
Abstract
Background:
Observational studies in Parkinson’s disease (PD) have focused on relatively small numbers of research participants who are studied extensively. The Molecular Integration in Neurological Diagnosis Initiative at the University of Pennsylvania aims to characterize molecular and clinical features of PD in every patient in a large academic center.
Objective:
To determine the feasibility and interest in a global-capture biomarker research protocol. Additionally, to describe the clinical characteristics and GBA and LRRK2 variant carrier status among participants.
Methods:
All patients at UPenn with a clinical diagnosis of PD were eligible. Informed consent included options for access to the medical record, future recontact, and use of biosamples for additional studies. A blood sample and a completed questionnaire were obtained from participants. Targeted genotyping for four GBA and eight LRRK2 variants was performed, with plasma and DNA banked for future research.
Results:
Between September 2018 and December 2019, 704 PD patients were approached for enrollment; 652 (92.6%) enrolled, 28 (3.97%) declined, and 24 (3.41%) did not meet eligibility criteria. Median age was 69 (IQR 63_75) years, disease duration was 5.41 (IQR 2.49_9.95) years, and 11.10%of the cohort was non-white. Disease risk-associated variants in GBA were identified in 39 participants (5.98%) and in LRRK2 in 16 participants (2.45%).
Conclusions:
We report the clinical and genetic characteristics of PD patients in an all-comers, global capture protocol from an academic center. Patient interest in participation and yield for identification of GBA and LRRK2 mutation carriers is high, demonstrating feasibility of PD clinic-wide molecular characterization.
INTRODUCTION
Parkinson’s disease (PD), the second most common neurodegenerative disease, was first clinically described over 200 years ago [1]; Lewy bodies, the characteristic neuropathological brain lesions in PD, were first described over 100 years ago [2]. The clinical and neuropathological criteria for diagnosis of PD still rely largely on these definitions in modern-day medical practice. However, people carrying a diagnosis of PD encompass a broad spectrum of clinical symptoms and disease trajectories, most notably in the rate of motor progression and occurrence of cognitive decline [3]. This observed heterogeneity suggests that PD may be a syndrome encompassing multiple subtypes rather than one monolithic entity [4]. Molecular characterization at the individual level may allow clinicians and researchers to define subtypes of PD based on biological markers (or “biomarkers”) from patient blood, cerebrospinal fluid (CSF), saliva, or other fluids and tissues. Moreover, biomarker information may shed light on the pathobiology underlying individual differences and may enable development of novel disease-modifying treatments as well as tailoring of existing therapies at the individual level, allowing for precision medicine approaches in PD [5].
A number of longitudinal observational studies enrolling participants with PD have focused inten-sely on relatively small numbers of dedicated res-earch participants to better understand relationships between clinical symptoms and genetic, biochemical, and imaging biomarkers in PD [6–10]. A notable example of this approach is the international, multi-center, Parkinson Progression Markers Initiative (PPMI), which followed 424 PD subjects for over eight years and collected extensive amounts of clinical and molecular data [11]. While this “deep but narrow” approach is valuable for many research questions, focusing on relatively small cohorts of PD subjects with comprehensive molecular and genetic characterization may limit ability to capture heterogeneity and variability across the spectrum of PD presentations. Moreover, because of the significant effort and time involved for research participants, these types of studies can suffer from selection bias for those individuals most able to donate this time and effort—individuals who have relatively higher health and research literacy, have motivated and supportive caregivers, or live in areas readily accessible to large academic centers. Indeed, the frequent observation that minorities and women are under-represented in PD research cohorts may reflect this bias [12].
The recent advent of genetically guided clinical trials in PD requires a new approach to research recruitment and genetic characterization in PD. Here, we sought global capture of a PD clinic population by approaching every patient seen in a large academic movement disorders center at the University of Pennsylvania (UPenn). Each participant was consented and enrolled for a blood sample and ascertainment of PD clinical features. In anticipation of eventual translation of biological findings from research-based results to clinically actionable results, we designed our study to allow for genetic counseling and disclosure of genetic data to patients who wished to know their own results after clinical confirmation testing. In this report, we present our findings from the first 15 months of the Molecular Integration in Neurological Diagnosis (MIND) Study, in which 704 individuals were approached, and 652 individuals were enrolled and genetically characterized. Our study demonstrates the feasibility of a first-in-kind unbiased global-capture approach to the collection of clinical and biomarker data in PD.
