
Abstract
The observation that cysteine is the top gainer amino acid during evolution attracted the attention of scientists dealing with protein chemistry. The thiol group of cysteine, indeed, is a potential site for several types of reactions with variable specificity and strength. This feature proved to be promising also in the field of membrane transporters that represent boundary proteins fundamental for cell homeostasis. These proteins are classified, according to the driving force for transport, in primary or secondary active transporters. Another frequently used classification is nowadays based on phylogenesis. Two major groups are identified that take into account both criteria: the ABC and the SLC transporters, the second being much more numerous. The cellular localization of the transporters makes them very attractive for drug design. Moreover, the presence of at least one cysteine residue in all the annotated SLC transporters, so far, highlights the possibility of using the thiol (SH) residue for covalent drug targeting. Even if a delay exists in this research field due to the scarce knowledge of structure/function relationships, the setup of novel experimental tools for studying SLC proteins of plasma and organelle membranes opens an important perspective in pharmacology.
Introduction
The idea that membrane transporters are crucial for cell homeostasis is nowadays well acknowledged by the scientific community. This assumption is grounded on two basic experimental pieces of evidence: (1) roughly 10% of all human genes are related to proteins involved in transport function, and (2) knocking out transporter genes leads to alterations of metabolic processes. The trusting proof of the fundamental role of transporters is the continuously increasing evidence that inherited defects of genes encoding membrane transporters are associated with human pathologies with different degrees of severity.1,2 The molecular basis of the mentioned findings relies on the biochemical function of membrane transporters in catalyzing the exchange of nutrients, catabolites, and ions across cell membranes, which are impermeable to most molecules. Over the years, it became clear that membrane transporters also mediate the response to xenobiotics, ranging from toxic compounds to specifically designed chemicals and drugs.3–5 In this frame, membrane transporters are also very promising for industrial appldications,6–8 as demonstrated by the increased funding of transporter-related research by pharmaceutical and biotech companies.9,10 The studies on membrane transporters started much earlier than genome sequencing. Therefore, transporters were originally classified on the basis of substrate specificity and transport mechanism. In the postgenomic era, data deriving from both functional and genomic studies guided the grouping of these proteins in two major categories, the ABC (ATP binding cassette) and SLC (solute carrier), even though the former classification is still in use and often overlaps the novel one. This classification is mainly based on energetics: ABCs are mostly primary active transporters, while SLCs are secondary active transporters ( Fig. 1 ). 1 The ABC series includes 48 transporters grouped into seven distinct subfamilies. 11 Proteins belonging to this superfamily are characterized by ATP binding domains called nucleotide binding domains (NDBs). These domains, localized in the cytosol, contain Walker A and B motifs separated by 90–120 amino acids. ABC transporters are present in all living organisms: in prokaryotes, ABCs are mainly involved in the uptake of nutrients, while in eukaryotes ABCs work in both directions (uptake and efflux) according to cell needs. The driving force for transport reaction directly derives from ATP hydrolysis occurring at the NBD level. 11 This quasi-enzymatic feature allowed the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB) to recently include most ABCs in the list of a new class of enzymes, named “translocase” EC7 (https://www.qmul.ac.uk/sbcs/iubmb/enzyme/EC7/). The SLC group includes 52 families harboring about 400 genes, which encode more than 800 proteins as products of alternative splicing, according to data deposited in https://www.ncbi.nlm.nih.gov/protein/. This number might be higher due to the presence of many other translation products that are not yet validated. As a general rule, with few exceptions, proteins belonging to the same family share at least 20% of amino acid sequence identity. 1 The SLC classification was first developed for human genes, and later their rodent orthologues were classified similarly. The driving force for membrane transport, mediated by SLCs, derives from concentration gradients of the solutes involved in each reaction or from the gradients of specific ions, which are co-transported with the solutes in the same or opposite direction ( Fig. 1 ). Solute or ion gradients are directly or indirectly generated by reactions of ATP hydrolysis explaining the classification as secondary active transporters. Besides the well-defined SLC and ABC transporters, an additional 70 human genes encode for non-ABC transport-related ATPases, also called pumps, and about 320 genes encode for ion channels and ionotropic receptors (data from HUGO, HGNC, https://www.genenames.org/) ( Fig. 1 ). Even though these additional membrane proteins are not sharply classified in the SLC or ABC group, their function is associated with the flux of ions across the membrane. Several reviews focus on the role of the different transporter families in the maintenance of cell homeostasis and on the pathological implications of their alteration.12–15
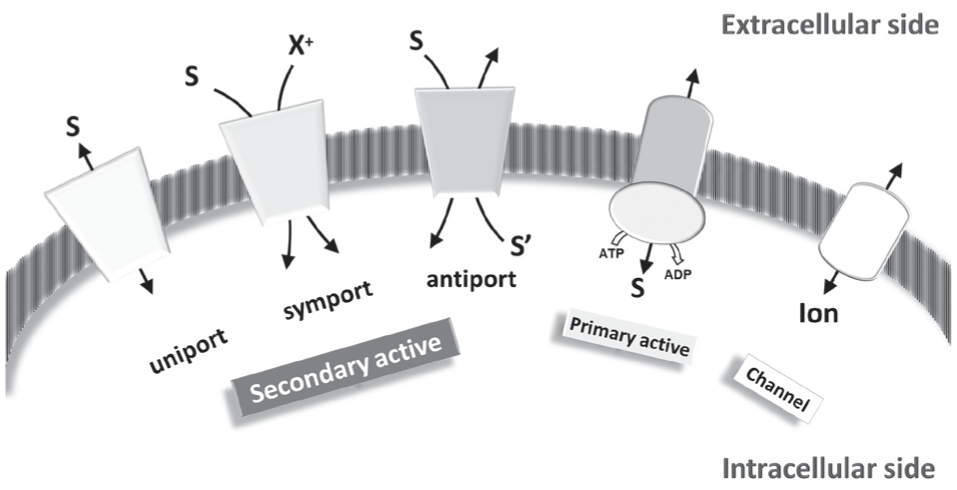
Functional classification of membrane proteins. Secondary active transporters (SLCs), primary active transporters (including ABC and pumps), and channels are sketched in a membrane. S (substrate), S′ (countersubstrate or co-substrate), and X+ (cations, such as Na+) representing the transported substrates in secondary active transporters follow the indicated transport paths according to the transport modes (uniport, symport, or antiport). Substrate (S) flux in primary active transporters requires ATP hydrolysis. The movement of ions catalyzed by channels is also sketched.
