
Abstract
Mutations in the gene encoding leucine-rich repeat kinase 2 (LRRK2), such as the G2019S mutation, are the most common cause of familial Parkinson’s disease (PD). The G2019S mutation impairs neurite outgrowth. We hypothesized that those effects could be related to an altered expression of pluripotency genes, which may provide a readout for a screening assay based on LRRK2 function. Here, we show that the G2019S mutation mediates a sustained aberrant upregulation of the transcription factors Nanog and Oct4 that in wild-type are downregulated after differentiation. The aberrant regulation of Nanog can be concentration dependently reversed by LRRK2 tool inhibitors. Building on this knowledge, we developed an assay for the identification and assessment of compounds that inhibit the aberrant pathophysiological activity of mutant LRRK2. Furthermore, the aberrant neural pluripotency is consistent with Parkinson’s pathophysiology and with the epidemiological association between the G2019S genotype and cancer risk.
Introduction
Mutations in the leucine-rich repeat kinase 2 (LRRK2) (PARK8, OMIM 607060) gene are the most common cause of familial Parkinson’s disease (PD). It has been demonstrated that at least in vitro, the frequently occurring pathogenic G2019S mutation is associated with an increase of LRRK2 kinase activity. 1
The full mechanistic understanding of how LRRK2 pathogenic mutations lead to an increased risk of disease is limited because the full pathway is still not known. LRRK2 is suggested to play roles in cytoskeletal dynamics, mitochondrial dysfunction, synaptic machinery, dopamine homeostasis, and autophagic processes.
A frequently described phenotypical alteration by G2019S LRRK2 is the early reduction of neurite ramifications and length, which has been described, for instance, in transgenic rodent models, 2 primary dopaminergic neurons, 2 and neuroblastoma cell lines. 3 However, it is not clear how this relates to dopaminergic cell death. In addition, LRRK2 affects neuronal progenitors: Tong et al. 4 described abnormalities on the nucleus of induced pluripotent stem cells (iPSCs) from PD G2019S LRRK2 patients. Consistent with a role in neuronal differentiation, retinoic acid receptor (RxR) signaling 5 has also been proposed to be modified by LRRK2 knockout. To increase our understanding of the above, we decided to further explore the pathophysiological mechanisms linking the G2019S LRKR2 hyperactive kinase to neuronal differentiation. Initially, it may appear counterintuitive that mechanisms underlying PD could be influenced by neuroplasticity and neurogenesis events, but evidence indicates that those pathways may play a key role in processes still active in the aged brain. Postmortem PD brains still exhibit proliferating capabilities suggesting the presence of adult neuronal stem cells even in 80-year-old patients. Moreover, in samples from both aged rodent models and aging humans, although brain volume is decreased compared with younger ages, the number of cells is known to increase in neurogenesis-competent regions such as the olfactory bulb. 6
We envisioned developing a mechanistic assay that explored the efficiency of compounds targeting LRRK2 in pathways related to neurogenesis and neuronal differentiation. As mutations in LRRK2 are the most common cause of familial PD, the protein continues to be a target for drug discovery programs. To expedite such efforts, a mechanistic cellular assay based on LRRK2 aberrant function is highly desirable. However, current high-throughput screening (HTS) LRRK2 assays are based on assessing LRRK2 kinase activity using an artificial substrate such as LRRKtide or a cellular LRRK2 target engagement marker such as the Ser935 site in LRRK2. Those approaches have the drawback that the chosen substrate may not be relevant for PD pathogenesis. Here, in addition to providing a mechanistic link between LRRK2 and PD, we report the development of a mechanistic LRRK2 assay measuring the effects of compounds on pathways affecting neurogenesis and differentiation.
Methods
Chemicals, Reagents, and Plasmids
Rabbit polyclonal anti-Nanog antibody was purchased from Abcam (ab21624, Berlin, Germany). Plasmids encoding wild-type LRRK2 under the pCMV6 promoter were obtained from Origene (Herford, Germany). The construct was used as a template for site-directed mutagenesis to generate a construct encoding G2019S LRRK2 under the pCMV6 promoter (pCMV6-G2019S-LRRK2). Plasmids encoding GFP-G2019S-LRRK2 and kinase-dead (KD) GFP-KD-LRRK2 were a gift from Dr. Matthew J. Farrer, Mayo Clinic. The latter was also used to subclone the open reading frame of KD LRRK2 (without the sequence encoding for GFP) into the pCMV6 vector to create pCMV6-KD-LRRK2. All constructs were verified by DNA sequencing. Plasmid constructs and controls used specifically in the luciferase assay are described in that section. Compound BMPPB-32 was identified as compound example 32 in a patent application (WO2011038572A1), synthesized, and then characterized in detail as described in Afsari et al.7
Culture, Transfection, and Differentiation of Neuroblastoma Cells
The protocol was the same for all experiments described here ( Fig. 1A ). A detailed description of the procedure, including the HTS protocol, is provided as supplemental material.