METHODS
Recruitment and enrollment process
All participants were recruited from the UPenn Parkinson’s Disease and Movement Disorders Center (PDMDC) between September 2018 and December 2019. The inclusion criteria were a clinical diagnosis of PD by a movement disorders expert, age greater than or equal to 21 years, ability to provide informed consent, and a previously scheduled visit at the PDMDC. The only exclusion criteria were inability to consent to research, or designation as a vulnerable population (e.g., pregnant and lactating women, prisoners, children).
The entire PD population seen at the PDMDC served as the potentially eligible participant pool. The electronic medical record (EMR) of each patient was screened to identify patients meeting eligibility criteria. Patients meeting eligibility criteria were approached by a clinical research coordinator in the waiting room prior to their clinician encounter to provide them with information about the study. Individuals showing interest were asked to remain in the office after their clinical visit. Participants were enrolled at the conclusion of the clinical visit after review of inclusion and exclusion criteria and after signing informed consent and HIPAA authorization. To minimize impact on clinic flow, only one to two research coordinators enrolled patients on any given day, and only one clinical examination room was utilized. This pragmatic approach allowed us to capture roughly one-third of eligible subjects each clinic day, distributed randomly among eleven movement disorders physicians in the office. Informed consent included three optional components allowing researchers to: (1) access the EMR currently and in the future; (2) contact participants for additional studies for which they might meet eligibility criteria; and (3) store patient plasma and DNA as part of the UPenn Center for Neurodegenerative Disease (CNDR) biobank [13]. If a participant opted to enroll with future recontact, he or she could additionally learn genetic results after genetic counseling and clinical confirmation testing under a separate protocol. Enrollment eligibility was not affected by responses to these optional components.
As a result of the SARS-CoV-2 pandemic non-essential, in-person research recruitment and laboratory experimentation was halted at the University of Pennsylvania on March 13, 2020. This unplanned interim analysis was conducted as a result, with the expectation that future participants will be recruited and enrolled via a mixture of in-person and virtual sample and clinical data collection. Subsequent analyses will compare in-person to virtual methods.
Standard protocol approvals, registrations, and patient consent
This study was approved by the University of Pennsylvania Institutional Review Board, and informed consent was obtained at study enrollment.
Blood processing and DNA extraction
Fifty mL of blood was drawn by peripheral venipuncture into sterile vacutainer tubes coated in EDTA by a trained technician. Blood was processed on the day of collection according to previously published protocols [13]. Briefly, two blood tubes were used for DNA isolation, while all remaining blood was centrifuged and plasma was aliquoted and frozen at –80°C for future studies. For participants who refused venipuncture, or for whom technical difficulties prevented venipuncture, a saliva sample was obtained in an Oragene-DNA OG 500 collection kit (DNA Genotek, Ontario Canada) and incubated at 55°C for at least 2 h up to overnight to inactivate nucleases. DNA was extracted from 4 mL blood or 2 mL saliva solution in a semi-automated QuickGene 610 L using the DNA whole blood kit following the manufacturer’s protocol (Autogen, Holliston, MA).
Clinical screening questionnaire
Participants completed a brief demographic and clinical questionnaire (Supplementary Material). The questionnaire was developed by authors TFT, JR, AS, AW, DW, and ACP. The questionnaire was designed to be completed in five minutes or less, self-administered or administered by a clinical research coordinator, and collect clinical information not routinely collected in the EMR to identify targetable groups for future research studies. Data were collected in real-time and managed using REDCap electronic data capture tools hosted at Upenn [14] or by paper at participant request. For participants who refused the questionnaire during the clinic visit, the option to complete the questionnaire at a later time by paper to be returned by mail or electronically via REDCap e-mail survey was offered.