Membrane Transporters as Target for Drugs
Over the years, a novel field of investigation that is the interaction between drugs and membrane transporters has taken hold. Starting from pioneering works in the 1980s, this issue expanded exponentially ( Fig. 2 ). From a philosophical point of view, the interaction of xenobiotics with transporters is expected owing to the localization of these proteins at the forefront between the external environment and intracellular body districts. Nevertheless, pollutants and drugs appeared in the environment later with respect to the evolution of transporters. Thus, their interactions with transporters must be considered a side effect. 16 From a systematic point of view, the interaction and distribution of xenobiotics is much more complicated due to the organization of multicellular organisms such as in the case of the human body. In fact, the boundaries among external and internal environments are more complex than expected. As an example, the epithelial barrier of the intestine is composed of a single cell layer and several molecules, including drugs, need to cross it during absorption. The molecules, moving from the lumen to blood, have to cross two types of membranes, characterized by a collection of different transporters on apical and basolateral sides, respectively. The same applies to kidney reabsorption and secretion phenomena ( Fig. 3 ). In other nonpolarized cell types, such as muscle or liver, circulating molecules leave bloodstream from capillaries and cross only one membrane type. The scenario becomes even more complex in the case of a fetus and the brain that are separated from blood by the impermeable placenta and blood–brain barrier (BBB). Metabolites must cross the two membranes of the barriers before reaching the final destination. The presence of specific plasma membrane transporters that may be different, or may work differently, on the two cell sides will characterize the permeability of these two barriers ( Fig. 3 ). Very little information is available on this issue so far. Last but not least, in all cell types, molecules have to also cross intracellular membranes to reach their definitive destination ( Fig. 3 ). Drug design, absorption, and disposition are therefore issues that need to be afforded considering different players at the same time to properly evaluate the efficacy of a new drug; in this respect, very often new drugs come from old ones or by serendipity. 17 From a pharmacological point of view, membrane transporters can play a dual role: (1) direct transport of a specific drug that does affect its disposition and dynamics or (2) being the target of a drug with consequent alteration/modulation of the transport of a set of physiological substrates. Due to the great importance of these issues in drug design and efficacy, industrial and academic scientists with expertise in drug metabolism make up the International Transporter Consortium (ITC).18–20 Over the years, this institutional society compiled a list of transporters responsible for influencing pharmacokinetics. This big effort is mainly devoted to understanding the most important SLC and ABC players in drug disposition in order to identify the fastest and most efficient way to improve the design of new drugs and reduce side effects. In this frame, the activity of the consortium also meets the second point above mentioned, that is, the possibility for a membrane transporter to be a target of either desired or undesired interactions with drugs. In fact, with respect to their membrane localization, membrane transporters can be considered first- or second-level targets. The first category includes the plasma membrane transporters, at the boundary with the extracellular milieu, and hence with the external environment from where drugs and xenobiotics come. The second category includes the membrane transporter of intracellular organelles, which are fundamental for the proper compartmentalization of cell metabolism and cell homeostasis ( Fig. 3 ). This picture, even if rough, may help in understanding the complexity of the pharmacological role of membrane transporters, which is still far from being systematically approached. 20 It is interesting to note that, notwithstanding the key role played by transporters and the link of most of them to human pathologies, very few are well-established examples of drugs targeting membrane transporters with significant commercial impact: proton pump inhibitors (PPIs), antidiabetics, and antidepressants/antipsychotics.21–23 The low number of Food and Drug Administration (FDA)-approved drugs whose efficacy is based on the interaction with transporters is a clear consequence of the delay in the study of membrane transporters with respect to soluble enzymes or receptors. Different from receptors, transporters are mostly embedded into the membrane and are thus highly hydrophobic proteins whose handling is challenging. Therefore, techniques employed for investigating soluble proteins could not be applied tout court to these type of molecules. 20 Indeed, one of the most controversial but important points in drug–transporter interaction studies is the identification of the proper experimental models to predict and validate interactions; several efforts have been made in this direction.18–20 So far, the combination of bioinformatics for high-throughput screening (HTS), transport assay in intact cells, and in the in vitro proteoliposome tool, represents the most suitable way to afford the drug–transporter interaction study. Thus, the studies on membrane transporters have largely increased in recent years, with a terrific enlargement of their knowledge ( Fig. 2 ). The information on the different ABC and SLC members is obviously not yet systematic, but the tendency to improve the characterization of kinetics, interactions, and regulation of activity/expression may increase the chances of finding effective interactions with old or novel drugs. In particular, some relevant progress has been made in terms of defining molecular determinants, that is, key amino acid residues, for the binding of substrates or allosteric regulators. In this respect, an amino acid deserving great attention is cysteine due to both its presence in most proteins and the versatility of chemical reactions that it can undergo.
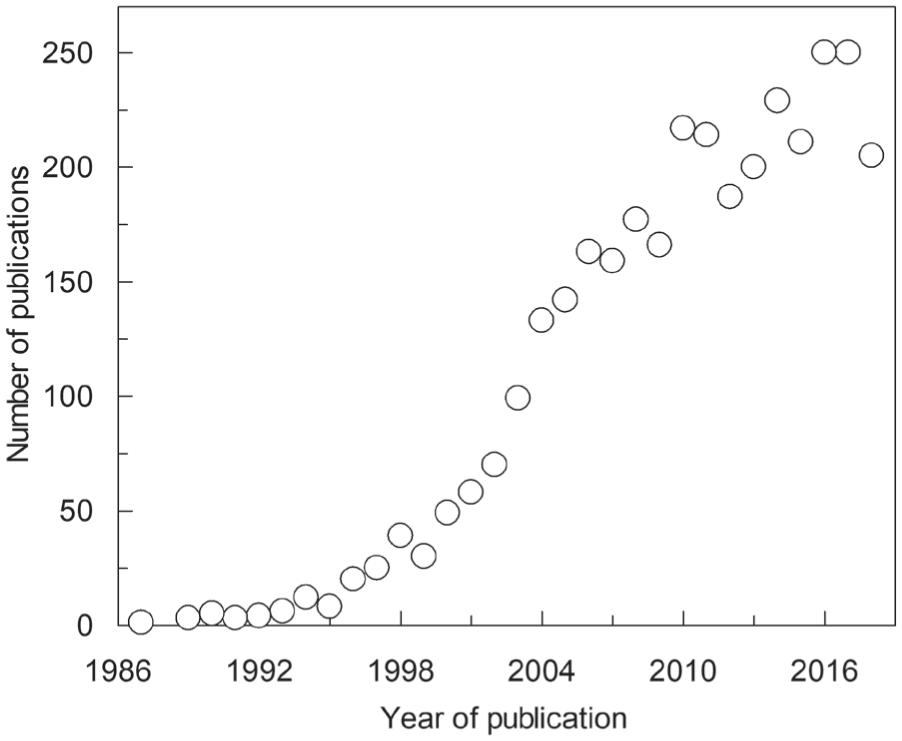
Trends of publications over time. The number of papers published in 1987–2018 according to a Web of Science database search using the wildcards “TI=(SLC*OR transporter*) AND TI=(drug*OR xenobiotic*)” in the search toolbar.