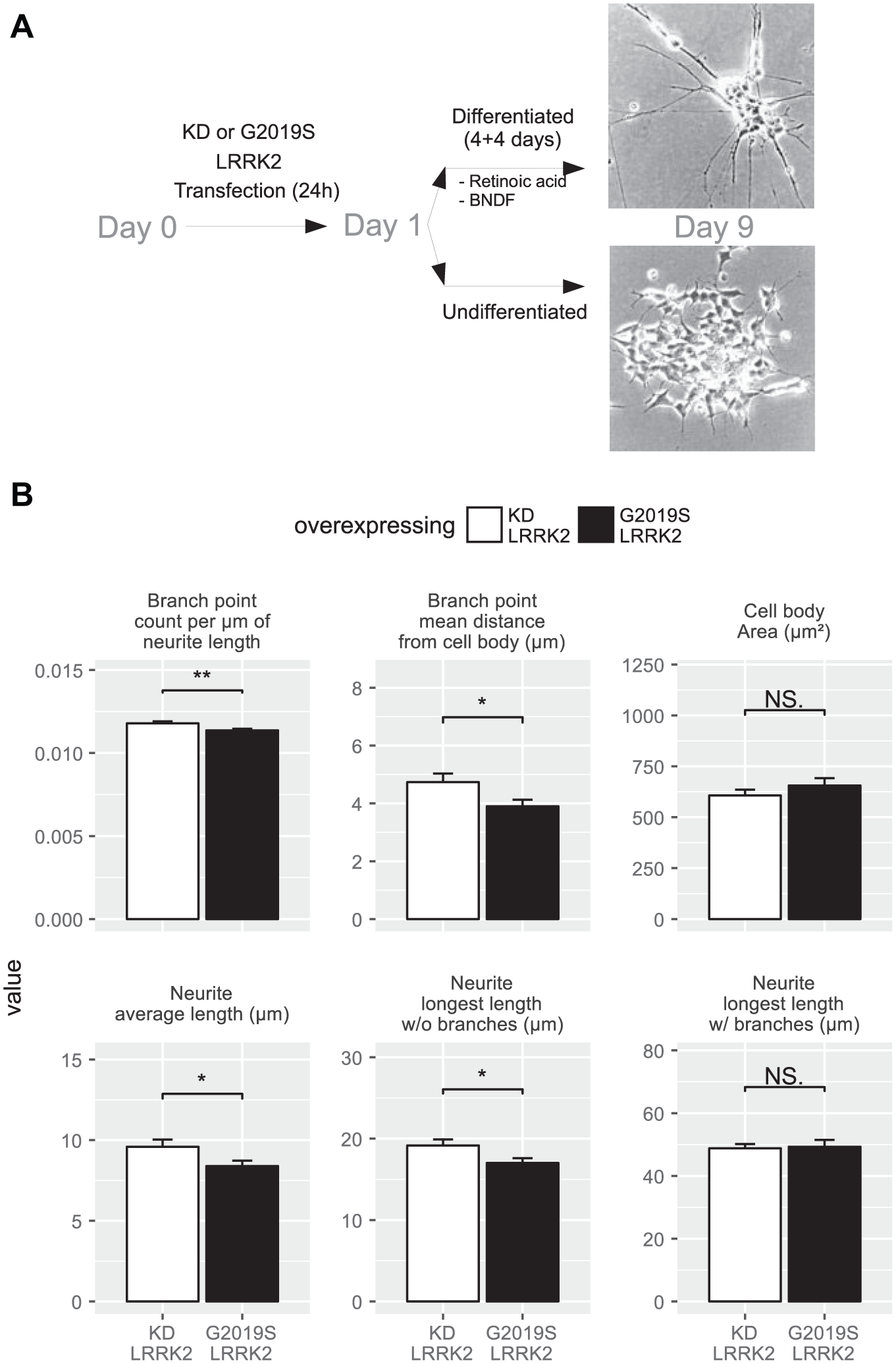
G2019S LRKR2-transfected neuronally differentiated SH-SY5Y neuroblastoma cells exhibit an altered neurite profile. (
Staining and High-Throughput Imaging in Retinoic Acid Differentiated SH-SY5Y Cells
SH-SY5Y cells were transfected with the fusion constructs GFP-G2019S-LRRK2 and GFP-KD-LRRK2 and subjected to the differentiation protocol as described in the supplementary material. At the end of differentiation, cells were fixed using 4% paraformaldehyde (PFA), and nuclei were stained with Hoechst 33342. Images from 96-well plates were automatically recorded using the Cellomics ArrayScan VTI HCS Reader (Thermo Scientific, Vedbæk, Denmark) with a 10× microscope objective and the built-in standard autofocus method. From each well a total of 40 nonoverlapping images were obtained. Images were analyzed using the Cellomics assay algorithm Neuronal Profiling v3.5. The segmentation was performed based on the nuclear staining and cells that exhibited green fluorescence around the nuclear area were positively selected for the analysis. Branch point counts per micrometer of neurite length, branch point mean distance from cell body, cell body area, neurite average length, neurite longest length without branches, and neurite longest length with branches were selected for posterior statistical analysis.