Genetic testing
All participants had research-based screening for GBA and LRRK2 mutations/disease-associated variants. Eight LRRK2 variants previously reported to associate with PD and four GBA variants based on reported frequency in PD populations were selected (Supplementary Table 1) [15, 16]. Twelve SNPtype assays were designed by D3 Assay Design tool (Fluidigm Corp., San Francisco, CA). Allele-specific PCR was performed using the Flex Six genotyping integrated fluidic circuits (IFCs) (Fluidigm Corp.), and genotyping was carried out using the Biomark™ HD system (Fluidigm Corp.) according to the manufacturer’s protocol. Briefly, specific target amplification (STA) was performed to enrich target sequences flanking the 12 SNPs using 40 ng genomic DNA and 50 nM of each STA and locus-specific primer pairs. Pre-amplified DNA was mixed with 2X Fast Probe Master mix (Biotium Inc., Fremont, CA), 20X SNP Type sample loading reagents, 60X SNP Type reagent, and ROX in 5μl. Sample mix and 10X SNP Type assay mix was added into assay and sample inlets on the IFC, respectively, and loaded and mixed in the IFC controller. PCR was performed on the BioMark HD System under the following cycling condition: thermal mix for 30 min at 70°C followed by 10 min at 25°C, hot start for 5 min at 95°C, touchdown PCR for 15 s at 95°C, 45 s from 64°C –61°C dropping 1°C per cycle, and 15 s at 72°C, and additional 34 cycles of 15 s at 95°C, 45 sec at 60°C, and 15 s at 72°C, and cooling for 10 s at 25°C. The genotype call data were analyzed using BioMark Genotyping Analysis software which calculated the FAM and VIC relative fluorescence intensities and automatically called genotypes by the k-means clustering algorithm. Automatic genotype calls were reviewed in individual plots after cluster analysis. The frequencies of GBA and LRRK2 variants among Ashkenazi Jewish and non-Ashkenazi Jewish participants were compared using Fisher’s exact test.
Role of the funding source
The funding sources did not have any role in the study design, collection, analysis, or interpretation of the data; writing the report; or decision to submit for publication.
Data availability
All data will be made available upon request.
RESULTS
Participants
