
Abstract
SLC6A14 (ATB0,+) is a sodium- and chloride-dependent neutral and dibasic amino acid transporter that regulates the distribution of amino acids across cell membranes. The transporter is overexpressed in many human cancers characterized by an increased demand for amino acids; as such, it was recently acknowledged as a novel target for cancer therapy. The knowledge on the molecular mechanism of SLC6A14 transport is still limited, but some elegant studies on related transporters report the involvement of the 12 transmembrane α-helices in the transport mechanism, and describe structural rearrangements mediated by electrostatic interactions with some pivotal gating residues.
In the present work, we constructed a SLC6A14 model in outward-facing conformation via homology modeling and used molecular dynamics simulations to predict amino acid residues critical for substrate recognition and translocation. We docked the proteinogenic amino acids and other known substrates in the SLC6A14 binding site to study both gating regions and the exposed residues involved in transport. Interestingly, some of these residues correspond to those previously identified in other LeuT-fold transporters; however, we could also identify a novel relevant residue with such function.
For the first time, by combined approaches of molecular docking and molecular dynamics simulations, we highlight the potential role of these residues in neutral amino acid transport. This novel information unravels new aspects of the human SLC6A14 structure–function relationship and may have important outcomes for cancer treatment through the design of novel inhibitors of SLC6A14-mediated transport.
Keywords
Introduction
Amino acids are the building blocks of protein synthesis. 1 They are essential components of all cells and are required for many metabolic pathways, nitrogen metabolism, neurotransmission, and cell growth.1,2 Due to their physicochemical properties, amino acids cannot pass freely across cell membranes but rather undergo carrier-mediated transmembrane (TM) movement by specific amino acid transporters. Mammalian cells express numerous amino acid transporters in their plasma membranes to enable intake and efflux of amino acids between each cell and the extracellular environment. Most amino acid transporters have a relatively narrow substrate specificity. However, there is one mammalian amino acid transporter that has an unusually broad substrate selectivity transporting 18 of the 20 proteinogenic amino acids. 1 This transporter was originally characterized in rabbit ileum and named the β-alanine carrier, although it also transports a wide range of zwitterionic and dibasic amino acids. 3 Separately, a broad-scope amino acid transporter was identified in preimplantation mouse blastocysts and named system B0,+. 4 The transporter cDNA was cloned from human mammary gland and named ATB0,+. 5 ATB0,+ is a sodium- and chloride-dependent neutral and dibasic amino acid transporter that possesses all of the functional characteristics of both system B0,+5 and the β-alanine carrier, 6 demonstrating that the early functional studies in intestine and blastocyst represent function of the same transport mechanism.
Mammalian membrane transporters are categorized (based on amino acid sequence identity) into solute carrier (SLC) families. 7 ATB0,+ is the 14th member of the SLC6 family and is also known as SLC6A14. 8 Although members of the SLC6 family show high levels of sequence identity between transporters, this family includes transporters with very different substrates where some are selective for amino acids and others are able to transport biogenic amines such as norepinephrine (NET/SLC6A2), dopamine (DAT/SLC6A3), or serotonin (SERT/SLC6A4). 8 The Transporter Classification Database (TCDB) of eukaryote and prokaryote transporters 9 catalogs the human SLC6 transporters in the neurotransmitter:sodium symporter (NSS) family (TCDB family 2.A.22), which is within the broader amino acid polyamine organocation (APC) superfamily. 10
SLC6A14 couples co-transport of two Na+, one Cl−, and one amino acid, and is a highly concentrative electrogenic transport system with inward TM amino acid transport being driven by membrane potential and Na+ and Cl− gradients. 1 SLC6A14 transports neutral amino acids and the dibasic amino acids arginine and lysine, and thus transports all essential amino acids and only excludes the anionic amino acids glutamate and aspartate. 1 Different studies showed that the Na+/Cl−/amino acid stoichiometry is 2:1:1 irrespective of whether the transported amino acid is neutral or a cationic amino acid.11,12 With this stoichiometry, transport of neutral amino acids (e.g., glycine) will be associated with the transfer of a single positive charge into cell per cycle, while the transport of cationic amino acids (e.g., arginine) will be associated with the transfer of two positive charges into cell per cycle. This broad substrate specificity also makes the SLC6A14 transporter accessible to a wide variety of amino acid-based drugs and prodrugs, including valacyclovir, 13 valganciclovir, 14 1-methyltryptophan, 15 nitric oxide synthase inhibitors, 16 and carnitine and analogs. 17
Abundant expression of SLC6A14 in healthy adult tissues is fairly restricted with expression mainly in the retina, and respiratory and distal intestinal (ileum and colon) tissues, where it is localized to luminal surfaces.1,13,18 In contrast, ATB0,+ expression is upregulated in many solid tumors including colorectal, 12 cervical, 19 estrogen receptor-positive breast, 11 and pancreatic 20 cancers. The high expression in cancer tissues alongside the broad substrate specificity make ATB0,+ a good target for anticancer treatments using small molecules.
SLC6A14 is known to be regulated posttranslationally, mainly by trafficking, that is, by a process translocating the protein to, or from, the plasma membrane. Change in SLC6A14 localization was correlated with activation of protein kinase C.21,22 Two consensus sites for protein kinase C phosphorylation were reported by Sloan et al. 5 at Ser40 and Ser261. The Ser40 site is also present in the amino acid transporters (e.g., SLC6A5 and SLC6A7), and the protein kinase C consensus site, located at Ser261, is highly conserved in the Na/Cl-dependent neurotransmitter transporter family. 5 For ATB00,+, a consensus site is also reported for phosphorylation by casein kinase II at Thr434. 5 Moreover, on the basis of SLC6A14 primary structure, Sloan at al. 5 hypothesized seven potential N-glycosylation sites in the second extracellular loop (EL2) and one on the third extracellular loop (EL3) of SLC6A14.