Quantitative RT-PCR
Total RNA was prepared using the Qiagen RNeasy Micro Kit (Qiagen). RNA quality was evaluated by running a 1.0% agarose/formaldehyde gel and quantified spectrometrically (NanoDrop ND-1000) before proceeding to subsequent steps. One-step real-time PCRs (RT-PCRs) were set up in a Biorad CFX-96 system using iScript RT, 10 µL of iTaq containing SYBR Green supermix with ROX, and 3 µL of primer mix (2.5 pmol/µL). The annealing temperature for all primers was 50 °C, and 40 cycles were performed prior to a melting curve starting at 55 °C. Relative expression levels for each gene were calculated using the delta-delta CT method. All points were determined in triplicate. Primers used for the RT-PCR are listed in Supplemental Figure 1 ; 2 different pairs of primers were used for each gene, designed to amplify distinct exons and always with an intron boundary between amplicons. This was done to ensure reproducibility, as at the time we did the experiments nobody had amplified Nanog or Oct4 in these cells before. GapDH and Cytochrome c were used for normalization. Results obtained with both sets of primers were similar in each experiment.
Western Blotting
Cell homogenates were prepared with RIPA lysis buffer containing protease inhibitor (50 mM Tris-HCl [pH 7.4], 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, one freshly added tablet of PhosSTOP [Roche, Basel, Switzerland], and one tablet per 50 mL of cOmplete Protease Inhibitor Cocktail [Roche]).
The protein concentration was determined by the BCA method (Pierce Biotechnology, Rockford, IL) and 20 µg of protein was resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; NuPAGETM Novex 4%–12% Bis–Tris Protein Gels), transferred to polyvinylidene fluoride (PVDF), and probed with mouse monoclonal anti-Nanog antibody (PR-2E11, Abcam ab95334, 1:1000). Quantification of protein levels by densitometry was conducted using LabImage 1D software (Kapelan Bio-Imaging Solutions, Leipzig, Germany) on Western blot images captured using a Fuji Film LAS-4000 Luminescent Image Analysis system.
Luciferase Assay
Sequences described in the next section were synthesized and cloned into pUC57 plasmid and subcloned into pGL2-control and pGL2-basic plasmid into the SacI-XhoI and XhoI-HindIII restriction sites, respectively. The sequences are fragments of the proximal promoters of the genes that were found altered in the quantitative PCR (qPCR) experiments, as described in the “Results and Discussion” section. These constructs were alternatively co-transfected with plasmids expressing G2019S LRRK2 or KD LRRK2, always together with pSEAP2-Control (Clontech, Saint-Germain-en-Laye, France), encoding for alkaline phosphatase, in a 1:20 ratio to provide a normalization value for the luciferase activity from pGL2. After differentiation and treatments, 25 µL of cell culture supernatant was saved and transferred to another multiwell plate; the rest of the media was replaced by HBSS-Mg. Steady-lite plus (PerkinElmer, Skovlunde, Denmark) was added 1:1 to the wells, plates were gently rocked for 10 min and sealed, and luminescence was read immediately in a Wallac Viewlux HTS microplate reader (PerkinElmer). To determine alkaline phosphatase activity, 75 µL of Clontech PT3954 dilution buffer was added to the supernatants and plates were sealed and incubated for 30 min at 65 °C, cooled on ice, and subsequently equilibrated to 20 °C. An equal volume of Clontech PT3954 substrate solution was then added; plates were sealed again and incubated for 30 min more at 20 °C and then read in the same Wallac instrument. Linearity of the secreted alkaline phosphatase (SEAP) measures in our range was confirmed by testing control plates with a series of different cell numbers. The effects of the different mutant forms of LRRK2 on cell proliferation resulted in a consistent and significant reduction of cell numbers after differentiation in G2019S LRRK2, which was reflected in the AP normalization values. To verify the accuracy of our normalization, we estimated the cell number via Cellomics, lactate dehydrogenase (LDH) release, and MTT assays, and the four methods resulted in consistent values between them.
Statistics
All quantified values are based on at least three independent cell culture experiments. Data are plotted as mean ± SD. For comparison between conditions, a two-tailed unpaired Student t test was used. p < 0.05 was considered significant. Statistical analyses were performed with R software using the CRAN public library for nonlinear fittings and dose–response analysis (http://www.r-project.org).