PD participants were recruited at their regularly scheduled clinical office visits. During the 15-month period reported here (September 7, 2018-December 17, 2019), 704 patients with a clinical diagnosis of PD based on movement disorders expert opinion were approached for the MIND study, and 652 patients (92.61%) were enrolled. Of the 52 patients who were not enrolled, 24 did not meet inclusion criteria when interviewed, and 28 (3.97%) declined to participate. The numbers of participants providing optional consent for electronic medical record access, future contact for research participation, and biobanking of plasma are shown in Fig. 1.
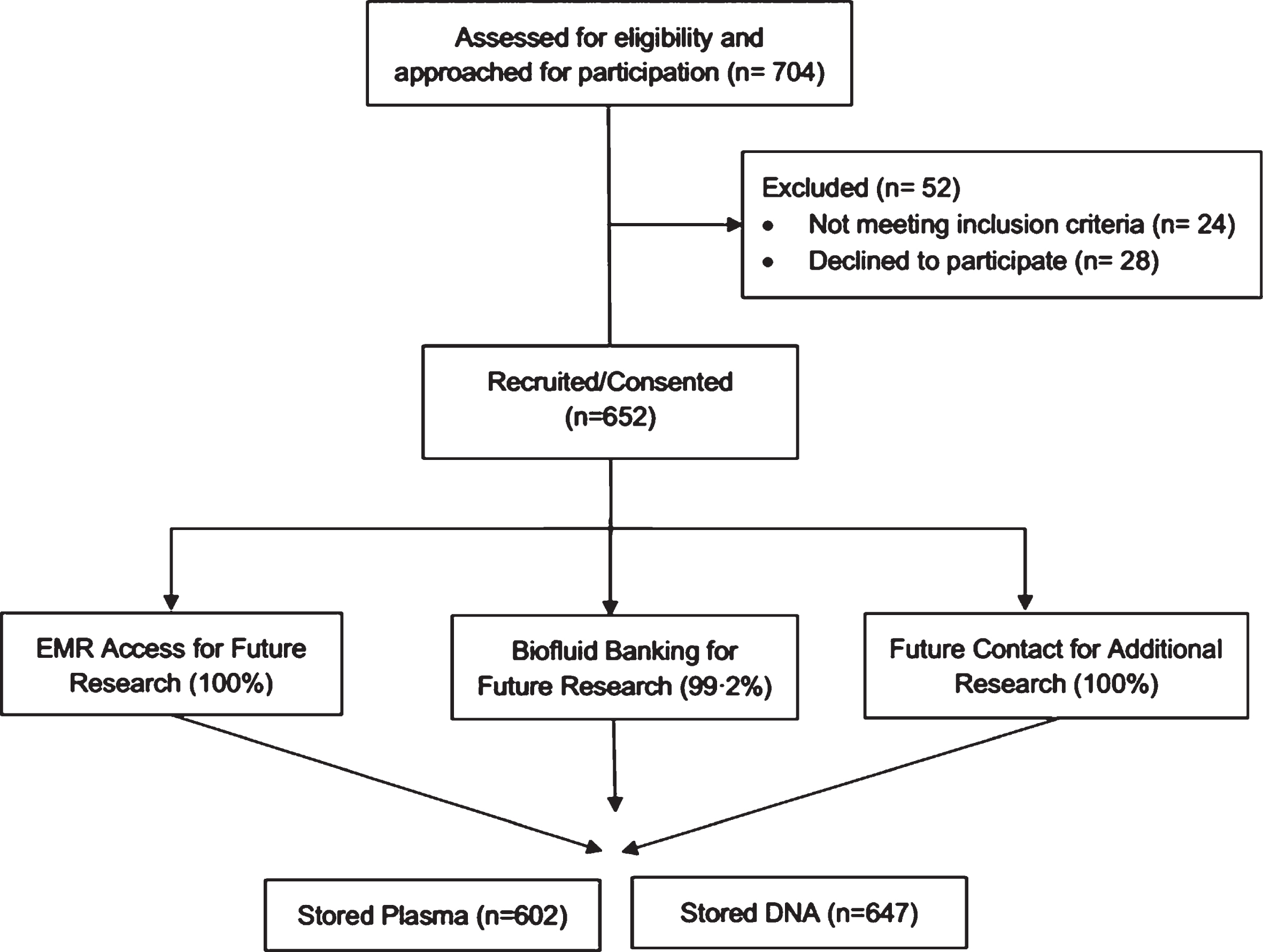
Molecular Integration in Neurological Diagnosis (MIND) Cohort Enrollment. Consort diagram outlining the Molecular Integration in Neurological Diagnosis (MIND) cohort enrollment. Percentages represent proportion of enrolled participants. EMR, electronic medical record.
Five participants declined consent for sample banking for future research, leaving 647 participants who consented to storage of blood plasma and DNA. Plasma samples were frozen and stored from 602 of 647 (93.04%) participants. Missing samples were the result of technical difficulty with venipuncture. For these participants, saliva samples were collected for DNA extraction only.
Cohort demographics and clinical characteristics
Demographics and clinical characteristics of the MIND cohort are shown in Table 1. Notably, only 88.90%of the MIND cohort self-report their race as white, compared with ∼96%in prior studies from our center that did not employ a global-capture design [17]. Moreover, 78 (11.96%) PD participants reported non-motor symptoms (including constipation, fatigue, anxiety, loss of sense of smell, sleep disorders, slurred speech, light-headedness, memory loss) as their first presenting symptom. Comparisons between GBA variant carriers vs. non-carriers, and between LRRK2 variant carriers vs. non-carriers are shown in Table 1.
Cohort Description. Values represent median (IQR) unless otherwise noted.
*Disease Duration is time from diagnosis to enrollment date
Motor, cognitive, neuropsychiatric, and general complications of PD reported in the MIND cohort are shown in Fig. 2. Notably, fatigue was a current symptom in 68.64%of the cohort, and impulsive behaviors in 11.40%. Rates of PD complications (currently or in the past) among the entire cohort, and in subgroups stratified by GBA or LRRK2 variant status, are shown in Table 2. Among all MIND cohort participants, data was missing in less than 2.40%for any PD complication.
