
Abstract
Single-particle cryogenic electron microscopy (cryo-EM) has been elevated to the mainstream of structural biology propelled by technological advancements in numerous fronts, including imaging analysis and the development of direct electron detectors. The drug discovery field has watched with (initial) skepticism and wonder at the progression of the technique and how it revolutionized the molecular understanding of previously intractable targets. This article critically assesses how cryo-EM has impacted drug discovery in diverse therapeutic areas. Targets that have been brought into the realm of structure-based drug design by cryo-EM and are thus reviewed here include membrane proteins like the GABAA receptor, several TRP channels, and G protein-coupled receptors, and multiprotein complexes like the ribosomes, the proteasome, and eIF2B. We will describe these studies highlighting the achievements, challenges, and caveats.
Introduction
Cryo-transmission electron microscopy (cryo-EM) has taken the field of structural biology by storm: 1 the number of near-atomic-resolution structures obtained with it is growing exponentially, 2 and its application has led to unprecedented structures of drug targets historically refractory to crystallography efforts (e.g., TOR, 3 ATR, 4 and NLRP35). Further, cryo-EM has surpassed some of its initial limitations: structures have been obtained for proteins with sizes as low as 52 kDa 6 and resolutions as high as 1.2 Å. 7 Typical resolutions range from 2.5 to 7 Å, and analysis of the Electron Microscopy Data Bank (EMDB) maps deposited this year reveals that most maps are resolved to higher than 4 Å.
The key advantages of cryo-EM over crystallography relate to lower sample requirements, no need for crystal formation, and the fact that cryo-EM allows for an extent of in silico purification of different compositional and/or conformational states of the sample. For instance, key aspects of the regulation of ATM (ataxia-telangiectasia mutated) were unveiled by cryo-EM as the kinase displayed two markedly distinct dimeric arrangements: one autoinhibited and one active. 8
A number of hurdles need to be overcome for the technique to turn into a regular player on structure-based drug design (SBDD). Solving a structure by cryo-EM relies on the averaging of thousands (or even millions) of images of identical macromolecular particles. The existence of multiple states is thus a double-edged sword, since it can limit resolution. Extensive optimization of protein preparation and sample freezing remain crucial for success.
The most transformative developments that propelled the technique to yield high-resolution structures routinely became widely available less than a decade ago, and progress has been steady and fast. The biologics drug discovery field was an early adopter of single-particle EM. AbbVie investigated the formation of high-order complexes between tumor necrosis factor α (TNFα) and therapeutic monoclonal antibodies, adalimumab and infliximab, demonstrating that they formed structurally different complexes by 2D classification of negative stain data. 9 More recently, Roche-Genentech characterized the complex between CD20 and therapeutic monoclonal antibody rituximab (RTX), establishing that CD20 forms a dimer and each dimer binds two RTXs. With NS-EM, they observed a circular RXT-CD20 assembly, which led them to raise a novel hypothesis for the mechanism of action: RTX-induced cell-surface CD20 clusters would be assemblies specifically predisposed to recruit complement. 10 Importantly, the technique has also made crucial contributions to the understanding of emerging epidemies: cryo-EM was used to reveal the structure of the Zika virus and the epitope to a neutralizing antibody binding to it. 11 This year, the technique was applied to investigate the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike 12 and to establish the structural basis for the recognition of SARS-CoV-2 by human angiotensin-converting enzyme 2 (ACE2). 13 In the same fashion of earlier antibody–antigen studies, Robianni et al. used negative stain EM to determine how converging neutralizing antibodies from COVID-19 convalescent individuals bound to the receptor binding domain of the spike protein of SARS-CoV-2. 14 Finally, cryo-EM has demonstrated that patient-derived structural biology is possible: in 2017, Fitzpatrick and coworkers published high-resolution structures of tau-paired helical and straight tau filaments purified from an Alzheimer disease brain, paving the way to investigate a range of neurogenerative diseases in a molecular context. 15
The advances in cryo-EM and the expectations on its impact in drug discovery have been comprehensively reviewed elsewhere.16–18 In this paper, we aim to provide a review of the impact of single-particle cryo-EM in drug discovery in diverse therapeutic areas. Targets that have been brought into the realm of SBDD by cryo-EM include membrane proteins like the GABAA receptor, several TRP channels and G protein-coupled receptors (GPCRs); and multiprotein complexes like the ribosomes, the proteasome, and eukaryotic translation initiation factor 2B (eIF2B). We will review those cryo-EM studies below and comment on the state of fragment-based drug design (FBDD) using cryo-EM.
