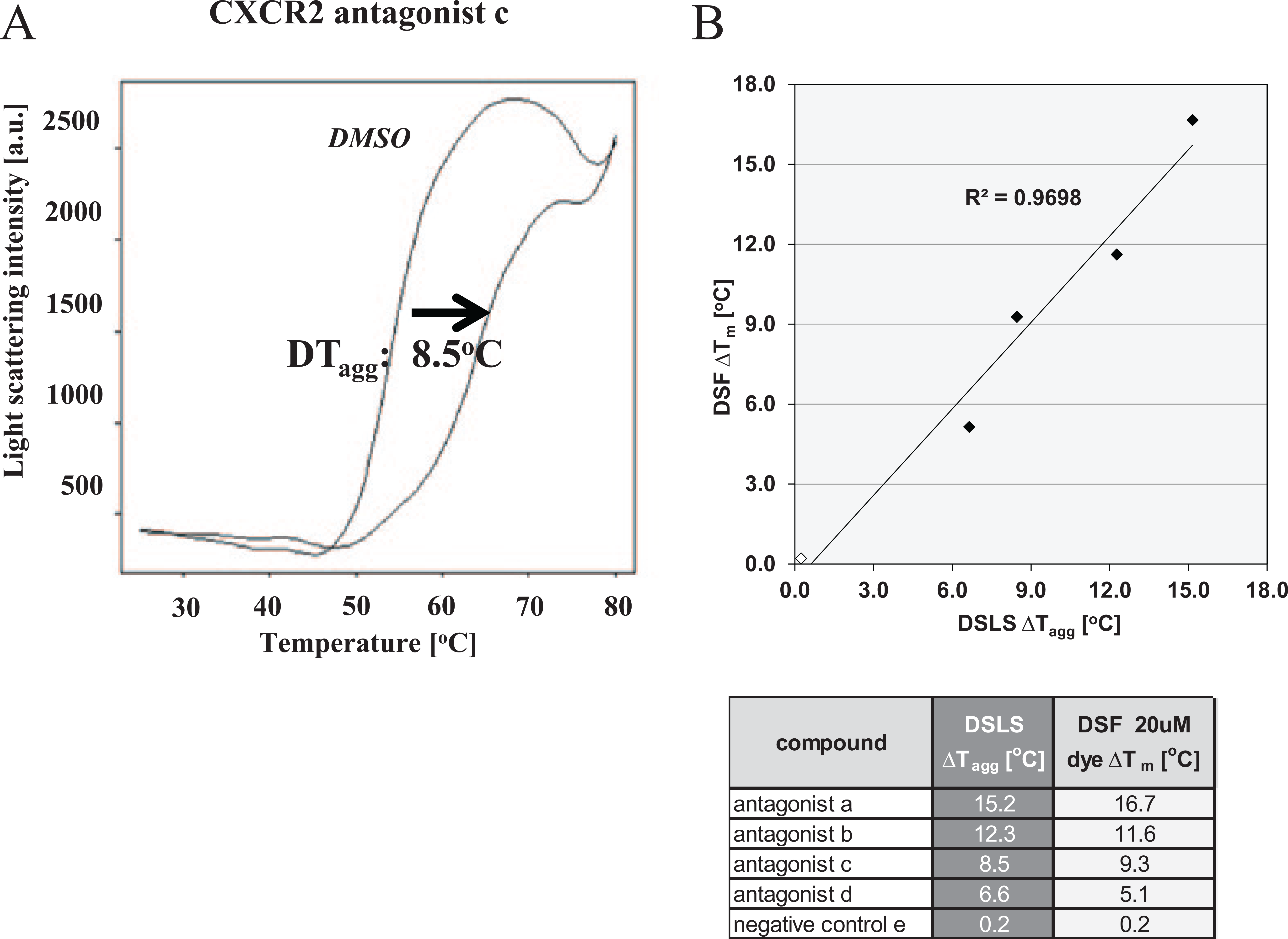
Comparison of CXCR2 ligand interaction data from differential scanning fluorimetry (DSF) and differential static light scattering (DSLS). (
Thiol-Reactive BODIPY FL-L-Cystine: A Versatile Dye to Monitor Membrane Protein Stability
TSA assays are characterized by their simplicity and robustness to follow unfolding of soluble proteins. 3 Thiol-reactive BODIPY FL-L-cystine offers the opportunity to broaden the TSA application to detergent-solubilized membrane proteins and hydrophobic proteins. In contrast to environmentally sensitive dyes such as SYPRO, the fluorescence properties of thiol-reactive dyes are not disturbed by unpolar solvents (e.g., detergents), resulting in low background fluorescence levels. This plate-based assay format can be used (1) to optimize buffer, additives, and detergent conditions for membrane protein purification/crystallization trials; (2) to guide mutation approaches to generate stabilized membrane protein species; and (3) to study ligand-protein interactions. Especially in the case of orphan GPCRs, this assay format could be an entry point to identify first ligands if no functional assay (e.g., GTPγS) is available. Furthermore, ligands found in functional cellular screens (e.g., second-messenger assays) can be validated for the direct physical interaction with the target of interest. The spectral properties of BODIPY FL-L-cystine (ex. 505/em. 513 nm) reduce compound autofluorescence artifacts and are compatible with most commercially available real-time PCR systems in a 384-well format, which are prerequisites to run the assay in a medium- or high-throughput mode. In addition, the assay volume can be miniaturized to 5 to 10 µL using low protein concentrations (0.025–0.1 mg/mL), being an attractive alternative for optical light-scattering methods.8,13 The assay robustness is reflected by highly reproducible apparent Tm values with standard deviations of 0.1 to 0.3 °C. This setup enables the reliable identification of potential ligands stabilizing the target of interest >1 °C.
Footnotes
Acknowledgements
We thank Peter Fürst and Mark Dowling for discussion.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.