The structure of the transporter is unknown, but like most members of the APC superfamily, 10 SLC6A14 is predicted to possess a LeuT-fold core structure, named after the amino acid transporter LeuT, the first transporter crystallized from the APC superfamily. 23 Other APC superfamily members have since been crystallized and have broadly similar folds, including the human serotonin transporter SERT (SLC6A4) 24 and the Drosophila melanogaster dopamine transporter (DAT; SLC6A3). 25 ATB0,+ is predicted to have 12 TM α-helices. On the basis of available APC superfamily crystal structures, TMs 1–5 and TMs 6–10 are predicted to be involved in the transport mechanism; an antiparallel symmetry in their secondary structures gives the α-helices a discrete mobility within the cell membrane and defines binding sites for transported substrates. The binding sites, S1 and S2, are located in the central binding pocket and in the extracellular vestibule, respectively. 26 The central binding pocket must also contain sites that allow the binding of two Na+ and one Cl− ion. TMs 11 and 12 are homologous to structures required for the homodimerization of other transporters, but currently no evidence is available regarding ATB0,+ dimerization. For LeuT-fold proteins, a model of transport has been proposed: 27 in the initial phase of the process, the protein lies in an “outward-facing” (OF) conformation and receives the transported substrate (and the ions) in binding site S2. The interaction between protein, ions, and substrate promotes the translocation of transported solutes to the S1 binding site and closure of the gate. This process starts with the rotation of a specific conserved aromatic amino acid, which in turn leads to a conformational change in the whole protein, promoting its transition to an “inward-facing” (IF) conformation.27,28 Afterward, the substrate is released by the transporter in the intracellular space and the protein returns to the OF conformation, passing through intermediate transitory occluded states.
Therefore, the purpose of this investigation was to present for the first time the SLC6A14 three-dimensional (3D) structure, using homology modeling procedures to build a chimera model. Moreover, with this investigation, we meant (1) to study in silico the structural features of the transporter, (2) to identify in silico the key residues for the molecular recognition mechanism in the S1 binding site, and (3) to gather information about substrate binding.
Materials and Methods
Comparative Modeling of SLC6A14
The sequence of human SLC6A14 (Uniprot ID: Q9UN76) was retrieved from the Uniprot knowledgebase database. 29 After a protein Blast 29 search in the Protein Data Bank (PDB) for a human SLC6A14 homolog, the crystal structure of the Drosophila melanogaster sodium-dependent DAT (PDB ID: 4M48) 25 was set as the main template. A multiple alignment among the NSS family was produced with the Clustal Omega software 30 and used for the homology modeling procedure. Since the crystal structure of DAT lacks the EL2, a second local Blast search was set on residues S205-W270 of SLC6A14, identifying the crystallographic structure of phosphofructokinase (PFKA1) from Saccharomyces cerevisiae (PDB ID: 3O8O) 31 as an adequate template to fix the EL2 gap. The 3D structures of the two templates and of SLC6A14 were also investigated through both SWISS-MODEL 32 and I-TASSER 33 servers. Moreover, the TM prediction program TMHMM was used to determine the putative topology of SLC6A14, strengthening the alignment between SLC6A14 and DAT from a structural point of view. PROTTER was also used to build the prediction of a secondary structure.
Selected 3D structures were first optimized and refined via further computational steps by correcting crystallographic-related errors through the MOE Structure Preparation Module of the Molecular Operating Environment 2018 (MOE, Chemical Computing Group, Montreal, QC, Canada).
The selected portion (residues from L620 to R665) of the phosphofructokinase template was placed carefully in the corresponding position to that in EL2 of DAT. Three amino acids from both crystal structures were deleted at the point of connection between the two templates in order to have more flexibility to shape their junction in the chimeric model of SLC6A14, using the MOE Homology Model tool. Ten different intermediate models were built and submitted to energy minimization (EM) to release internal constraints. The top-scoring model, according to the GB/IV scoring function, was submitted to further EM with the FINE option until the root mean square (RMS) gradient reached a value below 0.5 kcal/mol/Å.
The Amber10:ETH 34 force field with the reaction field model for electrostatics was applied for the whole modeling procedure.
Equilibration and Cluster Extrapolation
Molecular dynamics (MD) simulation and frame clustering procedures were carried out with the Schrödinger Small-Molecule Drug Discovery Suite 2018-01 (D. E. Shaw Research, New York, NY; Schrödinger, New York, NY).
The Desmond System Builder tool was used to place the apo-model of SLC6A14 into a POPC membrane bilayer. Protein orientation was set up according to the OPM server, 35 which provides spatial arrangements of membrane proteins with respect to the hydrocarbon core of the lipid bilayer. The N- and C-termini of the protein were capped. The system was solvated with 10,174 SPC water molecules in a cubic box with 90 Å edges. Adding chloride ions neutralized the exceeding positive charge; sodium chloride was further added up to a 0.15 M concentration. The system was energy-minimized to relax the assembly and remove clashes between protein, membrane, and solvent in the new setup.
To produce an equilibrated model of SLC6A14, the system was submitted to a 500 ns MD simulation using the Desmond Molecular Dynamics tool. Periodic boundary conditions (PBCs) and the following parameters were set: 300 K and Nose–Hoover thermostat for temperature coupling, 1 bar and Martyna–Tobias–Klein piston for pressure coupling, and 2 fs as the integration time step. Coordinates and velocities of each atom were saved every 0.5 ps. The trajectory was then analyzed using both the Desmond Simulation Event Analysis tool and VMD. 36
The Desmond Trajectory Frame Clustering tool was used to cluster the whole 500 MD simulation in order to select the most representative frame (the centroid) for each cluster. Distances between clusters were computed from the root mean square deviation (RMSD) matrix of alpha carbons with respect to the first frame of the MD simulation. Two different runs were performed to set the number of generable clusters according to the dendrogram.