TRP Channel Family
Transient receptor potential TRP channels are cation channels widely expressed in various tissues and cell types, where they are engaged to mediate physiological processes as diverse as the perception of stimuli and ionic homeostasis. 19 Dysfunctions in the activity of TRP channels have been linked to several pathologies, including chronic pain, type 2 diabetes, and cancer. 20 Within this context, compounds that target TRP vanilloid 1, 3, and 4; TRP ankyrin 1; and TRP melastatin 8 have all entered clinical trials.
Their functional assemblies are tetramers usually made up by identical individual subunits featuring six transmembrane segments that comprise the voltage sensor-like and pore domains and a variable size cytoplasmatic moiety that contains the TRP, the pre-S1, and the ankyrin repeat domains. While the intramembrane topology of six transmembrane helices is highly conserved within the family, they differ in the sensory and cytosolic moieties. Accordingly, the TRP channels have been classified into seven subfamilies: TRPC (canonical), TRPV (vanilloid), TRPM (melastatin), TRPP (polycystin), TRPML (mucolipin), TRPA (ankyrin), and TRPN (NOMPC-like).
Cryo-EM of TRP Channels
The structural understanding of the family of TRP channels was greatly advanced by the progress on cryo-EM. This family has been especially resistant to crystallization efforts; to date, only TRPV6 has been crystalized to a resolution higher than 4 Å. Cryo-EM enabled the structure determination of TRP channel proteins from all seven subfamilies. The first major leap in the field came in 2013, when the structure of a deletion mutant of rat TRPV1 prepared in nanodiscs was solved to a resolution of 3.4 Å. 21 TRPV1 has a strong link to pain-related mechanisms and therefore has attracted investigative efforts from both academia and industry. The overall architecture of TRPV1 resembles that of voltage-gated ion channels in which the protomers are arranged in fourfold symmetry around a central cation permeation path. Furthermore, this structure confirmed that the six transmembrane helices (S1–S6) span across the lipid bilayer and revealed that the first four helices (S1–S4) establish the voltage sensor-like domain (VSLD) and the last two transmembrane helices form a pore module lining a pore loop between S5 and S6. The main closed–open transition is controlled by the S6 bundle-crossing intracellular gate. The pore module contains a conserved hydrophobic region and an aspartate residue that confers ion selectivity. A concomitant study also published by the Cheng group further highlighted TRPV1’s broad outer pore domain, which was hypothesized to enhance the accessibility of diverse ligands; for example, the pore loop neighboring parts of S5 and S6 closer to the extracellular environment forms a site that binds spider toxin DkTx. 22 It also revealed a curious domain swap arrangement in which the VSLD of one protomer closely interacts with the pore domain of its neighbor, similar to the one featured by the voltage-gated ion channels. 21 Furthermore, it showed that the conserved TRP domain, which is an exclusive feature of this family and thought to propagate allosteric modulation, interacts with the S4–S5 intracellular connection. The cytosolic ankyrin domain was resolved to a lower resolution than the rest of the structure, but the docking of the crystal structure of this isolated soluble domain was still possible. Structures in the presence of the agonist capsaicin and RTx revealed that they bind to the vanilloid pocket, contacting S3, S4, and S4–S5 linker residues. Upon agonist binding, there are structural rearrangements in the area lining the ion conduction region and the outer pore; these changes relieve all constrictions on the ion conduction path by opening the S6 gate. Both the selectivity filter and the lower gate undergo expansion in the presence of DkTx and RTx, while in the presence of capsaicin only the widening of the lower gate is observed.