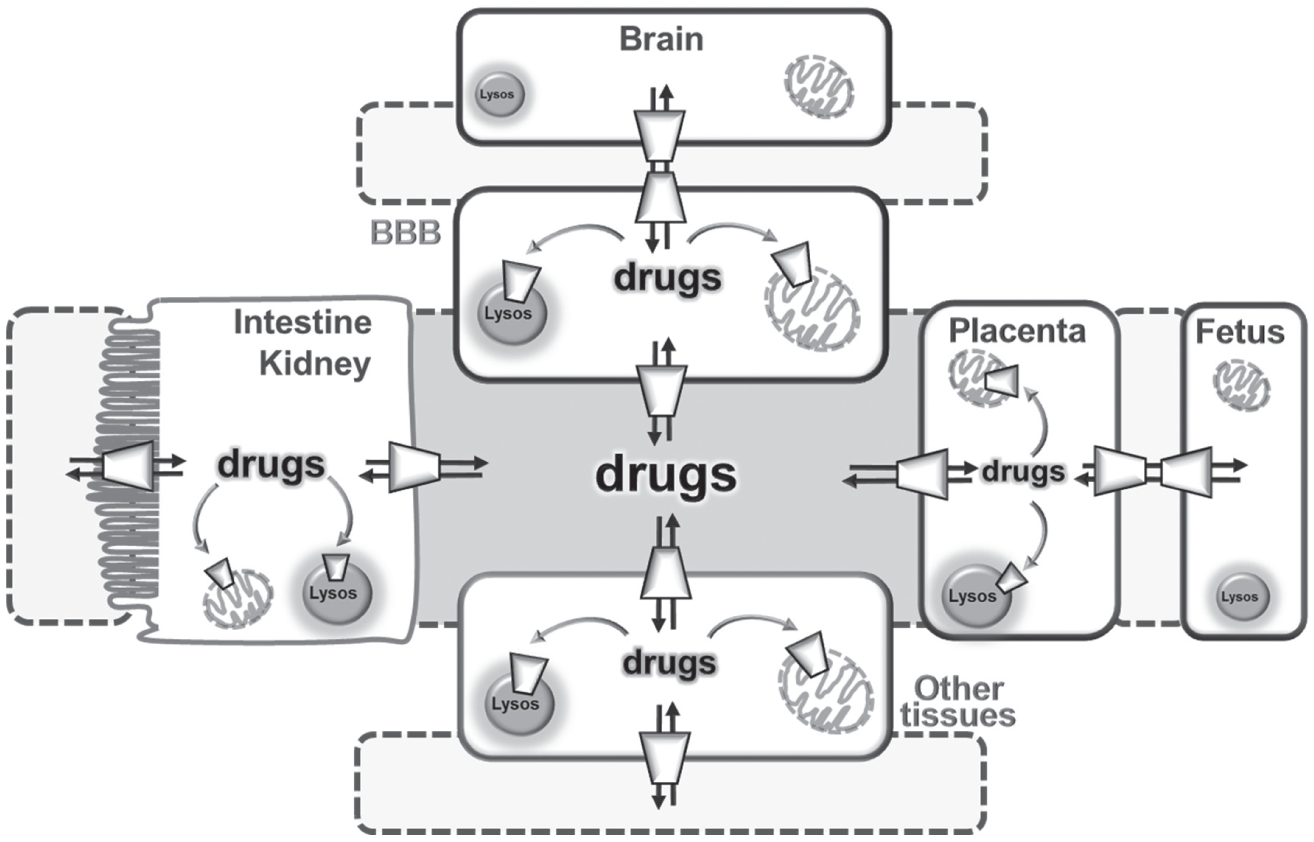
The drug distribution network through membrane transporters. Crosstalk among polarized cells of the intestine/kidney, placenta, fetus, BBB, brain, and other tissues. In polarized cells, the apical membrane is depicted as a brush-border facing intestinal/kidney lumen (light gray) and basolateral membrane in contact with blood (gray). Drugs are absorbed and reabsorbed or excreted through the intestine or kidney using transporters with different transport mechanisms (see Fig. 1 for a more detailed view). The drugs reach blood by other transporters localized in each cell. From blood, drugs can reach the brain through the BBB and the fetus through the placental barrier. Once in cells, drugs can reach mitochondria or other organelles (such as lysosomes) using specific membrane transporters. Fenestratus capillary membranes are depicted as dotted lines.