The OPLS3e 37 force field, suitable both for proteins and for small molecules, was applied both in the MD simulation and in the frame clusterization procedures.
Molecular Docking
The centroid of the most populated cluster was selected as the reference structure for molecular docking procedures. Water, ions, and the POPC membrane were washed out from the system and three pivotal ions (one Cl and two Na) from the DAT template were transferred to the SLC6A14 equilibrated model. The ions::SLC6A14 3D structure was then energy-minimized using the prime in order to fix structural issues in the ion binding domain.
Tested ligands were downloaded from PubChem and prepared for docking with Schrödinger Ligand Preparation using OPLS3e 37 as the force field.
The molecular docking procedure was carried out with Schrödinger Glide Docking in the “extra precision” (XP) mode in order to evaluate the ability of the tested ligands to bind the SLC6A14 binding channel, keeping only the five top-scoring poses and setting the rigid docking protocol.
The top-scoring solution for each ligand was submitted to Schrödinger Prime MM-GBSA, which integrates molecular mechanics energies with the generalized Born and surface area continuum solvation 38 in order to compute ligand binding and ligand strain energies for a set of ligands and a single receptor.
To study ligand::protein interaction and verify the stability of the ligand placement, we submitted the top-scoring solution for each ligand to 50 ns of MD simulation with the Desmond Molecular Dynamics tool. PBCs and the following parameters were set for each MD simulation: 300 K and Nose–Hoover thermostat for temperature coupling, 1 bar and Martyna–Tobias–Klein piston for pressure coupling, and 2 fs as the integration time step. Coordinates and velocities of each atom were saved every 0.5 ps. The trajectories were then analyzed using the Desmond Simulation Interaction Diagram tool and inspected through VMD.
The OPLS3e 37 force field was applied both in molecular docking procedures and in MD simulations.
Results and Discussion
Secondary Structure Analysis and 2D Topology Prediction
None of the SLC6A14 homologous templates provide complete sequence coverage, as all of them leave out EL2. Not unexpectedly, the SWISS-MODEL and I-TASSER predictions on the whole protein are unsatisfactory because, in both cases, the EL2 is predicted at an exceedingly high distance from a theoretical conformation resulting in a very low local quality estimation. A very high number (>30) of outliers is observed in both Ramachandran plots.
Building a chimera model seems then to be the right strategy to study SLC6A14 at an atomistic level.
Taking the global alignment among members of the NSS family as the reference structure to model SLC6A14 in its OF conformation, we selected DAT, 25 with a sequence identity of 43% and a similarity of 59% (50% and 68% of identity and similarity, respectively, for the TM domain), as the most suitable template presently available. Moreover, unlike other suitable templates whose structure contains only one cation and one anion, DAT is co-crystallized with two Na+ ions and one Cl− ion. On the other hand, according to BLAST, PFKA1, with a local sequence identity of 28% and local similarity of 46%, appears to be the most suitable template for modeling EL2.
The second step toward building a chimera model of SLC6A14 was to compare its putative secondary structure and the two-dimensional (2D) topology predicted with three independent methods, while taking into account both the alignment and the topology of the DAT crystal.
25
The PROTTER and TMHMM predictions are compliant with the DAT secondary structure and its TM placement. In particular, the first three TM α-helices of DAT are more extended than expected from the prediction, while TM α-helices from 4 to 7 and from 9 to 12 are in good agreement with the expectations. TM α-helix 8 seems to be shifted toward the C-terminus by five amino acids with respect to the PROTTER prediction, while it complies with the TMHMM prediction. The alignment among SLC6A14, DAT, and PFKA1 was carefully verified taking into account the PROTTER and TMHMM predictions. The predicted topology of SLC6A14 is shown in Figure 1 , also confirming the Sloan et al. 5 hypothesis of six potential N-glycosylation sites in EL2 (N155, N163, N174, N189, N197, N202) and one in EL3 (N302) ( Fig. 1A ). Moreover, the PROTTER prediction places Ser40 in the intracellular domain (N-terminus) and Ser201 in the TM domain, as the first amino acid of α-helix 5. Thr434 is predicted in the intracellular loop 4 (IL4) by PROTTER, confirming the Sloan et al. 5 observation.