TRPM8 is an ion channel activated by cold temperatures and menthol. It is essential to detect changes in environment temperatures. Moreover, TRPM8 is being probed as a target to alleviate cold hypersensitivity that is followed by nerve damage and has been implicated in the mechanism of action of cannabinoid-mediated relief on focal epilepsy. 23 Cryo-EM structures of complexes of agonist–TRPM8 revealed that, in contrast to TRPV1, the natural ligand binding site of TRPM8 locates to the VSLD. Both icilin and the menthol analog WS-12 bind to a cavity in between the VSLD (S1 and S4) and the TRP domain, which enables direct control of the TRP domain in opening the intracellular S6 gate. 24 Importantly, two chemically distinct antagonists, AMTB and TC-I 2014, bound to the same pocket. 25
The activity of several pronociceptive agonists generated in diverse pathophysiological pain conditions culminates in the TRPA1 pathway. Inhibition of TRPA1 could thus be promising for the treatment of central hyperalgesia and several types of neuropathy. 26 The structure of nanodisc-reconstituted TRPA1 bound to electrophilic covalent agonists JT010 and BITC revealed a sensing site distinct from those of TRPV1 and TRPM8 and located at the cytoplasmic side. In this case, the activation is more indirect; agonist binding provokes a conformational change in β-sheet BCD and then the newly described interfacial helix, which finally interacts with the S4–S5 linker to open the S6 gate. 27
To sum up, structure determination of TRP channels by single-particle cryo-EM provided unprecedent details on different conformational states of gating for the different channels.21,22,28 Figure 1 highlights how cryo-EM revealed the distinct overall structures for the different subfamilies and opened up the possibility for SBDD campaigns.
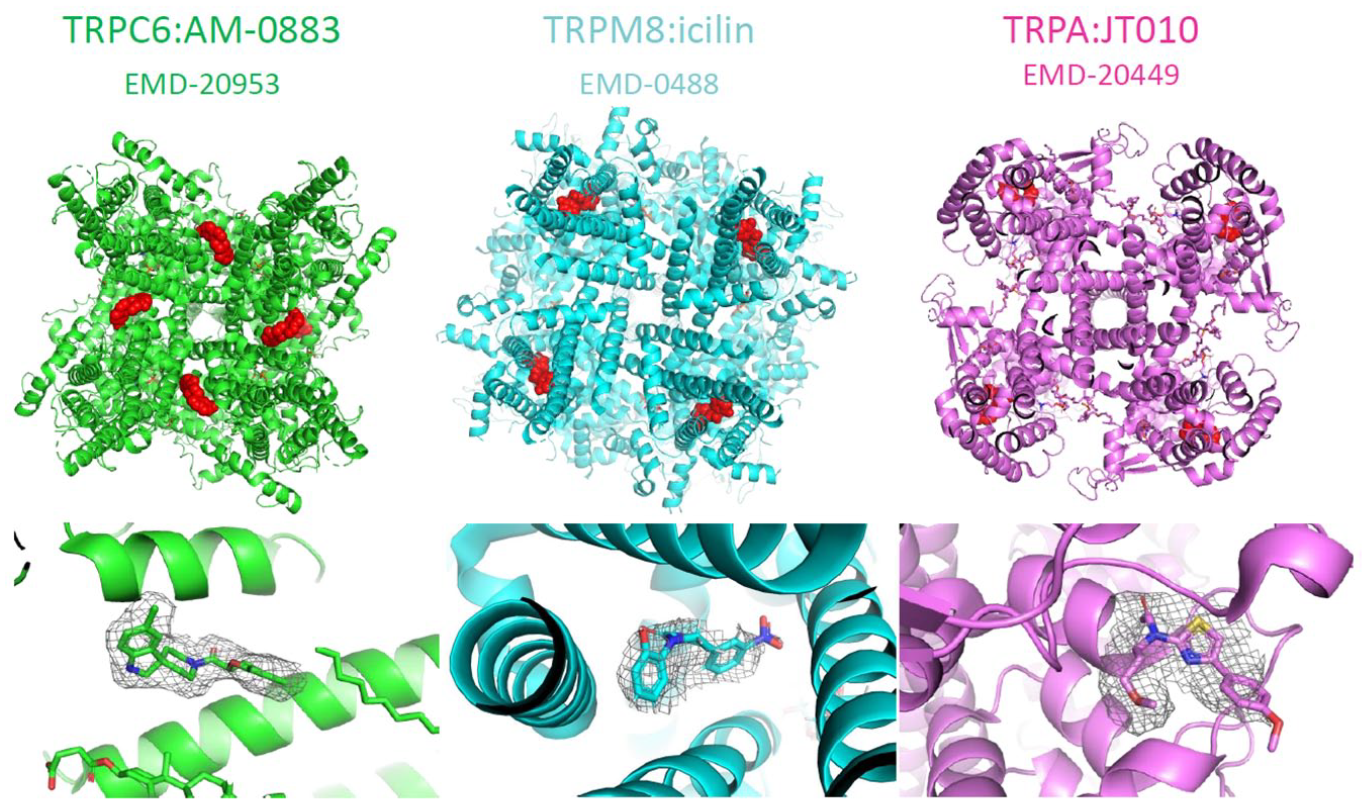
The cryo-EM structures of TRPC6 (green), 103 TRPM8 (cyan), 104 and TRPA1 (fuchsia) 27 in complex with their respective agonists, AM-0883, icilin, and JT010. The overall structures are shown in the first-row panel with the compounds pictured in red. The second-row panel shows the modeled compounds and the coulomb potential map on which the modeling was based.