Results and Discussion
G2019S LRRK2-Transfected Neuronal-Differentiated Cells Exhibit an Abnormal Neurite Profile, including Reduced Branching and Length
To examine the effect of constitutively active G2019S LRRK2 on neurite complexity, we transfected neuronal cells with plasmids encoding an in-frame EGFP-tagged LRRK2 with the G2019S mutation, or with mutations that result in KD LRRK2 as controls. After differentiation by retinoic acid and brain derived neurotrophic factor (BDNF), cells were fixed and imaged to visualize the cellular profile. We observed a statistically significant reduction (p < 0.05) on branching and neurite length (
Fig. 1
Pluripotency Genes Are Still Expressed after Retinoic Acid-Induced Neural Differentiation of G2019S LRRK2-Expressing Neuroblastoma Cells
To identify the mechanisms underlying the influence of G2019S LRRK2 on neuronal differentiation, we selected candidates of LRRK2-modulated retinoic acid-induced genes identified earlier in embryonic stem cells from LRRK2 knockout mice. 5 In neuronally differentiated cells, the mRNA levels of the three genes downregulated most by the LRRK2 knockout were Nanog, Oct4, and Ssp1 ( Suppl. Fig. S3 indicates the oligonucleotides used). We consistently obtained a fourfold induction of Nanog expression by G2019S LRRK2 overexpression and a twofold induction by wild-type LRRK2 overexpression ( Fig. 2A ). Similar changes were observed for Oct4 ( Fig. 2B ). For Ssp1, no changes were observed after any LRRK2 transfection. Cells overexpressing KD LRRK2 did not show aberrant upregulation of these transcription factors after differentiation.
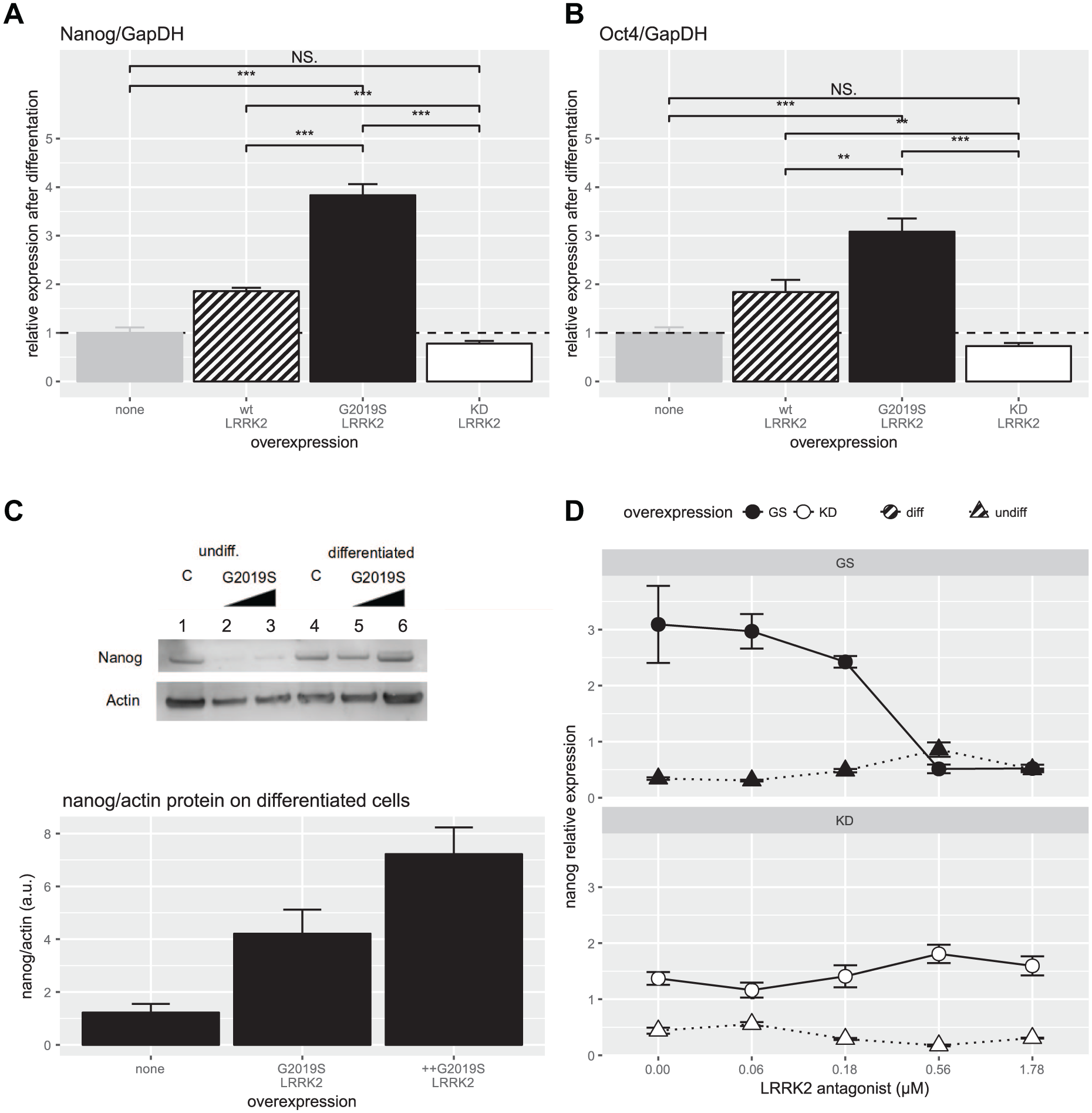
High levels of pluripotency genes are still expressed in G2019S LRRK2-transfected cells after retinoic acid-induced neural differentiation. (
To confirm LRRK2-mediated induction of Nanog after differentiation at the protein level, Western blotting was performed. A similar fivefold increase of Nanog protein levels was observed in G2019S LRRK2-expressing cells ( Fig. 2C ). Lastly, it was confirmed that G2019S-mediated induction of Nanog after differentiation could be reversed by treatment with the selective LRRK2 kinase inhibitor compound BMPPB-32. 9 Concentration–response experiments showed a reversal of Nanog induction in cells expressing G2019S LRRK2. As expected, in cells expressing KD LRRK2, the kinase inhibitor did not have any impact ( Fig. 2D ).