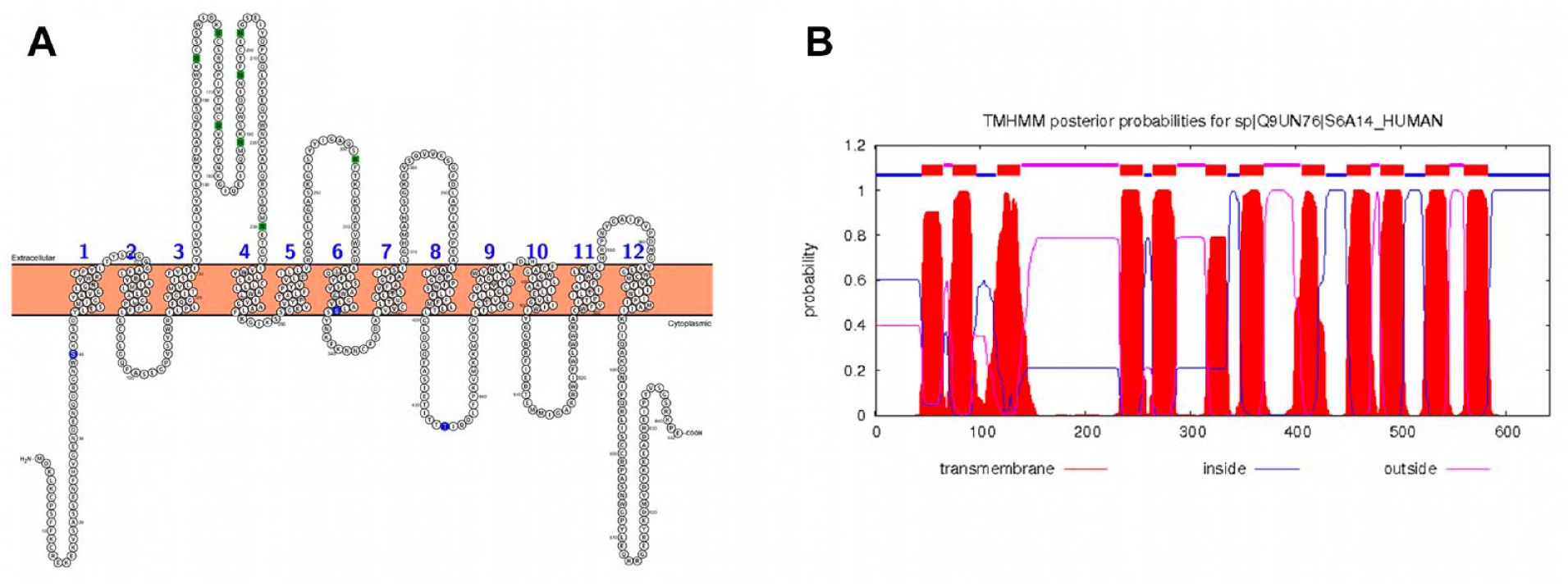
2D topology and transmembrane (TM) prediction for SLC6A14. Results with PROTTER are shown in
Homology Modeling of SLC6A14 and Its MD Equilibration
The Ramachandran plot for the chimera model of SLC6A14, based on both DAT and PFKA1, shows only six outliers, none of which are in the TM domains (
In order to equilibrate the chimera model of SLC6A14, we carried out a 500 ns MD simulation. Neither substrates nor ions were placed into the SLC6A14 transport channel in order to obtain the equilibrated OF model of apo-SLC6A14. The evaluation of the stability of our model during the simulation was based on both energetic and geometric parameters, as described in the following.
The general stability of the SLC6A14 model is confirmed by the tendency of the MD simulations to reach convergence. In detail, the tendency to reach an equilibrium state is suggested by the trend of the RMSD values, which, after 100 ns, reach a plateau around 3.5 Å ( Fig. 2A ); its fluctuations toward the end of the simulation are around some thermal average structure. The lowest root mean square fluctuations (RMSFs) are associated with α-helices, the highest with the loops, both intra- and extracellular, with maximum fluctuation in EL2 ( Fig. 2B ).
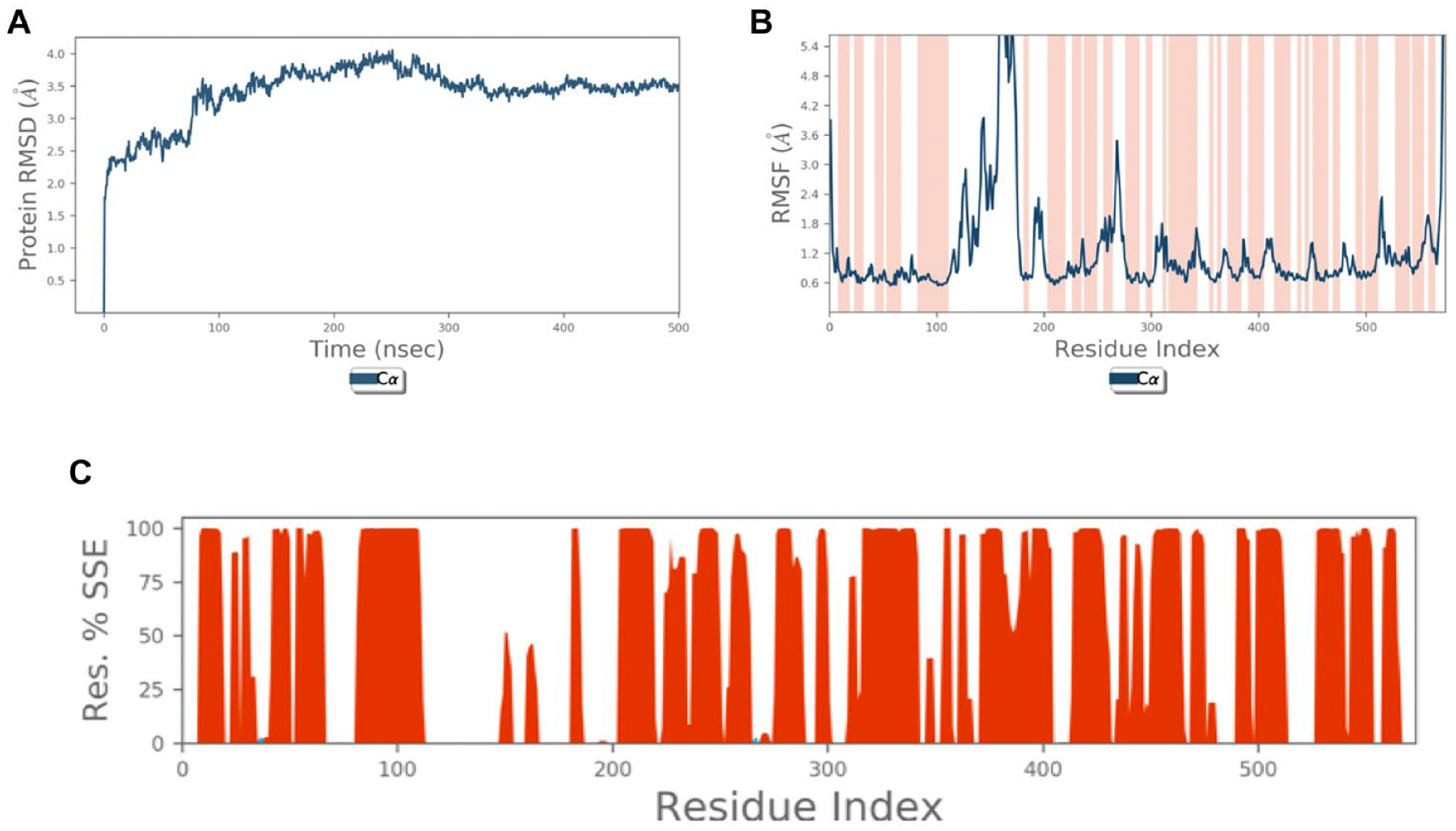
Stability of SLC6A14 model. (
Protein secondary structure elements (SSEs), that is, α-helices and β-strands, were monitored throughout the simulation.