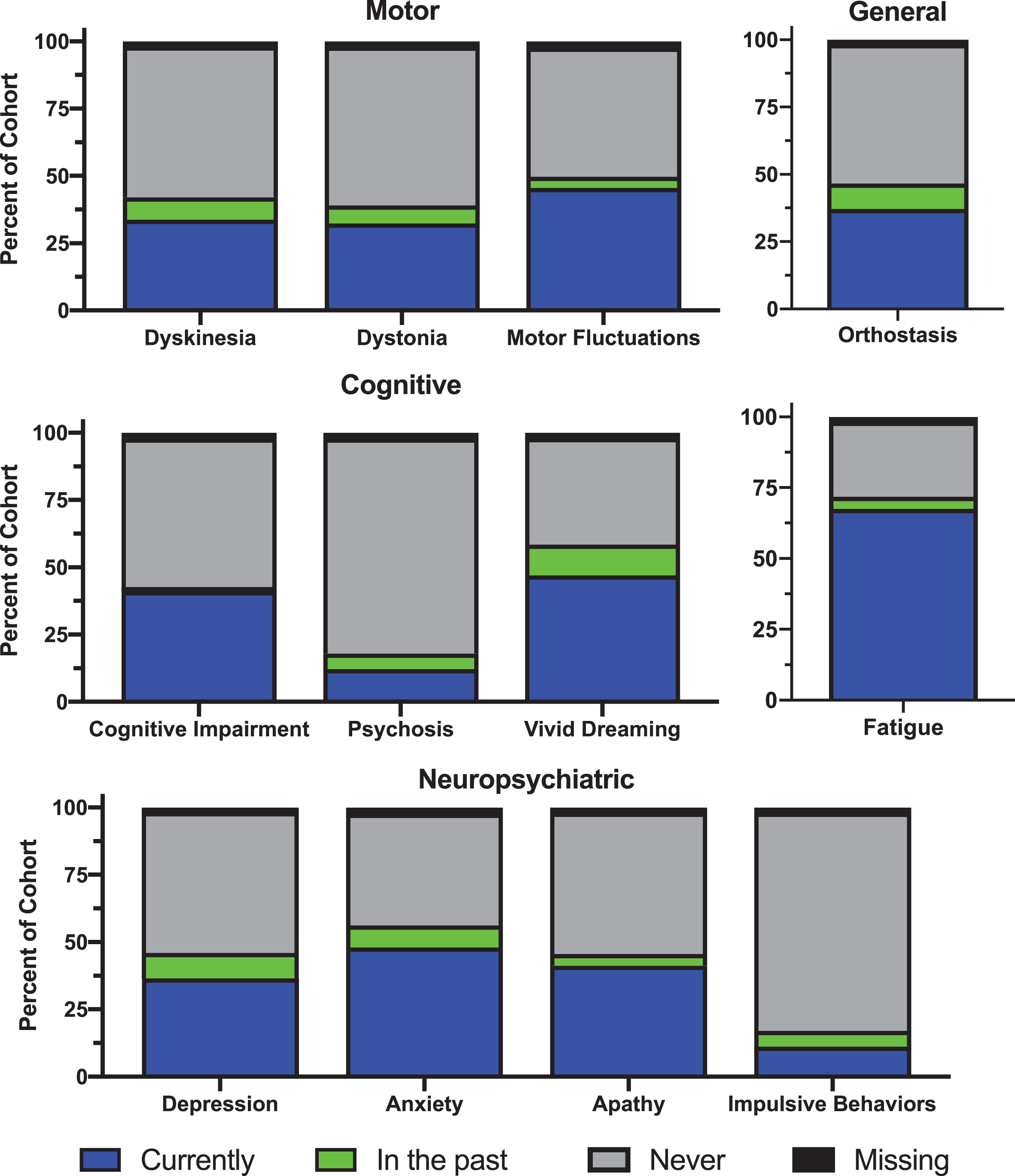
Clinical Characteristics of the Molecular Integration in Neurological Diagnosis (MIND) Cohort. Self-reported motor, cognitive, neuropsychiatric, and general complications of Parkinson’s disease in the Molecular Integration in Neurological Diagnosis (MIND) cohort.