These results suggest that aberrant G2019S LRRK2 activity prevents normal downregulation of the pluripotency transcription factors Nanog and Oct4 during neuronal differentiation. LRRK2 kinase inhibition can counteract this effect.
G2019S LRRK2 Expression but Not KD-LRRK2 Activates the Nanog Proximal Promoter during Neuronal Differentiation
To demonstrate that the mechanism of action was mediated by RxR and to develop a neuronal assay to screen for LRRK2 inhibitors, we constructed two reporter systems ( Fig. 3A ). A fragment of the human Nanog proximal promoter that encompasses a sequence of homology between Nanog and Oct4 promoters and also includes a putative retinoic acid response element was cloned in front of a heterologous basal promoter and a luciferase expression cassette (short promoter). Second, a 2 kb fragment upstream of the transcription initiation site of human Nanog was cloned 5′ of a luciferase expression cassette (long promoter). Subsequently, these reporter constructs (short or long) were co-transfected with either of the LRRK2 constructs (G2019S or KD LRRK2). To control for transfection efficiency and differential cell growth during the differentiation process, the reporter-LRRK2 mutation pairs were supplemented with a plasmid encoding SEAP as a normalization control.
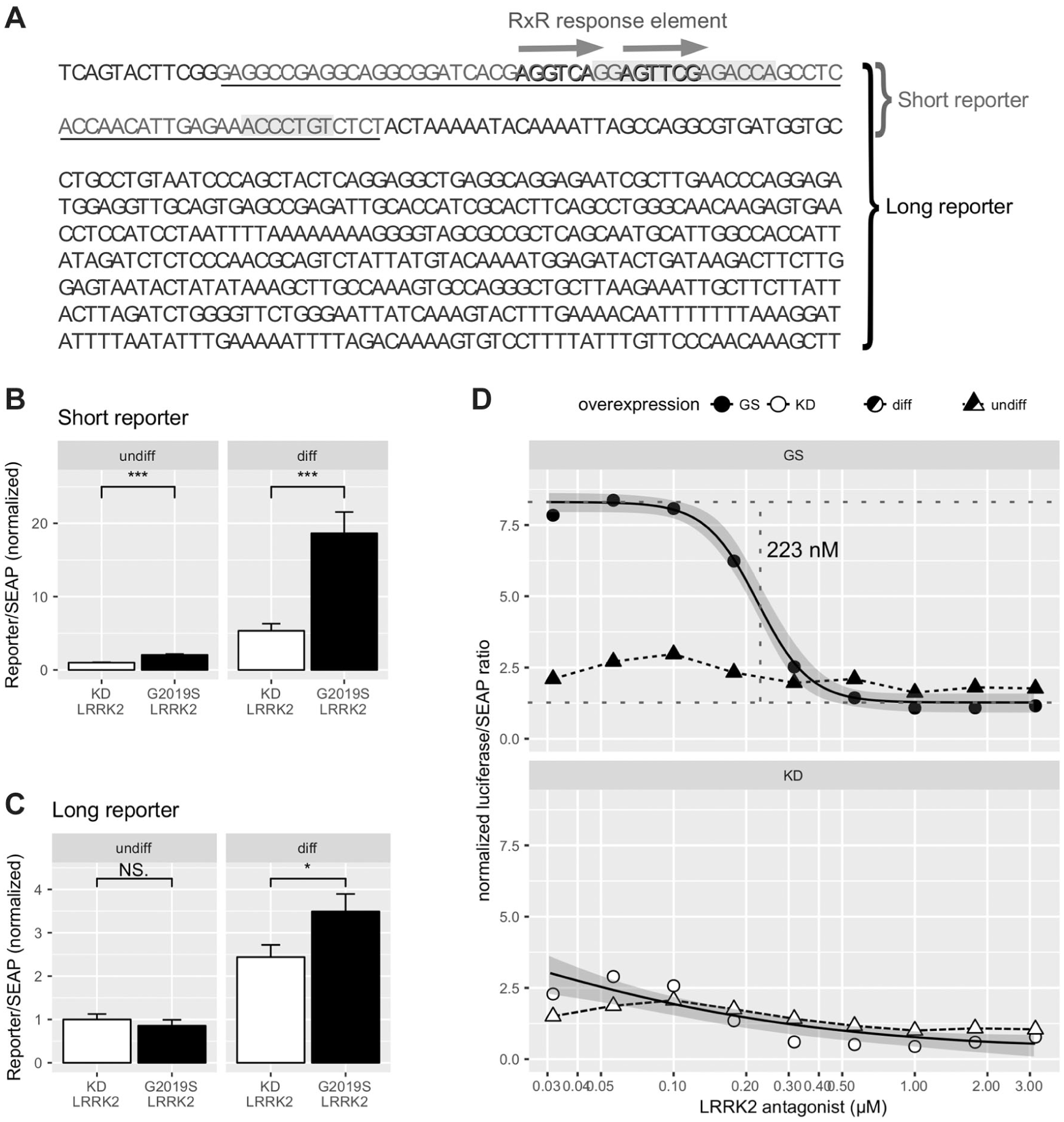
Reporter constructs with a fragment of the proximal promoter of human Nanog are activated by G2019S LRRK2 and inhibited by tool compounds. (
Following transfection, LRRK2-expressing cells were subjected to retinoic acid/BDNF-induced differentiation and promoter induction analysis. We observed a four- to fivefold induction of the short promoter construct activity in cells expressing G2019S LRRK2, when compared with cells expressing KD LRRK2 ( Fig. 3B ). In experiments using the long promotor construct, we observed a twofold induction when comparing G2019S with KD LRRK2-expressing cells ( Fig. 3C ). To demonstrate assay stability and reproducibility, we produced a series of control plates with KD LRRK2 and G2019S LRRK2 and the short version of the reporter ( Suppl. Fig. S4 ). In these layouts, we did not observe major plate drifts; row-wise and column-wise typical coefficients of variation were in the 10%–15% range and residuals in the ±2 (−3) SD range independently of the well position. We estimated an interplate variability with Z′ > 0.4 and a strictly standardized mean difference (SSMD) of around 6–7 in all control plates. Supplemental Figure S5 shows two representative control plates assayed from independent cultures and assayed during separate weeks.