Figure 2C
reports SSE distribution by residue index, while
The final model was identified generating the centroid of the most populated cluster, which is characterized by a very low internal Cα-RMSD value (1.4 Å). This procedure allowed us to assume that this cluster is populated by timeframe representative structures of the equilibrated part of our simulation. Moreover, as reported in
The Ramachandran plot of the selected frame shows only one outlier (
Moreover, in the selected SLC6A14 conformation, we found a Na+ ion in the S2 ion binding site. Analyzing the whole MD simulation, the Na+ ion landed in the S2 binding site after approximately 250 ns and remained there until the end. A second Na+ ion landed in the SLC6A14 transport channel at 400 ns and stayed at the bottom of the S1 substrate binding site until the end of the MD simulation.
Finally, in the selected SLC6A14 conformation, the side chains of both Ser40 and Ser261 are exposed to the solvent, as for Thr434, suggesting that these residues represent potential phosphorylation sites. Also, all the N-glycosylation sites, predicted by PROTTER, belong to a region accessible to glycosylation enzymes.
Structural Features of the Binding Sites
To study the structural features of the SLC6A14 transport channel, and its binding sites, we imported two Na+ and one Cl− ion from the DAT crystal structure to our SLC6A14 equilibrated model; then we relaxed the protein to avoid atom clashes. Figure 3 shows the SLC6A14 equilibrated structure after the local minimization of the ion pocket.
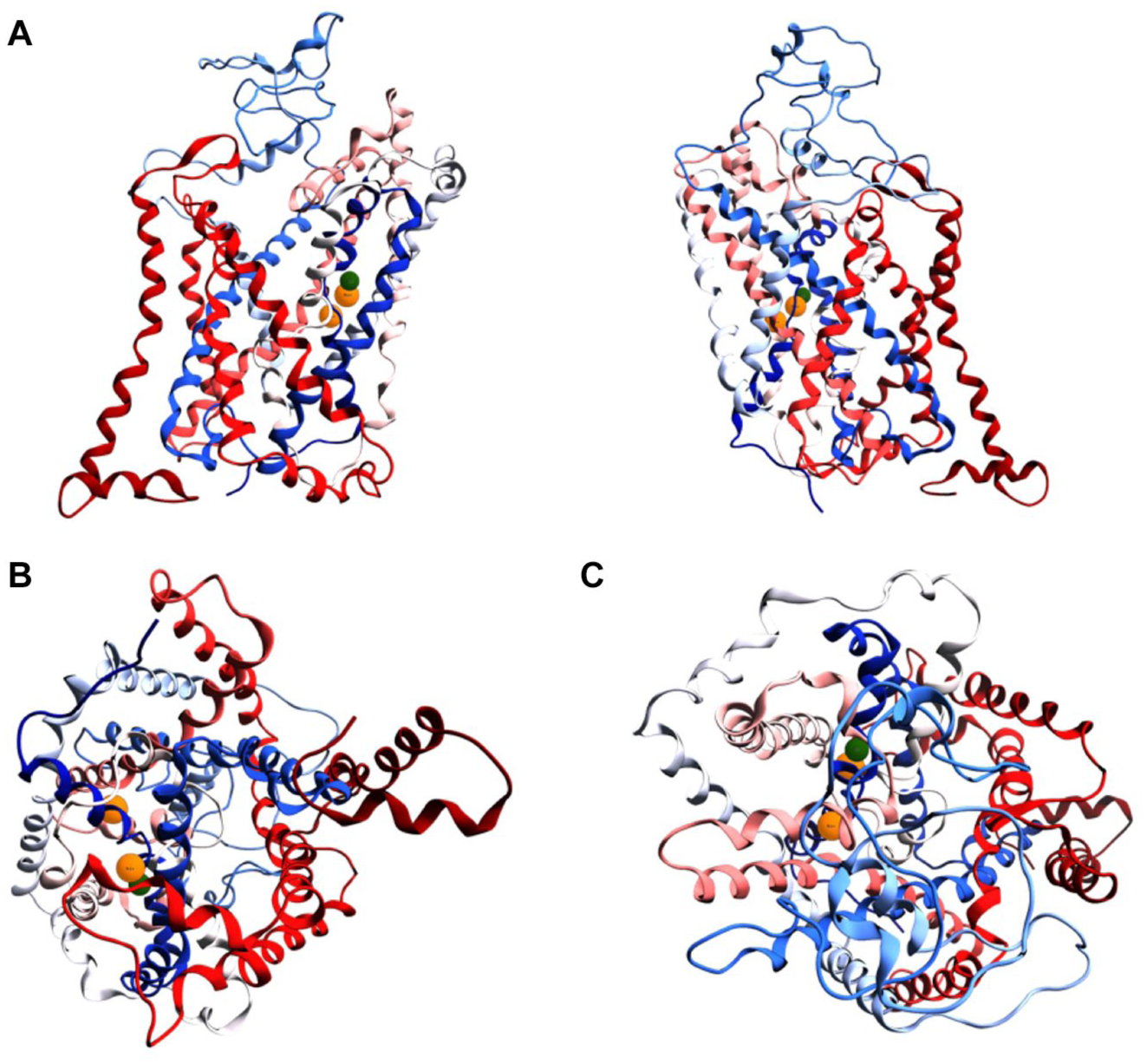
SLC6A14 equilibrated model. (
As mentioned in the introduction, it is well known that, when in the OF conformation, SLC6 family members have a binding site in the bottom of their transport channel. This binding site includes two specific regions: the first for the substrate and the second for the co-transported ions, which are a fundamental feature of all the SLC6 transporters.