GABAA Receptors
γ-Aminobutyric acid type A (GABAA) receptors play a central role in the inhibition of neurotransmission in the vertebrate central nervous system (CNS). 29 Located at synapses, GABAA receptors enable millisecond neuronal inhibition as the target of γ-aminobutyric acid (GABA), an endogenous agonist that acts as inhibitory neurotransmitter. 30 Incorrect functioning of GABAA receptors can lead to a spectrum of neurological disorders, including anxiety, 31 memory loss, 31 schizophrenia, 31 insomnia, 32 and epilepsy, 33 and GABAA receptors are also involved in forms of depression34,35 and alcohol dependence. 36 This rich pharmacology makes GABAA receptor targeting by ligands of large interest to the pharmaceutical industry, with the goal to modulate their activity with sufficient affinity and high specificity.
GABAA receptors form heteropentameric chloride-permeable ion channels with a molecular weight of ~260 kDa. With 19 subunits available, they can display wide structural diversity, 29 but the prototypical form in the human brain is α1β2/3γ2.37,38 Each subunit contributes to the pore-forming transmembrane domain (TMD) and the extracellular domain (ECD); GABA binds the β+ (principal)/α– (complementary) interfaces in the ECD. General intravenous anesthetic drugs39–41 exert positive allosteric modulatory (PAM) effects 42 on GABAA receptors. Ethanol debatably binds δ subunits.43,44 Benzodiazepines such as alprazolam (Xanax) and diazepam (Valium), classic drugs tackling insomnia and anxiety, among other neurological conditions, 45 act as allosteric PAMs. 46 Benzodiazepine overdoses 47 can be treated with the antagonist flumazenil.48-51 Structural insight into the binding modes of these ligands is highly beneficial for SBDD; however, as GABAA receptors are membrane proteins, they proved to be challenging crystallization targets. Using cryo-EM, breakthrough structures have been obtained of human receptors in near-physiological lipid environments in which the density for ligands enabled unambiguous placement. 45
Crystallography of GABAA
Pioneering structural work on receptors in this superfamily was done by Nigel Unwin and colleagues on the nicotinic acetylcholine receptor from Torpedo marmorata.52,53 GABAA receptors themselves proved recalcitrant to crystallization efforts for a long time, before Miller and Aricescu determined the crystal structure of a homomeric β3 GABAA at a resolution of 3 Å. 54 This crystal structure provided a framework for future structural studies. More detail of the TMD domain of GABAA receptors was provided by x-ray crystallography by Laverty et al. 55 and Miller et al., 56 who crystallized engineered homomeric chimera receptors to a resolution of 3.8 Å.55,56
Cryo-EM of GABAA
The real breakthrough in the structural work on GABAA receptors, however, has been enabled by cryo-EM. Three groups recently reported cryo-EM structures of near-physiological GABAA receptors in complex with several ligands, followed by a breakthrough in resolution using a homomeric GABAA receptor. 57 The reader is referred to an excellent review by García-Nafría and Tate for an extensive table highlighting available structures of GABAA isoforms with ligands. 58 All research groups employed analogous strategies to perform particle alignment on the pseudosymmetric triheteromeric GABAA receptor. Zhu et al. 37 and Phulera et al. 59 generated an antibody, binding the α1 subunit. Aricescu’s lab38,60 modified an α1 subunit-specific nanobody with a large, stable domain to form a so-called megabody, 38 which randomized the particle orientation in ice and assisted in particle alignment.