Percentage of cohort by GBA or LRRK2 variant carrier status reporting current or past history of Parkinson’s disease complications
Family history
Self-reported family history of (1) any neurodegenerative disease or (2) Parkinson’s disease is shown in Fig. 3A, by degree of genetic relatedness. In the entire cohort, 172 (26.38%) participants reported a family history of any neurodegenerative disease (i.e., PD, Alzheimer’s disease, amyotrophic lateral sclerosis, multiple system atrophy, dementia with Lewy bodies, progressive supranuclear palsy, frontotemporal dementia, corticobasal syndrome, or other dementia) or tremor in a first-degree relative. The frequency of family history of different neurodegenerative diseases is shown in Supplementary Table 2, by degree of genetic relatedness. Among those self-identifying as Ashkenazi Jewish, 15.31%reported a family history of PD in a first-degree relative, compared to 12.45%among those not identifying as Ashkenazi Jewish (Supplementary Table 3).
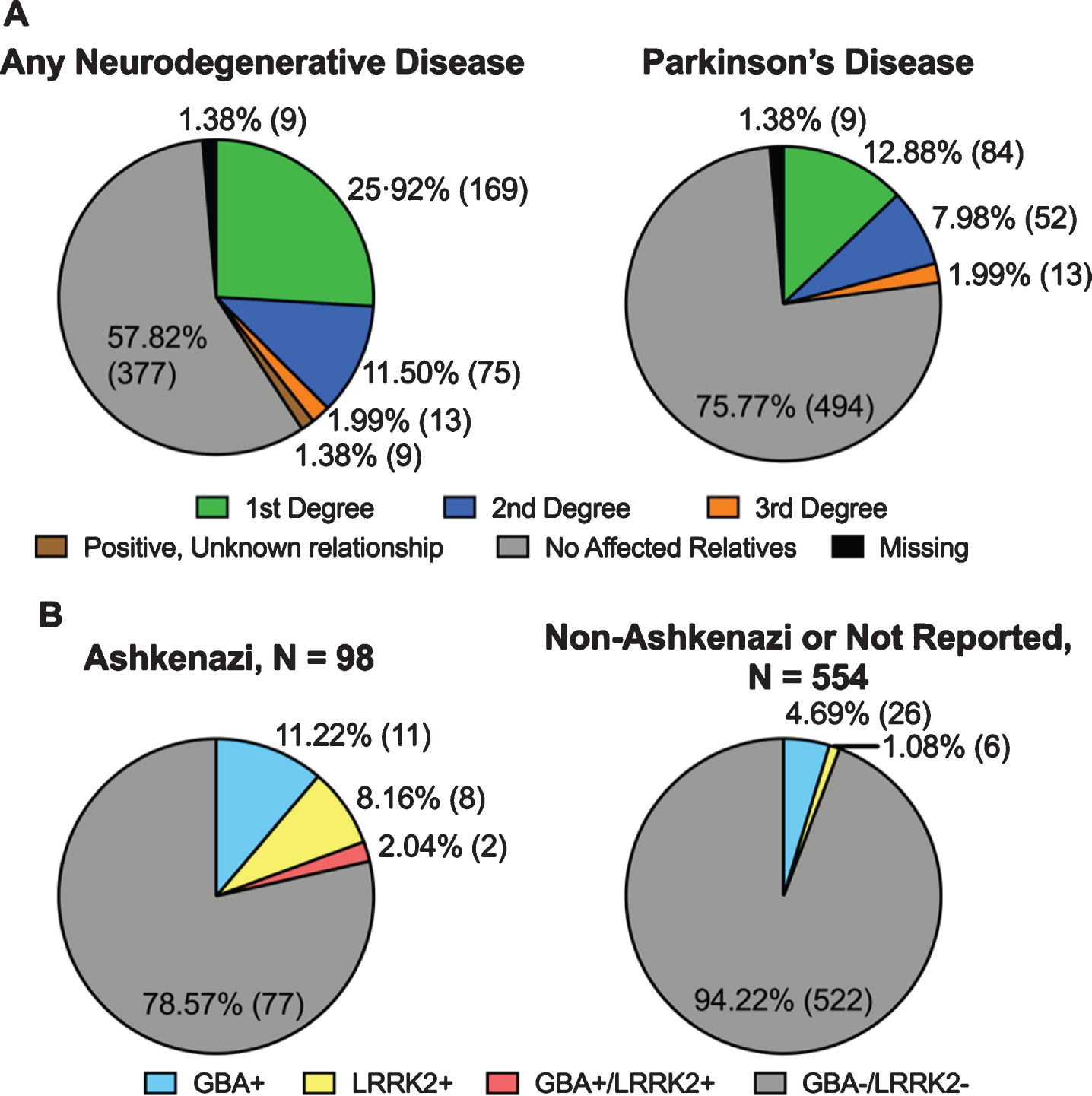
A) Family history of neurodegenerative disease in the Molecular Integration in Neurological Diagnosis (MIND) cohort (N = 652) by degree of relatedness. B) GBA and LRRK2 variant frequency among ethnicity groups in the MIND cohort. Numbers represent percent of ethnicity group (N).