In SLC6A14 we found two distinct sites in the S1 pocket that are unambiguously occupied by a Na+ ion, designated Na1 and Na2. 39 Both Na+ binding sites are believed to play a key role in stabilizing TMs 1 and 6 in the presence of a substrate.
The Na1 binding site is close to the Cl− binding site and is separated from Na2 by TM 1, sharing some amino acids with it. Residues composing the Na1 and Na2 ion binding sites are conserved among the transporters of interest and, in general, are highly conserved across the whole SLC6 family. 26 As suggested by Kristensen et al., 26 the evidence of contiguous surfaces between Na2 and the substrate binding site can be the basis for a coupled translocation mechanism for Na+ and substrate. On the other hand, the placement of Cl− in the Na1 binding site supports the idea that Cl− is translocated together with the Na+ ion along the SLC6A14 channel during the transport of the substrate. The same ion placement has also been reported for the crystallographic structures of DAT (SLC6A3) and human SERT (SLC6A4).24,25 These data confirm that our equilibrated SLC6A14 model maintains its ion binding site ( Fig. 4A ); in particular, the placement of Na+ into the Na2 binding site throughout the MD simulation is comparable to the situation in DAT.
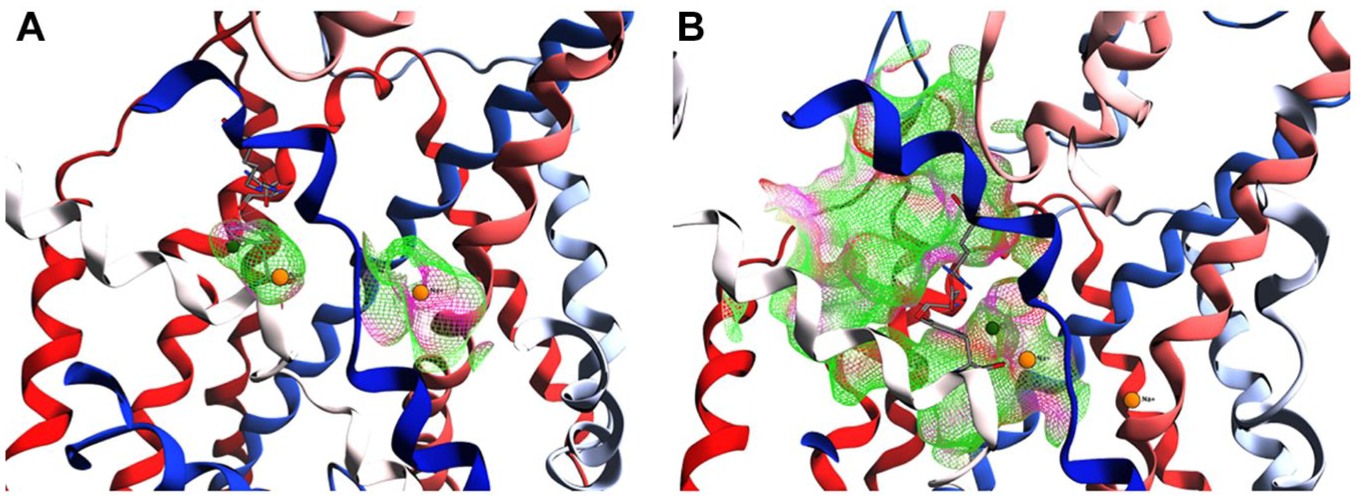
SLC6A14 binding sites. (
According to Kristensen et al.,26 the S1 substrate binding site is defined by polar, aromatic, and aliphatic amino acid side chains provided by all four TMs (TMs 1, 3, 6, and 8) that surround the binding site, in addition to the backbone amide groups from the unwound regions of TMs 1 and 6. 39 The S1 pocket can be divided into two regions: a polar region formed exclusively by the unwound regions of TMs 1 and 6 and a hydrophobic pocket formed by aliphatic side chains from TMs 1, 3, and 6 ( Fig. 4B ).
In the SLC6A14 equilibrated model, we can also detect the S2 binding site in the gating region, 26 which is partially defined at the bottom by three amino acids: Tyr321, Arg61, and Asp478, which are partially conserved among SLC6A14, DAT, and SLC6A4.
Arg61 and Asp478 establish an H-bond, fixing the Arg61 in a conformation stretched toward the inside of the transport channel. Tyr321 in SERT is stretched toward the transport channel, unlike its corresponding amino acids Phe319 and Phe335 in DAT and SLC6A14, respectively.
From a structural point of view, this particular aromatic amino acid seems to assume the same function of Trp202, the gating residue of the arginine/agmatine antiporter AdiC. This gating mechanism is also supposed to be conserved for the SLC6 family, 40 and it was simulated for LAT1 (SLC7A5) via target MD. 41 To evaluate the gating arrangement, we compared the placement of Phe319 in two DAT crystallographic structures in OF conformation, with gate open (PDB ID: 4M48) 25 and closed (PDB ID: 4XPA), 42 via a structural superposition. As shown in Figure 5A , Tyr321 of the equilibrated model of SLC6A14 is in an intermediate position between DAT Phe319 open and closed conformations, suggesting that this amino acid also has an intrinsic flexibility in the absence of substrates/inhibitors. This intrinsic flexibility is also observed during the MD simulation, during which Tyr321 oscillates between open and partially closed conformation, with respect to DAT as a reference ( Fig. 5A ), suggesting that this amino acid has many degrees of freedom while it explores the conformational spaces in its surroundings.