Cysteine: An Intriguing Amino Acid with Multifaceted Functions
Cysteine is considered a conditionally essential amino acid due to its involvement in pivotal cell functions and its biosynthetic pathway that requires the availability of the essential amino acid methionine as a sulfur donor in the trans-sulfuration pathway. 24 Furthermore, cysteine can be absorbed from one’s diet and derived from endogenous protein degradation. In the extracellular environment, cysteine is mainly present in its oxidized form, cystine. 25 Upon entry in cells, cystine is converted to cysteine due to the reducing environment of the cytosol that breaks the disulfide bond of cystine. The cysteine/cystine couple, together with ox/red thioredoxin and GSH/GSSG (the reduced/oxidized tripeptide glutamyl-cysteinyl-glycine called glutathione), interacts with thiol-containing macromolecular targets. Interestingly, these systems are independent of each other and are likely to be under kinetic control to maintain redox homeostasis in both intra- and extracellular milieus. 26 It is worth noting that the major player for cystine uptake in cells is the plasma membrane transporter SLC7A11, referred to as xCT. This protein is an obligatory antiporter that exchanges cystine with glutamate, and its relevance in controlling redox homeostasis is demonstrated by the occurrence of a specific type of cell death called ferroptosis. 27 This phenomenon has been recently discovered and is currently under active investigation to define the molecular determinants and the specific effects due also to the great overexpression of xCT in several human cancers. In this frame, ferroptosis stimulation can be one of the ways of inducing specific cancer cell death.28,29
Cysteine as a Druggable Protein Residue
A peculiarity of cysteine is its relevance as both a free amino acid and a targetable protein residue. The reacting functional group underlying the unique features of cysteine is the thiol (–SH) of the side chain. Thiol gives rise to a sizable number of different reactions because it is unique in terms of nucleophilicity and sensitivity to oxidative modification. The thiol group, as a negatively charged species (–S–), is reactive under nonextreme, physiological conditions of temperature and pH. Several processes can take place at the level of the SH group: (1) redox potential regulation, (2) the formation of disulfide bonds (S–S) leading to intra- and intermolecular covalent interactions affecting the three-dimensional structure of proteins, (3) the coordination of metals and metalloid cofactors with relevant outcomes in physiology as well as in toxicology, (4) alkylation by electrophiles, (5) oxidation by reactive oxygen species (ROS) and the formation of intermediates like sulfenic acid (reversible) and sulfonate (irreversible), and (6) reactions with gaseous signaling molecules such as NO and H2S or with amines/amides (sulfenamide/sulfinamide/sulfonamide).30–32 Such a huge number of possible reactions has a great impact on cell physiology. In fact, cysteine is involved in redox control and regulation acting as a signaling sensor; furthermore, cysteine residues are a potential source of posttranslational modifications of the proteins responsible for regulation of their activity. 33 Nowadays, the importance of these aspects is also emerging for transport proteins due to evidence that redox control is at the basis of several physiopathological states. Moreover, the large number of cysteine-based posttranslational modifications leads to a huge number of possible protein forms that enlarge the complexity of the cell proteome that, indeed, is still far from being determined. According to the reversible nature of many ligand–thiol modifications in vivo, the resulting proteome is dynamic and variable according to cell types, cell metabolism, cell cycle, and environmental conditions. 34 It is worth noting that the wide range of interactions to which cysteine residues can be subjected makes the ranking of cysteine in a hydrophobicity scale hard. Thus, in site-directed mutagenesis experiments, cysteine cannot be unambiguously substituted with a single amino acid residue. In some cases, the hydrophilic serine can be employed; in other cases, the hydrophobic alanine is more appropriate. The choice depends on the location of the cysteine in the protein structure and often cannot be predicted on the basis of sequence or structure analysis by bioinformatics. 32 A very important aspect of making cysteine an appealing target derives from evolutionary observations. Cysteine residues appeared later in evolution, together with glycine, proline, and tryptophan; notwithstanding, cysteine is a gainer amino acid. 35 Indeed, the cysteine frequency in proteins increases, with evolution being maximal in proteins of higher organisms. Despite this frequency trend, cysteine is at present one of the less abundant amino acids in proteins, comprising about 2.3% of the residues in the human proteome. Nonetheless, cysteine residues are tremendously involved in protein function: roughly 50% of cysteine residues play crucial roles for cell life. 36 This feature impacts the interaction with xenobiotics and drugs as well. In fact, cysteines represent interesting and specific targets, and their low frequency in proteins limits the number of potential off-target reactions. A peculiarity of cysteine localization is the clustering in the three-dimensional structure of proteins.32,37,38 This correlates well with the fast and reversible SH/S–S exchange among vicinal residues. This phenomenon may have an evolutionary implication in both structure stabilization and function regulation. Notwithstanding, the organization of cysteine residues does not always respond to canonical sequence organization; therefore, the setup of methodologies devoted to the characterization of cysteine reactivity is not straightforward, even though hyperreactive cysteines have been annotated following pKa measurements. 33 A nonstandard amino acid with a structure very close to that of cysteine is selenocysteine, in which the sulfur is posttranslationally replaced by a selenium atom. This substitution still maintains the reactivity of the classical cysteine. Then, the reactions occurring at the level of selenocysteine are similar to that of cysteine. 32 It is important to highlight that some of the features described for cysteine are common also to other amino acids, such as lysine, which is a protein building block but is also involved in protein regulation due to its ability to bind cofactors in enzymes. At difference with cysteine, lysine is much more abundant in proteins, and therefore it is less suitable as a specific target.37,39 Even though some current research also focuses on exploiting tyrosine, tryptophan, histidine, glutamic and aspartic acids, and methionine as covalent targetable residues,39,40 cysteine is the most interesting in drug discovery due to the combination of the high reactivity of thiol/thiolate groups and the low abundance in proteins. In particular, the SLC human membrane transporters display an average of roughly 12 cysteine residues and at least one cysteine present in all proteins ( Fig. 4 ) with only one exception, the zinc transporter SLC39A11. Importantly, if the cysteine residues are close enough, it is necessary to evaluate whether they are in a disulfide or thiol state. In the second case, cysteines can be predicted as a potential drug/xenobiotics target. Importantly, many data are available for the role of cysteine residues in soluble proteins or in some receptors, while its role in SLCs is still underneath. Notwithstanding the lack of knowledge, the location of transporters in cell membranes strengthens the role of cysteine as a potential target for drugs.
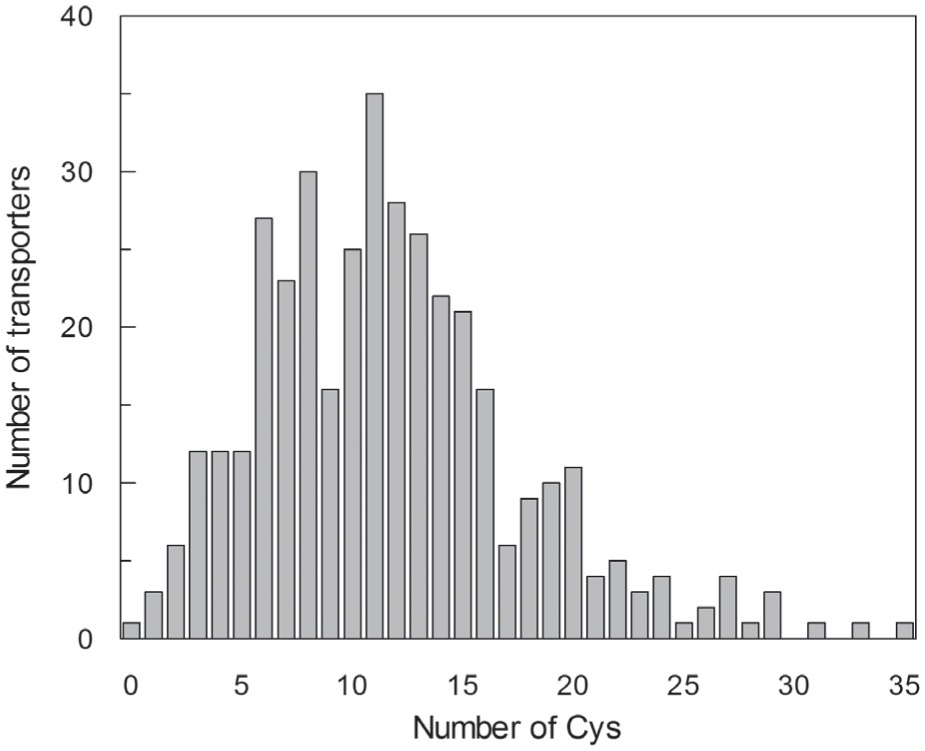
Distribution of SLC transporters versus the number of cysteine residues. The primary structures of the validated members of SLC families have been analyzed for cysteine residue counting. The graph follows an approximately Gaussian trend of distribution with only one SLC member (SLC39A11, a zinc transporter) with no cysteine residue. The average number of cysteine residues is 11.