The first cryo-EM structure at a resolution of 3.9 Å was published by Zhu et al. 37 and revealed the structure of the human triheteropentameric α1β2γ2 GABAA receptor. One of the TMD structures resembled a partly collapsed pore at the γ2 location ( Fig. 2C ), possibly due to presence of detergent. GABA was bound at its β+/α– subunit interface and is held in position by two electrostatic interactions at either end of the molecule, in addition to interactions with conserved aromatic residues. The binding site for the benzodiazepine antagonist flumazenil in the ECD is also resolved ( Fig. 2F ). Obtaining the structure of a triheteromeric receptor provided insight into the modulator specificity of subunit interfaces.
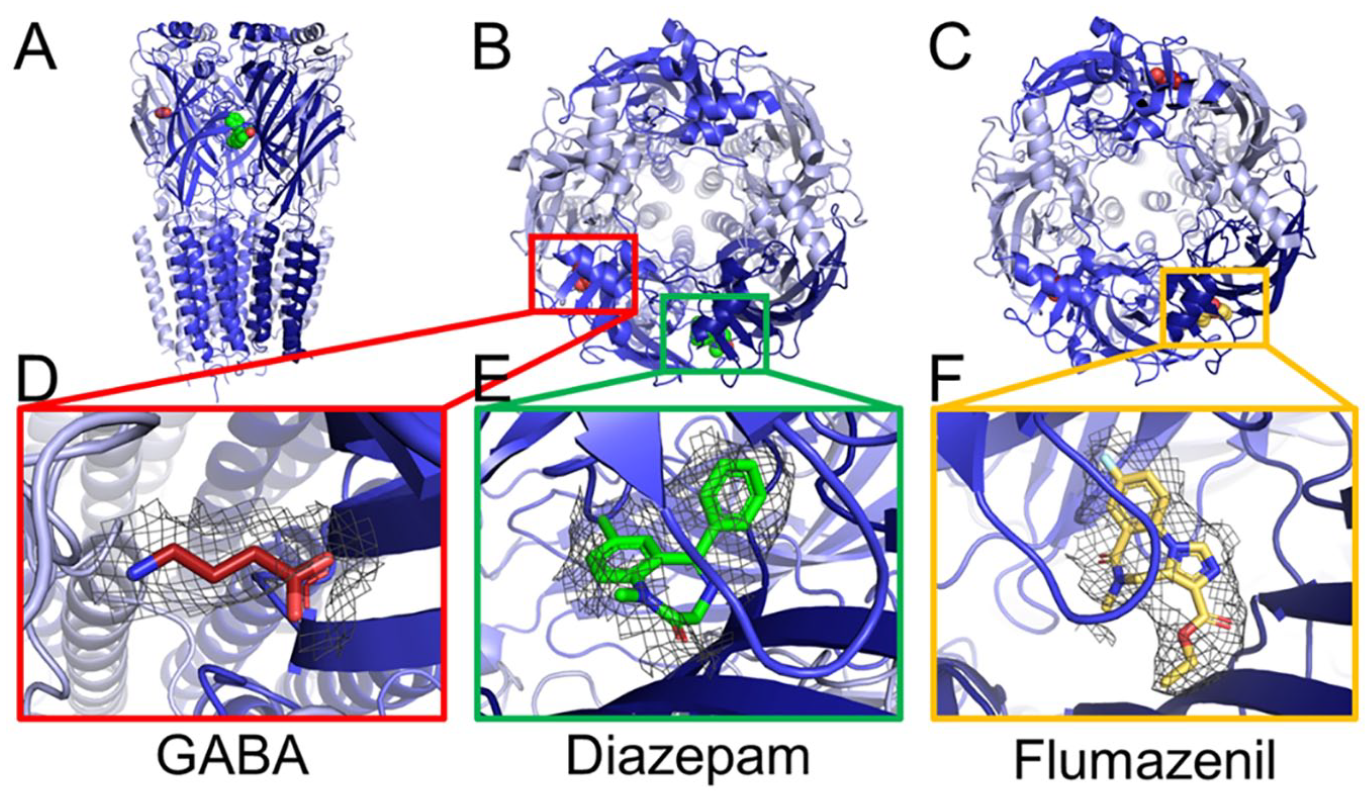
The cryo-EM structure of the human GABAA receptor isoforms α1β3γ2L (
Phulera et al. 59 solved the structure of a rat α1β1γ2 triheteropentamer by cryo-EM to ~3.8 Å resolution and the ECD domain to ~3.1 Å in the presence of GABA. The receptor was solubilized in detergent and grids were prepared on graphene oxide to achieve the desired particle concentration on the grid; however, a preferential orientation issue remained. Using a similar GABA concentration as Zhu et al., 37 but a 300-fold larger GABA concentration than Masiulis et al., 60 they observed an additional putative binding site for GABA at the ECD α+/β– subunit interface with EM density that fitted GABA.