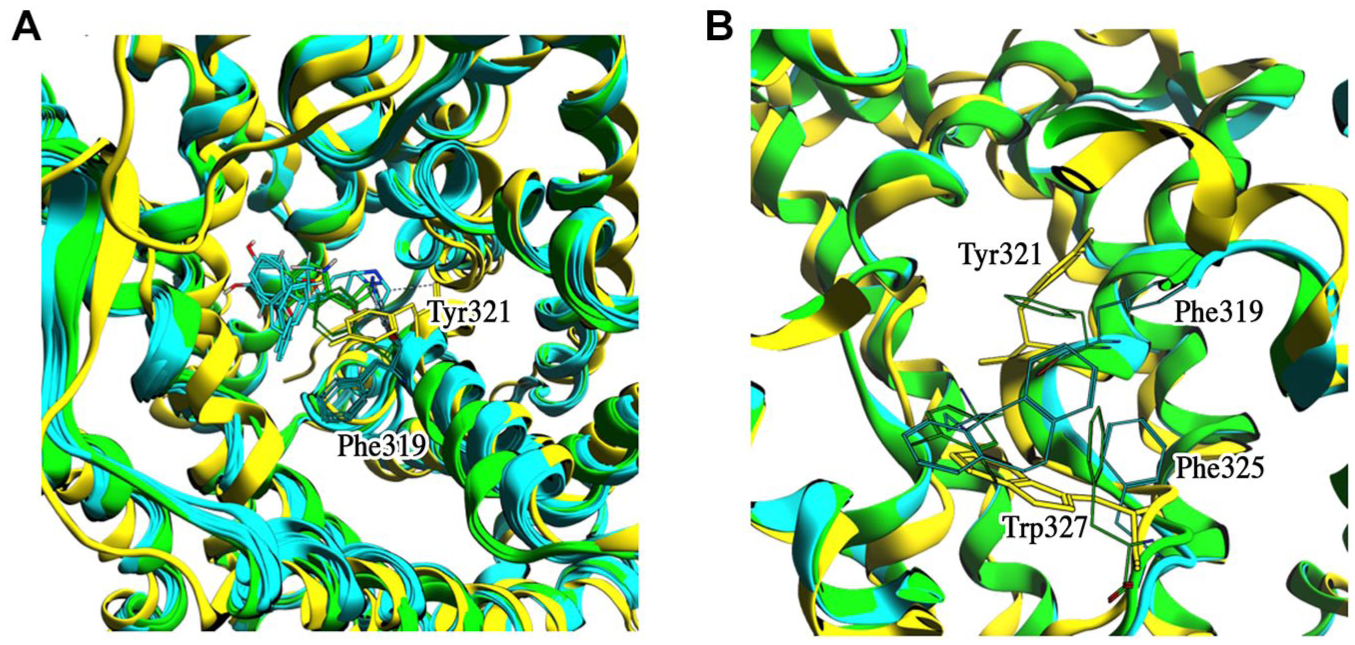
(
Moreover, we also identified Trp327 as a second putative inner gate of SLC6A14. In particular, this amino acid rotates from a DAT Phe325-like conformation to an occluded one, defining the bottom of the S1 substrate binding site. Evidence that Trp327 can be involved in the inner gating mechanism can be obtained from structural analysis of the DAT Phe325 behavior in the presence of transported molecules. As shown in Figure 5B , DAT Phe325 has some degrees of freedom to coordinate itself with the transported molecules. In the two reference crystallographic structures of DAT, Phe325 is oriented by the inhibitors and it is alternately open or closed, with Phe319 behaving as the outer gate.
Molecular Docking
SLC6A14 transports 18 of the 20 proteinogenic amino acids and only excludes glutamate and aspartate. 2 Molecular docking simulation was applied to place the substrates into the S1 binding site, appraising substrate::carrier interactions and computing the relevant docking scores.
Rigid docking successfully generated poses characterized by negative binding free energy values for all the tested substrates. No issues (bumps, failures, etc.) connected with the selected docking pipeline were found. Moreover, as shown in
On the basis of rigid docking results, each complex substrate::carrier was submitted to a 50 ns MD simulation to evaluate the stability of the interactions and to assess the molecular recognition mechanisms, looking for pivotal amino acids involved in the recognition of the different substrates.
Tryptophan and 1-methyl3-
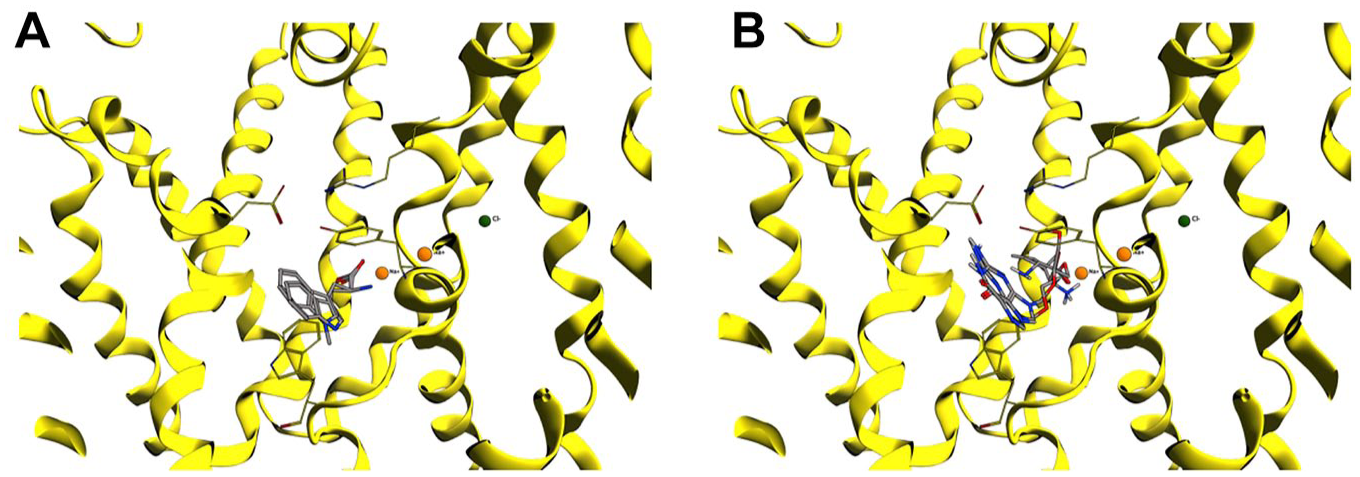
(
Tyrosine overlaps the tryptophan docking pose, with an associated docking score of −5.4 kcal/mol, mainly interacting with Ala326 and Ser324. The MD simulation also highlights H-bond interactions with Val54, Leu56, Ser324, and Ser 423 and other important hydrophobic interactions with Tyr132, Tyr321, and Trp327.