Covalent Interaction in Drug Targeting
The main goal in pharmacology is designing drugs with a high affinity for the targets and, hence, a high potency in order to be more efficient and minimize off-site reactions. The most common mechanism for drug interaction is the noncovalent binding in which the drug makes contact with its target exploiting hydrophobic and/or hydrophilic interactions. Targeting can occur at the catalytic or noncatalytic sites. Over the years, a strategy for reaching more stable molecular targeting with improved therapeutic efficacy emerged. This goal can be achieved by covalent drugs. It must be noted that most of the covalent drugs are covalent inhibitors, that is, pharmacological compounds that inactivate the protein. The covalent interaction can be reversible or irreversible depending on the reverse reaction on biological scale timing.
41
It is worth noting that drugs triggering covalent interactions are quite old in pharmacology. For example, acetylsalicylate (aspirin) was discovered in 1899 and is still one of the most widely administered drugs; acetylsalicylate leads to acetylation of a serine residue of cyclooxygenases I and II. Another example of a covalent drug is the well-known antibiotic penicillin, one of the blockbuster drugs that is able to bind to a serine residue of the catalytic binding site of the bacterial
Pioneering Use of Cysteine Targeting for Inhibition of the Gastric Proton Pump
A classic example of drug targeting cysteine residues of a membrane protein are the PPIs, whose prototype is omeprazole. The FDA approved six PPIs as a first-choice drug for treating a wide range of gastric diseases: esophagitis, nonerosive reflux disease, peptic ulcer diseases, and functional dyspepsia. PPIs are also used in combination with antibiotics for the eradication of Helicobacter pylori and in the prevention of ulcers associated with the use of nonsteroidal anti-inflammatory drugs. 21 This large use is due to their quite high safety for patients in comparison with other antiacids, such as H2 receptor antagonists, prostaglandin analogues, or anticholinergics. Omeprazole, introduced in 1989, is a heterocyclic molecule containing pyridine and benzimidazole rings linked by a sulfur atom able to interact with cysteine residues of the gastric K+/H+ pump. This protein is a member of the P-type ATPase family that mediates the electroneutral exchange of K+ and H+ coupled to a phosphorylation/dephosphorylation cycle. 51 The molecular mechanism of action is based on acidic activation of omeprazole after accumulation in gastric canaliculi. The strong acidic environment of this body district induces a rearrangement to a cationic sulfenamide that is virtually membrane impermeant. In this form, the drug binds to extracellular cysteine residues of the catalytic domain of the K+/H+-ATPase.52–54 Due to this strong reactivity, omeprazole and its derivatives are also able to interact with other proteins with the same molecular mechanism, that is, formation of a mixed disulfide. This phenomenon may constitute a typical case of off-target interactions. In this respect, omeprazole has been reported to inhibit few SLC transporters: the carnitine transporter of the mitochondrial membrane CACT (SLC25A20), the carnitine transporter of the plasma membrane OCTN2 (SLC22A5), and the organic cation transporters OCT1, 2, and 3 (SLC22A1, 2, and 3).55–57 In the case of CACT, the cysteine residues targeted by omeprazole have been unequivocally identified by a combined experimental approach based on bioinformatics, site-directed mutagenesis, and transport assay ( Fig. 5 ). Omeprazole is a strong covalent inhibitor of both OCTN2 and CACT; the inhibitory effect can be reversed by the addition of nonphysiological or physiological reducing agents. The effects of omeprazole can be classified as off-target interactions and may negatively affect the carnitine homeostasis: a lower cell carnitine uptake caused by inhibition of the plasma membrane transporter OCTN2 and a lower acyl-carnitine supply to mitochondria for β-oxidation, caused by inhibition of the inner mitochondrial membrane transporter CACT. These effects may be potentially dangerous in those tissues relying mainly on fatty acid utilization for energy production. 58 Interestingly, the side effects of omeprazole mimic, in a milder form, the symptoms described for the carnitine deficiency (http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?id=4761); this is also common for other pharmacological interactions that resemble the symptoms of carnitine deficiencies. 59 In the case of OCTs, the effect of PPIs has been evaluated in terms of drug–drug interactions. In fact, omeprazole and its derivatives are able to inhibit metformin uptake mediated by OCT1, 2, and 3 with IC50 values in the micromolar range without being transported. Metformin is a widely used drug to treat diabetes; therefore, inhibition of its uptake, due to off-site interaction of PPIs with OCTs, may have clinical relevance for diabetic patients. 57 Another example of omeprazole off-target is constituted by targeting an ABC transporter, the P-glycoprotein.60,61 This is a well-known protein overexpressed in several human cancers and involved in drug transport; P-glycoprotein is responsible for resistance to chemotherapy agents, which are actively extruded from cancer cells. Therefore, it can be argued that in the case of P-glycoprotein, the covalent interaction with omeprazole may be an example of a “positive” off-target effect.62,63 Then, selective and covalent inhibition of P-glycoprotein could be “novel” employment for an “old” drug. Indeed, the fact that a drug may show interaction with alternative targets, or that different drugs may interact with the target with different binding profiles, can be important in developing next-generation drugs. 17