Laverty et al.
38
and Masiulis et al.
60
determined structures of the α1β3γ2L isoform, in complex with GABA and therapeutic drugs (
Cryo-EM structures with the classical benzodiazepines alprolaxam (3.3 Å) and diazepam (3.6 Å) (
So while crystallography of GABAA receptors was challenging and yielded structures of homomers 54 and homomeric chimeras55,56 where detergents caused the pore to collapse, cryo-EM has proved instrumental in the quests for high-resolution structural insight into receptor architecture and ligand binding and resolved more structures than x-ray crystallography. In the reported breakthrough structures, human, full-length GABAA receptors assume a near-physiological conformation in lipid nanodiscs.38,60 At ~3 Å, the map quality of cryo-EM is similar to that in x-ray crystallography and assists unambiguous placement of several ligands, revealing critical interactions with their binding pocket. Using new techniques (see below), a homomeric GABAA receptor structure was reported at a resolution of 1.7 Å, showing an unprecedented level of detail. 57 These studies help to understand ligand specificity for receptor subtypes, 60 and such information will contribute toward the rational design of compounds, optimizing affinity and selectivity.
GPCRs
GPCRs are a particularly important class of protein target for the pharmaceutical industry, with roughly one-third of approved drugs acting on GPCR family members. Indeed, GPCR small-molecule modulators have impacted a diverse range of indications, including neurological, inflammatory, cardiovascular, respiratory, and gastrointestinal disease, and remain a focus of the industry. 65
Crystallography of GPCRs
Significant success has been had in the determination of high-resolution crystal structures for many GPCRs, and these structures have transformed our understanding of receptor pharmacology and, through structure-based design methods, supported the delivery of several clinical stage drug candidates. However, obtaining crystal structures of GPCRs remains extremely challenging. Typically, receptors are thermostabilized by the introduction of mutations that stabilize the solubilized receptor in a particular predetermined conformational state. 66 Nearly all crystal structures of GPCRs are of nonactive conformations, a notable exception being the active-state structure of the A2A receptor. 67 From these studies it was learned that to gain active-state GPCR structures, capturing the receptor in a complex is required; these complexes are formed with the heterotrimeric G protein signal transducer units. These larger complexes have proved suitable for cryo-EM structural studies, with several active-state GPCR-G protein structures being reported.