Arginine, leucine, and isoleucine share the same binding pose and have similar docking scores of −4.9, −4.6, and −4.4 kcal/mol, respectively. Arginine mainly interacts with Tyr132, Phe320, and Asp478, while leucine and isoleucine establish H-bonds with Tyr132, Phe320, Val325, and Ser324. The MD simulations show that Asp478 is the pivotal binder for arginine, and it is probably also involved in the recognition mechanism, while for leucine and isoleucine Tyr321 is the pivotal amino acid, interacting for ~94% of the MD simulation. Arginine interacts with Cl−, while leucine interacts with Na+ in the Na1 ion binding site.
As reported in Scalise et al., 2 glutamine is compressed between Val128 and Tyr321, with a docking score value of −3.8 kcal/mol. Glutamine also interacts via H-bonds with Tyr132, Phe320, and Asp478 in its binding pose, while the MD simulation detects ionic interactions with Ala53, Tyr321, and Ser324. Glutamine also interacts with Na+ in the Na1 ion binding site.
Proline and cysteine have very similar binding poses and docking scores of about −3.1 kcal/mol. These tested amino acids share the same interaction with Tyr132 and Phe320, establishing four H-bonds during the MD simulations also with Ser324 and Val325. Contacts with Tyr132, Phe320, and Ser324 are conserved for both substrates for more than 90% of the total time of the MD simulations.
Carnitine shows a completely different binding pose with respect to the one observed with the tested amino acids, with an associated docking score of −1.4 kcal/mol. From the ligand interaction diagram of molecular docking, it interacts with Leu56 and Ser423 via H-bonds, while during the MD simulation it shifts in the S1 binding pocket from Ser478 to the S1 ion binding site, interacting with Na+. In this context, carnitine interacts with Tyr95, Leu99, Gly100, Tyr176, Trp381, Tyr375, and Ser378.
The two prodrugs valganciclovir and valaciclovir have dissimilar behavior in their binding poses. Despite their binding poses partly overlapping ( Fig. 6B ), the docking score is −5.2 kcal/mol for the former and −1.2 kcal/mol for the latter. Both substrates interact with Asp478, Tyr321, and Tyr132, but valganciclovir also establishes contacts with Tyr52, Leu56, and Asp477. The MD simulations show for both substrates that those with Tyr132 and Asp478 are the most conserved H-bond interactions, together with Leu56, Ala537, and Ser324. None of them interact with Na+ or Cl− ions.
For all the tested substrates, Val128 is also always involved via a hydrophobic interaction. In fact, this amino acid is part of the S1 binding site, and it sterically occupies part of the pocket with its side chain. Moreover, arginine, glutamine, and both the tested prodrugs are coordinated by Asp478 that is placed in the upper part of the S1 binding site, together with Tyr321 that is pivotal in all the interactions. These data suggest that Tyr321 can act as a gate, involving also Asp478, while other aromatic and aliphatic side chains of amino acids in S1 are involved in the recognition and transport mechanism of substrates through the SLC6A14 channel.
To conclude, we propose here the first atomistic model of the SLC6A14 transporter based on a chimeric approach: the two templates selected for modeling the OF SLC6A14 state are the most suitable so far available, and one of them has also been profitably used in a recent publication. 2 Thanks to 500 ns of MD simulation, we identified a specific behavior of Tyr321, highlighting its role in the outer gating mechanism, which is coordinated with the involvement of Arg104 and Asp478. We also identified Trp327 as a putative inner gate, in analogy to DAT, our reference template. These two amino acids define the S1 binding site, while Tyr321 is at the same time at the bottom of the S2 binding site. From the cluster analysis, we selected a reference 3D structure of SLC6A14 and we observed that ion binding sites are conserved across the SLC6 family.
Natural amino acids and some other well-known substrates were tested for interaction via molecular docking simulation. For all of them, the docking scores were evaluated, and the molecular recognition mechanism was characterized, highlighting that both gates are essential for binding. In a broader way, Tyr52, Gly57, Val128, Ser322, and Ser324 compose an ensemble of amino acids that orientate the substrates, confirming the structural and modeling observations of others.2,21,37
Supplemental Material
DISC867317_SupplementalMaterial – Supplemental material for SLC6A14, a Pivotal Actor on Cancer Stage: When Function Meets Structure
Supplemental material, DISC867317_SupplementalMaterial for SLC6A14, a Pivotal Actor on Cancer Stage: When Function Meets Structure by Luca Palazzolo, Chiara Paravicini, Tommaso Laurenzi, Sara Adobati, Simona Saporiti, Uliano Guerrini, Elisabetta Gianazza, Cesare Indiveri, Catriona M. H. Anderson, David T. Thwaites and Ivano Eberini in SLAS Discovery
Footnotes
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by grants from MIUR “Progetto Eccellenza” and MIUR “Finanziamento annuale individuale delle attività di base di ricerca.” I.E. gratefully acknowledges departmental Linea 2—Azione A 2017 funding.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.