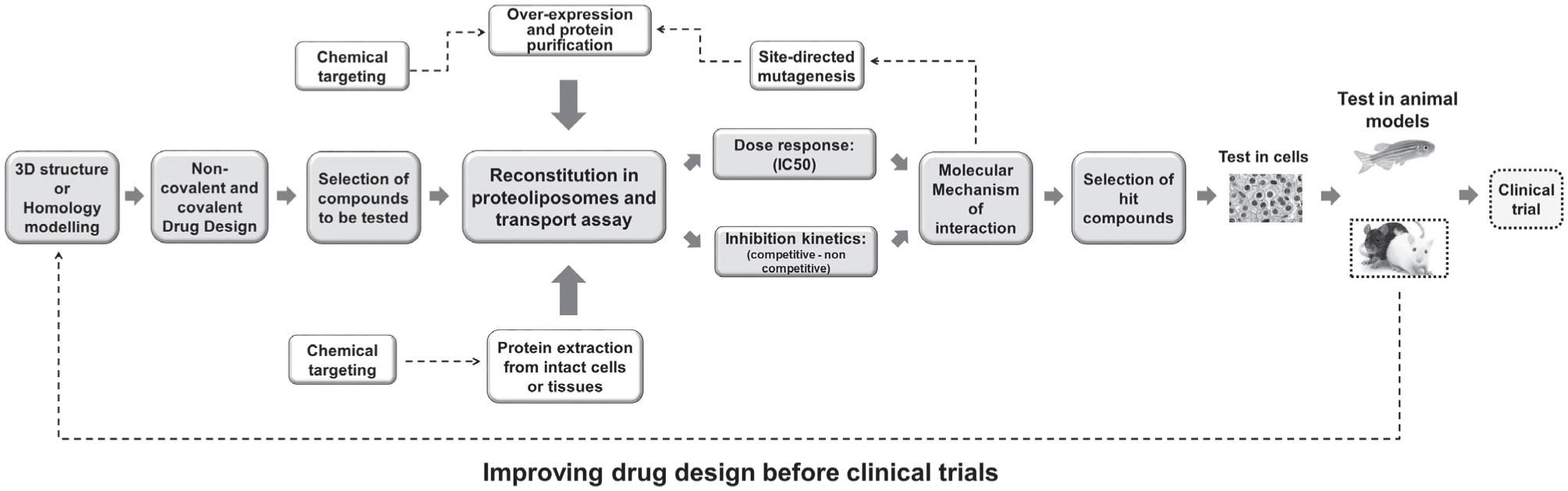
A typical workflow based on bioinformatics and molecular transport assay before trials. The workflow depicts the key steps of designing and testing potential drugs for identifying hit compounds with effects on SLCs. The best compounds are then validated on cell and animal models before clinical trial. The dotted lines represent more advanced stages with respect to those performed in our studies.
Prospective Use of SLC Cysteine Targeting for Anticancer Therapy
Membrane Transporters in Cancer
Investments for research applied to novel anticancer drugs account for a large segment of total investments. 9 SLCs are acknowledged as novel and promising molecular targets for this type of drugs. Transporters, in fact, can be considered the cell “doors” of several nutrients that become essential for cancer growth and development. Indeed, cancer cells rewire their metabolism and, hence, their specific nutrient needs. 64 In particular, cancer cells are characterized by much increased utilization of glucose and some amino acids such as glutamine, leucine, and arginine. The altered metabolic state relies mainly on ATP produced by an anaerobic like catabolism even in the presence of adequate oxygen supply. This phenomenon, known as the Warburg effect, was described in the early 1920s and, after some debates, has been updated and demonstrated at the molecular level.65,66 In line with these observations, the glucose transporter GLUT (SLC2A family) and the amino acid transporters ASCT2 (SLC1A5), LAT1 (SLC7A5), and ATB0,+ (SLC6A14) are overexpressed in virtually all human cancers, becoming a hallmark for this disease.64,67 This feature is relevant in the frame of designing novel drugs that could block glucose and amino acid uptake, thus specifically impairing cancer cell growth. From the first thought in this direction, several studies have focused on searching for new molecules able to specifically target these proteins.
Targeting Plasma Membrane Transporters of Glucose: Noncovalent Interactions
Regarding the glucose transporter GLUT, only in silico and in vitro studies have been performed and two small molecules have been tested in cancer models in vivo.68–71 These compounds are competitive inhibitors of GLUTs able to decrease the anaerobic glycolysis occurring in the cytosol of cancer cells at a high rate to sustain the need for energy and reducing equivalents typical of these peculiar cells. 64 The designed competitive inhibitors of GLUTs are still under evaluation for ameliorating their efficacy and potency; it is worth noting that no covalent inhibitors have been designed so far for GLUTs. The three-dimensional structure of GLUT1 has recently been determined; 72 therefore, drugs with improved structures are expected to be designed with the ability to specifically target cancer cells to impair their metabolism without affecting normal cells. 68
Role of Plasma Membrane Transporters of Amino Acids in Cancer: Noncovalent Drug Targeting
The overexpression of the amino acid transporters ATB0,+, LAT1, and ASCT2 is functional to the need for glutamine, leucine, and arginine typical of cancer cells. These amino acids, besides proteogenic function, play other roles in cells. In particular, glutamine is a nonessential amino acid that becomes conditionally essential in highly proliferating cells.
73
This phenomenon is typical of either physiological and pathological conditions, such as activated inflammatory cells/stem cells and cancer cells, respectively.74,75 The requirement for glutamine is linked mainly to energy production and signaling function: cells that undergo a high proliferation rate show a metabolic switch characterized by a peculiar utilization of carbon skeleton deriving from glutamine; after removal of the amino group, the residual carbon scaffold enters the TCA in mitochondria for oxidation and ATP production at the substrate level. However, the TCA is slowed down because malate exits the cycle and leaves mitochondria to continue oxidation in cytosol to produce the reducing equivalents necessary for anaerobic glycolysis, fatty acid synthesis, and other cell pathways.64,73 Leucine is an essential amino acid required for protein synthesis but also for allosteric regulation of glutamate dehydrogenase, a key enzyme in the metabolic switch occurring in highly proliferative cells, responsible for the conversion of glutamate-derived glutamine into α-ketoglutarate.73,76 Leucine, glutamine, and arginine are also required for sensing amino acids’ availability in cells through a mechanism involving peculiar molecular systems; in particular, two serine/threonine kinases responsible for responding to the presence/absence of amino acids in cells are GCN2 (general control non-derepressible 2) and mTOR (mammalian target of rapamycin). GCN2 is responsible for inhibiting the protein synthesis de novo when one of the 20 amino acids is missing or the concentration is very low; the activation or repression of mTOR is due to the presence/absence of glutamine, arginine, and leucine.
77
In this mechanism, the plasma membrane transporter SLC38A9 residing in the lysosomal membrane plays a fundamental role. This protein is considered a “sensor” of glutamine and arginine availability in cells due to a specific molecular mechanism in which SLC38A9 translocates the amino acids from the internal to the external compartment of lysosomes and triggers a conformational change allowing interactions with the mTOR complex for downstream pathway activation/repression.78,79 Moreover, the plasma membrane transporters for glutamine and arginine might also be indirectly involved in the sensing phenomenon.