Genetic analysis
We screened for four GBA variants and eight LRRK2 variants (Supplementary Table 1), covering the most frequently reported PD-associated GBA variants and all LRRK2 variants reported to associate with PD. All screened variants would confer eligibility for a genetically guided clinical trial in PD.
In total, 53 PD participants (8.13%) were found to harbor a GBA or LRRK2 variant: 39 participants (5.98%) were found to carry a GBA variant, while 16 participants (2.45%) were found to carry a LRRK2 variant. Two un-related participants carried both a GBA (c.1226A>G) allele and a LRRK2 (c.6055G>A) allele; both reported Ashkenazi Jewish descent. The two most common GBA variants found were c.1093G>A (often referred to as the E326K variant) and c.1226A>G (often referred to as the N370S mutation), while the most common LRRK2 variant found in the MIND cohort was c.6055G>A (often referred to as the G2019S mutation). GBA or LRRK2 variants were more commonly found among participants reporting Ashkenazi Jewish descent compared to non-Ashkenazi Jewish participants (GBA, p = 0.004; LRRK2, p < 0.001). Frequencies of GBA and LRRK2 variants are shown in Fig. 3B.
DISCUSSION
Here we report the interim results of the ongoing Molecular Integration in Neurological Diagnosis (MIND) Study, which aims to characterize molecular and clinical features of every PD patient in a large academic movement disorders center at UPenn. Of 704 PD patients approached in the first 15 months, 652 (92.61%) enrolled in MIND, with >99%of enrollees consenting to access to the EMR, contact for future studies and possible genetic disclosure, and use of biosamples for future studies. Among enrollees, 53 of 652 (8.13%) participants harbored one of the GBA or LRRK2 variants screened.
Our all-comers approach is highly efficient for enrolling participants. In the first fifteen months, we approached 704 patients, representing approximately one-third of our total PD clinic population, and only 28 of 704 (3.98%) declined to participate. The low participant burden is a likely factor in the high participation rate and the demographics of enrolled participants. Notably, > 10%of participants identify as non-white, a much higher proportion than the <4%non-white participation in genetic research studies from our same clinical center pre-dating this all-comers approach [17]. Moreover, the MIND cohort has a higher proportion of women (35.58%vs. ∼32%) than prior studies from our clinical center [17]. Multiple prior studies have noted that women and minorities may be under-represented in PD research cohorts [12]. Our current findings suggest that a global approach to participant capture in a “gateway” protocol that allows for contact for future studies may be effective in combating selection bias.
Data and biofluid capture in the MIND study has been similarly efficient. Specifically, DNA was extracted for 647 of 652 (99.23%) participants, plasma was obtained in 602/652 (92.3%), and clinical complications were captured in 616 of 652 (94.47%) participants.