We also tested the ability of LRRK2 inhibition to revert the G2019S LRRK2-induced activation of the reporter. G2019S LRRK2-expressing cells were treated with the selective LRRK2 tool inhibitor BMPPB-32 during neuronal differentiation. We observed a concentration-dependent reversal of luciferase activity to a similar extent and in the same concentration range as observed for the changes in qPCR-measured Nanog expression, estimating an IC50 for BMPPB-32 of 0.22 µM (95% CI, 0.183–0.271 µM) in the luciferase assay, which is statistically not different from the qPCR experiment value (p = 0.25 in a t test; Table 1 ). However, besides the better technical suitability for HTS, the luciferase assay also showed better precision in the evaluation of the parameters ( Table 1 ); for example, the coefficient of variation of the IC50 (parameter e in the equation) was 9.7% in the luciferase versus 19% in qPCR assay.
Estimated Values and Errors of the LRRK2 Inhibitor Compound BMPPB-32 Curves by the qPCR Assay and the Luciferase Assay.
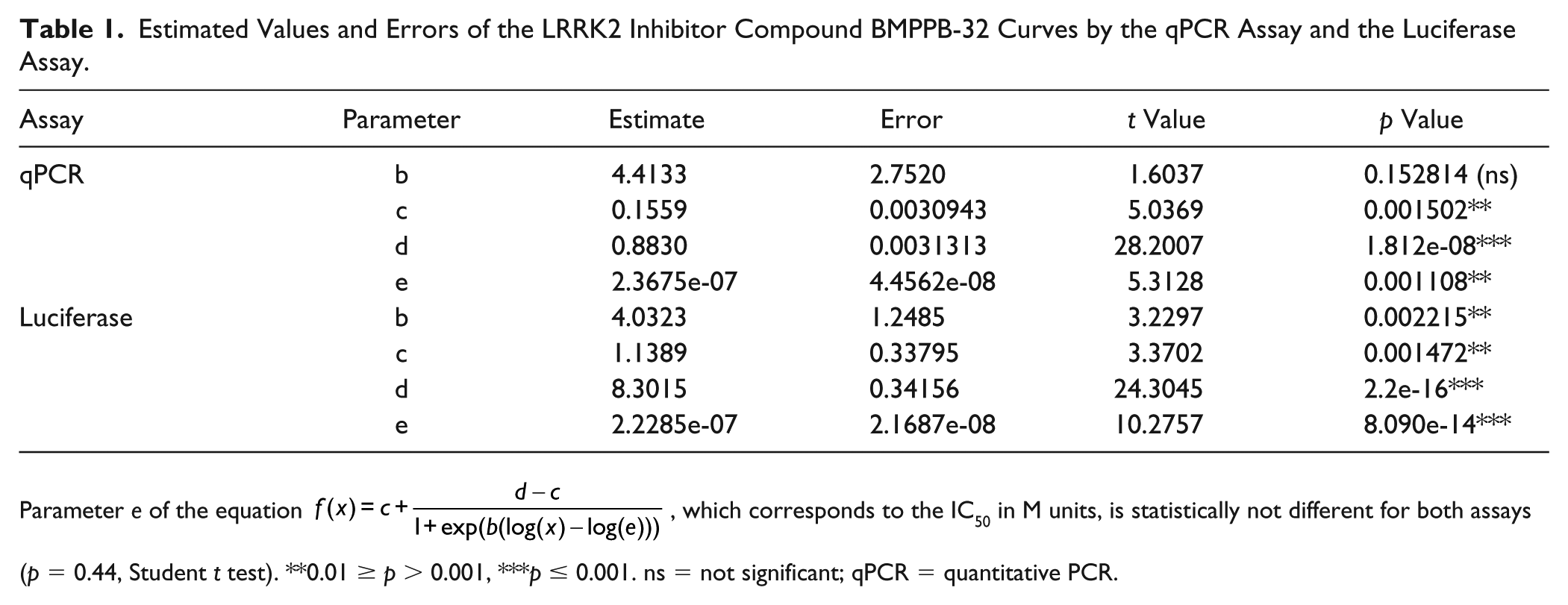
Parameter e of the equation
Cells co-transfected with KD LRRK2 and treated with BMPPB-32 showed a concentration-response profile on luciferase expression similar to the one measured by qPCR ( Fig. 3D and Table 1 ). Controls present in the plates used for BMPPB-32 inhibition experiments rendered Z′ values of 0.4−0.7, compatible with the values observed when characterizing the plates.
Our results suggest that active LRRK2 and hyperactive G2019S LRRK2, but not KD LRRK2, activate Nanog expression via an 86 bp fragment in the proximal promoter that also contains the RxR response element.
Finally, the intra-assay variability (Z′ factor) for the different assays was estimated, and we evolved from statistically significant changes—Z′ is not compatible for screening purposes in the morphological assays (estimated Z′ = −6.6 for the average neurite length and Z′ = −6.3 for the max length)—to Z′ being negative in the qPCR assays (Z′ = −0.7 for times of Nanog expression change by differentiation, Z′ = −0.4 for relative changes just in differentiated cells), to Z′ being larger than 0.4 for the luciferase assay. Thus, we provide for the first time a functional mechanistic assay for HTS exploiting the effect on pluripotency gene expression.