Cryo-EM of GPCRs
The recent structure of the glucagon-like peptide 1 receptor–G protein transducer (GLP1-Gαs,β,γ2) complex bound to a nonpeptidic activator, termed TT-OAD2, is a significant milestone in the use of cryo-EM in GPCR-focused drug discovery. 68 GLP-1 receptor activation induces glucose secretion from β cells, and peptide agonists are established drugs for the management of type2 diabetes. The structure at 3.0 Å resolution allows unambiguous assignment of the TT-OAD2 binding site, fitting of the ligand pose, and accurate modeling of protein side chains ( Fig. 3 ). Some regions of the protein complex distant to the ligand binding site were not resolved at such high resolution, an observation that is consistent with the higher mobility for these units. The structure defines an unpredicted agonist binding pocket, distinct from the peptide binding pocket, and links the induced receptor conformation with the specific pharmacology observed for TT-OAD2. The structure follows on from peptide agonist-bound structures, and not only does the ligand site have little overlap with the GLP-1 peptide site, but also conformational differences are observed between the structures. 69 Although the key features of activated peptide-bound B class receptors are conserved, principally the interaction with the heterotrimeric G protein unit, changes in orientation are seen for the ECD. The structure demonstrates that receptor conformational plasticity can be manipulated by ligand binding and that a rational approach to tuning (biasing) receptor signaling may be possible in the near future. The same group followed up the GLP-1 receptor structure with further B class GPCR cryo-EM structures, the pituitary adenylate cyclase peptide receptor (PAC1), the corticotropin-releasing factor receptor 1 (CRF1), and the corticotropin-releasing factor receptor 2 at 3.0, 2.9, and 2.8 Å resolutions, respectively. 70 These structures are complexes with different activating peptides plus the Gαs,β,γ2 unit and represent the agonist state; the resolution allows for ordered water molecules to be modeled. These structures collectively give a comprehensive view of B class GPCR signaling complexes, demonstrating that while peptide recognition can involve different residues and features, the interface involved in G protein coupling is conserved. Significantly, they demonstrate a consistent method for the determination of high-resolution B class GPCR cryo-EM structures.
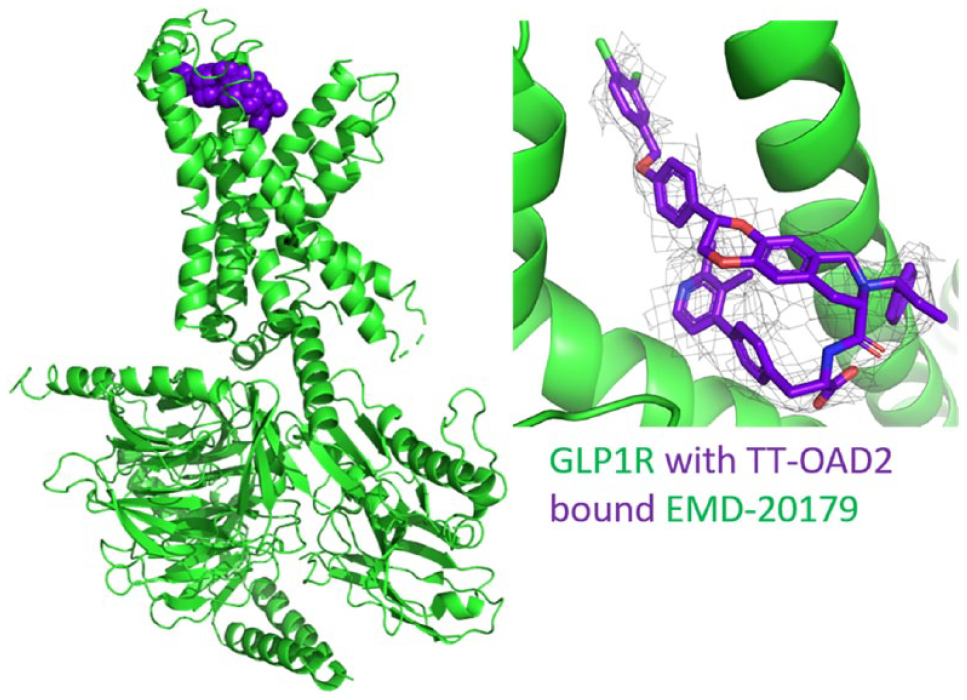
EM structure for GLP1 receptor with nonpeptide bound agonist TT-OAD2. The fitted model displayed is 6ORV (green), with the ligand shown as purple spheres. The inset view shows the modeled compound and the coulomb potential map (mesh) on which the modeling was based from EMD-20179.