Finally, among the 652 MIND enrollees to date, we have identified 39 (5.98%) carriers of GBA variants and 16 (2.45%) carriers of LRRK2 variants. Among the 16 LRRK2 variant carriers, 2 (12.5%) also carry a GBA variant, which may have implications for trial eligibility, highlighting the importance of appropriate genetic testing in PD. So far, we have approached approximately one-third of our total clinic population in the first 15 months, and we estimate that by the end of the MIND study, we will have identified ∼120 carriers of GBA variants and ∼50 carriers of LRRK2 variants in a single clinical site. In comparison, since initiation in 2010 the PPMI study has enrolled 165 LRRK2 mutation carriers with PD and 85 GBA mutation carriers with PD from 22 US sites and 10 international sites [11]. The PDGENEration study plans to enroll PD participants from many US-based PD centers and conduct genetic testing; however, the results of this study are not yet available. We note that the GBA variants screened here are not comprehensive, and one variant (c.1093G>A, or E326K, variant) does not cause Gaucher’s disease in its homozygous form. However, all carriers identified would be eligible for genetically guided disease-modifying trials in PD. We further note that the MIND study does not cover the genetic counseling and disclosure of genetic information to participants. However, research participants can opt in to potentially learning if they carry a GBA or LRRK2 mutation in a companion, ongoing study from our group after clinical confirmation testing (see clinicatrials.gov identifier NCT04527146 for more information). Although we demonstrate high interest in future recontact, it remains to be seen how many people will consent to participate in the clinical genetic testing program after appropriate genetic counseling. Anecdotally, many of the MIND participants cited a wish to possibly learn if they carry a GBA or LRRK2 mutation as a reason for enrollment, suggesting that there is an unmet need in the PD patient community for molecular data.
We acknowledge some limitations of our study. First, we have only approached one-third of the 2100 + PD patients who receive clinical care in our center. As a consequence, we could be biased still towards the subset of our clinic population most likely to participate in research, resulting in skewed estimates of “yield” in genetic variant carriers and an inflated participation rate. However, we approached PD patients randomly, and five different research coordinators enrolled subjects, suggesting that our findings do not rely on the identity of the treating physician or the enrolling coordinator. Moreover, with the ongoing worldwide SARS-CoV-2 pandemic, and its accompanying uncertainties, we felt that an interim analysis of the pre-pandemic phase of MIND was warranted due to expected changes in enrollment methods.
Second, clinical features described in this study are by self-report only, which may decrease accuracy. However, the most commonly reported clinical complications of PD were fatigue (reported as a current symptom by 67.48%of the participants) and anxiety (reported by 48.01%of participants). Both are by their very nature subjective, but important, symptoms for PD patients. That said, future studies validating self-reported symptoms against neuropsychological testing batteries or validating self-reported Ashkenazi Jewish ancestry against data from ancestry informative genetic markers, would be a valuable addition to the work shown here. Differences in clinical characteristics between genetic carrier and non-carrier groups are important, although the small sample size in this study limits these comparisons. Notably, the occurrence of cognitive impairment in GBA-variant carriers is lower than non-carriers (35.90%vs. 43.07%), which differs from prior reports, [18] highlighting the importance of additional enrollment and validation of the self-report instrument.
Third, as a single-site study, it is unclear whether the high efficiency seen here will be reproducible in other clinical settings. In this regard, though, we note that the UPenn clinical site encompasses the practices of 11 movement disorders specialists, whose clinical effort ranges from 5%to 80%of total academic effort, offering some confidence that similar efficiency would be feasible across a range of clinical practice styles. Moreover, for three months of the 15-month period reported here, the entire clinical operation was moved to another physical location within our hospital due to an unexpected elevator breakdown, suggesting that even in the face of logistical difficulty, rates of uptake for the global capture approach reported here are very high.
Keeping these limitations in mind, we demonstrate that an all-comers approach capturing genetic and clinical data at the time of the clinical visit is feasible in a busy academic clinical setting. As such, MIND offers a model for how molecular information might enter the clinic at scale, paving the way for a precision medicine approach to the care of PD patients, and serving as a model for future neurological clinics beyond movement disorders.
CONFLICT OF INTEREST
All authors report no conflicts of interest related to the work presented in this manuscript.
Footnotes
ACKNOWLEDGMENTS
The authors would like to acknowledge our patients for their generous participation in this study, and Travis Unger and Sam Rudovsky for technical support, and the clinical research associates at the University of Pennsylvania. This research was supported by the NIH (R01-NS115139, P50-NS06284, U19-AG062418), and the Penn Center for Precision Medicine. ACP is additionally supported by the Parker Family Chair. TFT is additionally supported by the NIH/NINDS (K23 NS114167).