Both Nanog and Oct4 are considered core elements of the pluripotency transcriptional network, and they are normally downregulated upon differentiation. 10 But in presence of the pathogenic G2019S mutation their shutdown is incomplete, and this can be the mechanism underlying the partial differentiation phenotype, such as shorter and less ramified neurites. Pharmacological LRRK2 inhibition counteracted the G2019S-induced effect on Nanog gene expression.
We also localized a short sequence of the proximal Nanog promoter containing RxR response elements responsible for this mechanism, and we used this sequence to design a suitable HTS-friendly cell-based mechanistic reporter assay for screening LRRK2 inhibitors. This short sequence also contains other conserved motifs besides RxR ( Suppl. Fig. S5 ), but most of these cis elements are characteristic of nuclear receptor binding, where RxR heterodimers are frequent. Moreover, RxR-alpha is effectively phosphorylated in vitro by LRRK2. 5 Differentiation is induced by retinoic acid, which supports the assumption that the transcriptional effects of LRRK2 on Nanog are mediated by RxR. Furthermore, the IC50 values obtained for the BMPP-32 tool inhibitor were similar when analyzing Nanog mRNA expression by qPCR and when analyzing LRRK2 effects via the short reporter construct, which is consistent with the hypothesis that LRRK2 exerts its effect on Nanog expression via this sequence.
Additionally, regarding the IC50 values obtained in our assay for the tool inhibitor BMBP-32, 220 nM is consistent with the reported 279 nM in a pSer935 autophosphorylation assay using brain homogenates as substate. 9
Previously, IC50 values of 6.2 and 94 nM were reported 7 using a cell-free autophosphorylation assay and an HEK293-based assay, respectively. The value was 2.5 µM using a Drosophila visual response in another unique phenotypic outcome reported as an LRRK2 assay. Differences between cell types (HEK293 vs neuronal differentiated SH-SY5Y) can explain the different IC50 values; plus it is common to obtain larger values for IC50 differentiated cells compared with fast-growing tumor cells.
This assay correlates drug effectivity with reversal of incomplete differentiation, a known phenotypic outcome measure in neurons expressing hyperactive G2019S LRRK2.2,3 While the effect of the G2019S mutation on Nanog and Oct1 has been substantial, those transcription factor changes do not appear to be driving a dramatic change in morphology. Incomplete and defective differentiation has been associated with an increased demand of ATP, and reactive oxygen species (ROS) production that results in an increased cellular vulnerability that could explain the role in PD pathophysiology.