80
The sensing of leucine for mTOR regulation is mainly mediated by LAT1, which is primarily responsible for leucine uptake in cells; it is worth noting that LAT1 has also been localized in lysosomes.
81
On the contrary, the cytosolic sensor for leucine, sestrin 2, has been well described and recently crystallized.
82
Given the mentioned overexpression of ATB0,+, LAT1, and ASCT2 in cancer cells, several studies have focused on searching inhibitors for their activity with the scope of reducing the supply of fundamental amino acids to cancer cells. The majority of those works relies on the use of substrate-like molecules that exert competitive inhibition of amino acid uptake. This is the case of α-methyl-
Targeting Amino Acid Transporters by Covalent Inhibitors Reacting with Cysteine
In the above-summarized reports, the competitive nature of the inhibition is characterized by a noncovalent reversible interaction. This feature may have the negative consequence of lowering the efficacy of a drug in the treatment of cancer. This is particularly true in the case of amino acid transporters that are characterized by redundancy, that is, by overlapping substrate specificities. Therefore, when the concentration of an amino acid increases above the Ki for the inhibitor, the pharmacological compound can be displaced from the substrate binding site abolishing the pharmacological effect. 93 This negative consequence of the competitive noncovalent interaction could be partially avoided by using covalent inhibitors. In this case, the inhibitor binds covalently to the substrate binding site or to an alternative site with a stable inactivation of the transporter. The design of molecules interacting with cysteine residues has been approached for ASCT2 and then for LAT1 using the typical workflow summarized in Figure 5 . In the case of ASCT2, the rat isoform has been detergent solubilized from the kidney brush border and inserted in liposomes for transport assay. 94 In the case of LAT1, the human isoform has been produced by heterologous expression in Escherichia coli and used for inhibitor screening according to Figure 5 . 95 The procedure of reconstitution in liposomes is an experimental model in which the protein of interest is inserted in an artificial membrane with the same orientation that it has in native cell membranes. This experimental setup allows the study of the protein of interest in an environment in which no interferences deriving from other proteins or enzymes are present. 96 The reconstitution in liposomes of both recombinant and native proteins is a powerful tool for measuring kinetic parameters and, hence, inhibition. The reconstitution in liposomes is an exquisite in vitro strategy that permits scaling up the experimental procedures reaching high-throughput levels that are required for large-scale screening of chemicals with potential pharmacological functions. In the case of both ASCT2 and LAT1, the reconstituted proteins have been tested for reactivity toward compounds with a dithiazole moiety containing sulfur atom able to form a mixed disulfide with cysteine residues of the proteins of interest. Interestingly, dithiazoles are compounds with antibacterial, antifungal, herbicidal, and antitumor activities (see Oppedisano et al. 94 and Napolitano et al. 95 and references therein). Concerning rat ASCT2, the molecular mechanism of action has been linked to the formation of mixed disulfide with cysteine residues of the protein that could not be identified. 94 Concerning human LAT1, the molecular mechanism of interaction has been well described and defined. 95 A set of hundreds of compounds have been tested to identify a small subset of hit molecules whose molecular mechanism has been deepened. The most potent inhibitors are able to interact with at least one cysteine residue that lies on the substrate binding site of LAT1, a typical example of TCI. This has been demonstrated by site-directed mutagenesis of the cysteine residue involved in the interaction with dithiazole. The most potent inhibitors have also been used in cancer cell lines that harbor high expression of LAT1; interestingly, by chemically knocking out LAT1 activity, cells undergo death. Since the interaction is covalent, the effect on cells is strong and also resistant after washing out the compounds. 95 The approach depicted in Figure 5 also includes a further element: the contribution of bioinformatics. In fact, in silico screening represents an important step in designing novel drugs, in the case of both competitive and noncompetitive inhibitors. To allow the prediction and/or design of novel drugs with high potency and less off-target effects, one must have knowledge of the three-dimensional structure of the target protein, which increases the chance of improving the efficacy of therapies. This aspect, while well established for soluble enzymes, is underdeveloped for mammalian membrane transporters due to the difficulties in obtaining three-dimensional structures. Interestingly, the three-dimensional structures of human LAT1 and ASCT2 have been determined very recently.97,98 This finding opens important perspectives for improving the affinity of inhibitors originating from the identified hits.
Cysteine as a Target for Xenobiotics in SLCs
Among reactions occurring at the level of cysteine residues, there are the S-metal bonds (see “Cysteine as a Druggable Protein Residue”). This is relevant in physiological conditions, as in the case of zinc binding proteins, which play fundamental roles in cell homeostasis. 99 However, in this review, we seek to stress that the ability of cysteine to interact with metals assumes great importance in toxicology. In fact, heavy metals are dangerous environmental pollutants and their ability to form stable bonds with proteins represents a further degree of danger. It is interesting to note that among heavy metals, mercury is one of the most abundant, and its concentration reaches values above the threshold considered dangerous for human beings. In fact, mercury has been listed by the World Health Organization (WHO) in the top 10 chemicals of major public health concern; notwithstanding, it has to be highlighted that the increasing presence of mercury in the environment is mainly linked to human activities (https://monographs.iarc.fr/wp-content/uploads/2018/06/mono58–8.pdf; https://www.atsdr.cdc.gov/toxprofiles/tp.asp?id=115&tid=24). In the environment, mercury is found as an element (metallic form), as an inorganic ion, and as an organic bound to aliphatic compounds, in air, soil, and water. This wide distribution triggers the existence of a mercury cycle that involves bacteria, plants, and animals including humans with a phenomenon known as biomagnification. The severity of intoxication by mercury depends on different parameters, including dose, time, and type of mercury compounds. Populations chronically exposed to mercury are subject to systemic damage exerted by mercury that is related to several organs, that is, lung, skin, brain, and immune system. It is important to highlight that mercury is life-threatening for the fetus since it is consumed as methylmercury in the diet and then reaches and crosses the placental barrier causing toxicity ( Fig. 3 ). Given these premises, it is not surprising that membrane transporters can also be considered key players in underlying toxicity exerted by mercury. In this respect, mercury has been shown to inhibit several membrane transporters belonging to different classes of proteins: the Na+/K+ pump, 100 the water channel aquaporin 1, 101 the glucose transporter GLUT1, 102 and the glutamine transporter SNAT3. 103 The workflow depicted in Figure 5 has been employed by our group to study, at the molecular level, the interaction of some plasma and mitochondrial transporters with HgCl2, methylmercury, and the preservative ethylmercury. The latter is known as a thimerosal and is used in human drugs because it is considered safe at the clinical dose.