Up to now, there have been no assays exploiting this G2019S LRRK2-induced phenotype. The assays available are autophosphorylation assays typically targeting pSer985 that evaluate total versus phosphorylated LRRK2, the same way it was described in the first molecular paper on LRRK2 in 2005.11 These assays are commercialized into very convenient solutions for HTS, as in the MSD U-PLEX platform. However, only to assay LRRK2 autophosphorylation is insufficient to drive LRRK2 drug discovery forward. Over the last 15 years, the understanding of LRRK2 biology has advanced substantially, which should also be taken into account when choosing an assay.11
Furthermore, the work reported here provides the first indication that G2019S LRRK2 restrains the pluripotency shutdown after neuronal differentiation, backed up by an earlier publication on genetic ablation of LRRK2 in stem cells, 5 conceptually the opposite paradigm, which it is consistent with our findings, as it shows alterations of Nanog and Oct4 when LRRK2 is absent. There are other studies that imply RxR, and by extension neuronal differentiation, is a putative target in PD. For example, mutations in the Nurr1 transcription factor (NR4A2), which makes functional heterodimers with RxR, have been associated with increased susceptibility to disorders related to dopaminergic dysfunction (including PD), and its expression is reduced in PD patients. 12 Interestingly, the highly selective RxR agonist IRX4204, primarily developed as an anticancer compound, improves the survival of dopaminergic neurons in rodent models of PD 8 and has been proposed as a monotherapy for PD. 13 However, these effects are not exclusive to LRRK2-associated PD, but RxR-mediated protection seems to work in other models in which dopaminergic neurons are selectively targeted, for example, via mitochondrial toxins or by alpha-synuclein. 14 We chose the Nanog promoter for the HTS optimized assay because it was the one exhibiting the largest response. Some evidence points toward a more complex regulatory network downstream of LRRK2, because Nanog seems to be dispensable for pluripotency under certain conditions, specifically if other factors, such as Oct4 and Sox2, are active. 15 Both Schulz et al. 5 and we identified Oct4 as having an active role in the LRRK2 phenotype, so at least it is also involved; therefore, the pharmacological targeting of Nanog instead of LRRK2 will not be very effective against LRRK2-associated PD.
Additionally, association between the G2019S LRRK2 mutation and lack of restraint of pluripotency on terminal differentiation does not seem to be exclusive to neurons. There is an increased risk of cancer (age adjusted) with an odds ratio of 3.38 in PD patients carrying the G2019S LRRK2 mutation when compared with noncarrier PD patients. 16 Consistent with our finding, Nanog has been reported to be broadly upregulated in human cancers. 17 The upregulation of Nanog occurred to a similar extent to what we observed in G2019S LRRK2 overexpressing neuroblastoma cells.
Neuronally differentiated SH-SY5Y cells are practical for use in large-scale screening, and they exhibit many neuronal markers, such as NeuN, MAP2, beta 3 tubuline, or GAP43, and changes coherent with young dopaminergic neurons, regarding dopamine metabolism, increased mitochondrial susceptibility, and kinome profile. 18 As more evidence points toward neurodevelopmental aspects of PD etiology, we think that our mechanistic assay has a good translational value.
Interestingly, the underlying work is a good example of the commonly accepted design rule in the scientific field, that the higher upstream the pathway is targeted, the less the variability and the better is the precision, and the more assay power that can be obtained. Due to the fact that similar inhibitor concentrations produce results in the luciferase reporter assay that are similar to those in RT-PCR approaches, the former provides a considerably more robust and suitable assay for high-throughput approaches; thus, it can be conveniently implemented in screens, as it displays a lower well-to-well variability (higher Z′).
In summary, we provide a novel mechanistic cellular assay that links pathological G2019S LRRK2 function and aberrant control of pluripotency genes that impact the terminal differentiation. In the context of LRRK2, this is the first description of an assay suitable for HTS beyond autophosphorylation.
Supplemental Material
DS_DISC864086 – Supplemental material for Novel Cell-Based Assay for Identification of LRRK2 Inhibitors Using Its Aberrant Regulation of a Pluripotency Gene
Supplemental material, DS_DISC864086 for Novel Cell-Based Assay for Identification of LRRK2 Inhibitors Using Its Aberrant Regulation of a Pluripotency Gene by David Ramonet and Gunnar P. H. Dietz in SLAS Discovery
Footnotes
Acknowledgements
We thank Jan Egebjerg and Kenneth V. Christensen for their continued support and many useful discussions and suggestions; Lara Buscemi for her comments and suggestions; and Lone Lind Hansen and Annette Bjørn for their expert technical assistance.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: While this work was in progress, all authors were employed by the pharmaceutical company H. Lundbeck A/S in Valby, Denmark, which focuses on the treatment of psychiatric and neurological diseases.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.