Targeting Plasma Membrane Transporters with Mercury Compounds
The molecular mechanism underlying mercury toxicity has been deepened using reconstitution in proteoliposomes of the rat isoform of the glutamine transporters ASCT2 and B0AT1, which harbor well-known metal binding motifs. In the first case, tested compounds exert a noncompetitive inhibition, probably interacting with the CXXC motif of the protein; in the case of B0AT1, the protein has two metal binding motifs, CXXC and CXXXC. The studied proteins derived from rat kidney brush border suggest that the inhibition exerted by mercury compounds may affect renal reabsorption of important amino acids, such as glutamine. In the case of CXXC motifs of both ASCT2 and B0AT1, this metal binding site is not close to the substrate binding site; in line with this observation, no prevention of the inhibition by the substrates could be observed.104,105 In the case of the CXXXC motif of B0AT1, this is closer to the predicted binding site for glutamine. In fact, when B0AT1 is preincubated with glutamine, the inhibition exerted by a mercury derivative is prevented. 105 The same experimental system has been employed for the carnitine transporter OCTN2 of rat kidney brush border, which does not harbor a metal binding motif. 106 Besides the inhibitory effect exerted on the carnitine uptake due to covalent interaction, transport of a conjugated form of mercury-cysteine by OCTN2 was also observed. In all the described works, the covalent inhibition could be reversed by the addition of physiological or nonphysiological reducing agents that could be suggested as mercury scavengers. Importantly, the measured IC50 values of all the proteins for mercury reagents are in a range of concentrations reached by mercury during high-level exposure, explaining a possible molecular mechanism of damage observed at the kidney level. We have exploited proteoliposomes for studying interactions of human transporters with mercury compounds as well. In particular, the human isoform of OCTN1 (SLC22A4, organic cation transporter novel) has been tested for reactivity toward HgCl2, methylmercury, and ethylmercury. In this case, the molecular mechanism of interaction has been revealed by using site-directed mutagenesis of the seven cysteine residues of the protein that are not organized in a canonical CXXC motif. This approach, combined with reconstitution in proteoliposomes, allowed us to discover that two residues, residing in an extracellular domain of the transporter, are responsible for the interaction with mercurials. 107 Furthermore, the human isoforms of LAT1 and ASCT2, which do not harbor a CXXC motif, have also been tested for their sensitivity to mercury compounds. Notwithstanding that LAT1 harbors in its active site two cysteine residues, C335 and C407, it is interesting to note that neither is targeted by mercurial agents that are still able to inhibit the site-directed mutants built for transport assays in proteoliposomes ( Fig. 5 ). 108 Regarding ASCT2, the eight cysteine residues have been substituted by alanine and mutants have been tested for sensitivity toward methylmercury. Only one mutant behaves differently from wild-type protein, indicating that this cysteine residue, C467, is the one mainly involved in the interaction with mercury. It is interesting to note that, in contrast to LAT1, this residue lies on the substrate binding site of human ASCT2. 109 In conclusion, several transporters residing in the plasma membrane of many tissues are targeted by mercury compounds. Even though the described phenomena occur at a relatively high mercurial level, they may have possible consequences on nutrient absorption.
A Mitochondrial Transporter Identified as a Novel Target of Mercury Toxicity
The molecular mechanism underlying mercury toxicity has also been explored on second-level targets: the mitochondrial transporters. The ornithine/citrulline (SLC25A15) and the carnitine/acylcarnitine (SLC25A20) carriers interact with mercury compounds. In the first case, the reactivity of the protein toward several heavy metals, including mercury, was tested on the protein extracted from rat liver mitochondria. 110 The interaction with cysteine residues was indirectly proven by the addition of reducing agents able to reverse the inhibition exerted by the metals. The rat protein harbors a CXXXC motif that was predicted as responsible for the interaction. 110 Later, the same approach was used for the human isoform of the ornithine/citrulline carrier. In this case, cysteine residues responsible for interaction have been clearly identified by generating site-directed mutants whose sensitivity toward mercury compounds has been evaluated. The cysteine residues responsible for the formation of the CXXXC motif are not those targeted by mercury reagents. Our results suggested that two cysteine residues, during the transport cycle, come close to the central cavity impairing translocation of the substrates. 111 As in the case of plasma membrane transporters, these interactions occur at a relatively high concentrations of heavy metals. Possibly, these interactions may cause impairment of the urea cycle in which the transporter is involved. 112
The most interesting data concern the CACT carrier (SLC25A20). A comprehensive study has been conducted in which the effects of HgCl2 and methylmercury have been evaluated employing the complete experimental approach depicted in Figure 5 , from analysis of the toxicity in vitro to in vivo animal model. First, the recombinant protein produced in E. coli has been used for the determination of the two cysteine residues involved in the interaction with mercury compounds by generating site-directed mutants. Then, a model human cell line has been used to evaluate the effect of HgCl2 on cell growth and viability. Transport activity of the CACT extracted from mitochondria of treated cells was impaired, and this alteration was reversed by using reducing reagents. Finally, zebrafish were treated with toxic doses of HgCl2 and mitochondria were analyzed showing bioaccumulation of the compounds. Also in this case, the transport activity of CACT extracted from zebrafish mitochondria was impaired and rescued by the addition of reducing reagents. The strong inhibition of CACT transport activity may have a terrific effect on β-oxidation of fatty acids, causing a lower ATP production and a higher ROS generation in mitochondria triggering cell damage that was revealed as cell death and impairment of fish activity. The described effects were caused on this transporter at very low mercury levels (IC50 of 90 nM). These data allowed us to identify the mitochondrial CACT as a novel target for mercury toxicity together with thioredoxin. 113
Conclusions
Exploiting cysteine residues of membrane transporters as a novel target is grounded on several observations and experimental data. The findings and perspectives described in this review justify the present and future efforts in this direction. In particular, (1) the localization of SLC transporters at the boundary between the intracellular and extracellular environment facilitates targeting, and (2) the presence of a limited number of cysteine residues in the majority of SLCs promises an increased specificity of the drug. These aspects give us an opportunity to design novel TCIs with improved efficacy and limited side effects. The last issue is, indeed, a crucial challenge in the drug design process and one of the first causes of novel drug failure even at the end of clinical trials, with huge loss of investments.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by funds from MIUR (Ministry of Instruction, University and Research): Programma Operativo Nazionale (01_00937)—“Modelli sperimentali biotecnologici integrati per lo sviluppo e la selezione di molecole di interesse per la salute dell’uomo.”