The class A cannabinoid receptors CB1 and CB2 offer an additional example of the impact cryo-EM is having on our understanding of small-molecule receptor interactions. Cannabis elicits its mood-enhancing and analgesic effects through the CB1 receptor predominantly expressed in the CNS, while the CB2 receptor is predominantly (though not exclusively) expressed in peripheral tissues such as the spleen and thymus. Cryo-EM structures of both receptors bound to small-molecule ligands are now available.71,72 A 3 Å resolution structure of CB1 bound to the synthetic cannabinoid MDMB-FUBINACA (FUB), a particularly potent compound linked to substantial drug abuse fatalities, has been determined. The agonist state is observed in complex with the heterotrimeric G complex (GiBG). The single-chain antibody fragment scFv16 was used to further stabilize the complex. The authors discuss the structure–activity relationship (SAR) of synthetic cannabinoids in light of the structure, run sophisticated molecular dynamics simulations on ligand and G protein binding, and define a structural basis for FUB affinity. The CB2-Gi signaling complex structure, at 3.2 Å resolution, is bound to the potent agonist WIN 55,212-2. The small molecule can be modeled in the EM map. Binding site analysis and comparison with the FUB-bound structure of CB1 demonstrate side chain movements and subtle changes in ligand receptor interactions; observations of this type are common with high-resolution crystal structures. In this case, however, cryo-EM is being used to gain the same detail and granularity of insight. WIN 55,212-2 is not a highly selective agonist for CB2; however, the authors present a strategy for the rational design of CB2-selective agonists based on the differences in residue conformation observed.
This bolus of high-resolution, small-molecule, agonist-bound GPCR structures is hugely encouraging for the field and indicates that cryo-EM-enabled, structure-based design of GPCR signaling modulators is now a reality.
Protein Synthesis and Protein Degradation
A protein can be targeted at many stages of its life in the cell, either during its synthesis, during its functional lifetime, or at the end of its life when it is degraded.
Targeting protein synthesis is an attractive strategy for preventing parasitic diseases, slowing or stopping the organism’s replication inside the human host. Such diseases affect a large proportion of the world’s population, around a sixth, but account for only a very small proportion of drug discovery research funding. Opportunities have recently arisen due to whole-genome sequencing of many parasitic species and alliances such as the Medicines for Malaria Venture and the Drugs for Neglected Diseases initiative. Whole-genome sequencing in many cases reveals that the parasite replication machinery has significant conservation with the human host machinery, which may hinder drug discovery. 73 However, there are now increasing numbers of protein structures, from both x-ray crystallography and cryo-EM, available for parasite and human species, and by comparing these, unique areas that could permit species selectivity can be identified.
On the other end of the spectrum, proteins that are at the end of their life or are misfolded when initially produced are targeted to the proteasome via the ubiquitin–proteasome system (UPS) for degradation. By inhibiting the proteasome from functioning, protein species build up in the cell, causing stress and eventually cell death. Compounds such as bortezomib, ixazomib, and carfilzomib have been approved for treating certain cancers. Due to their rapid growth and cell division, pathogenic organisms rely heavily on protein turnover, so the proteasome is an attractive target for bacterial and parasitic diseases as well. Existing knowledge from the development of bortezomib and carfilzomib can inform and aid the drug discovery process against the proteasome from other species.
Cryo-EM of the Protozoan Ribosome
One example of such structures has revealed the mechanism of action for the antibacterial agent paromomycin against the Leishmania parasite. Biochemical results indicated that the target of paromomycin was the cytosolic ribosome, not the mitochondrial ribosome as previously thought. The structure of the cytosolic ribosome from Leishmania donovani in complex with paromomycin was solved by cryo-EM, with some regions reaching a resolution of 2.2 Å (PDB: 6AZ1 and 6AZ3 EMD-7024 and EMD-7025). 74 A number of paromomycin molecules were resolved in the structure, but only three bind close to the functional regions, with one being designated the primary site close to where the A-site tRNA binds ( Fig. 4 , inset view). The apo structure had also been solved previously by two separate groups. This was to 2.8 Å resolution by the same group as that of paromomycin (PDB: 3JCS EMD-6583) 75 and 2.9 Å resolution (PDB: 5T2A EMD-8343) for a second group, who also published the human ribosome to 3.6 Å resolution by cryo-EM for comparison (PDB: 5T2C EMD-8345). 76 Comparison of Leishmania and human structures highlighted areas of the Leishmania binding site that are unique and could provide species selectivity. The structure of a complex including the E. coli ribosome has also been solved by cryo-EM with the inhibitor compound kirromycin bound (PDB: 5AFI, EMD-2847). 77 All these structures will allow areas for potential selectivity to be explored.
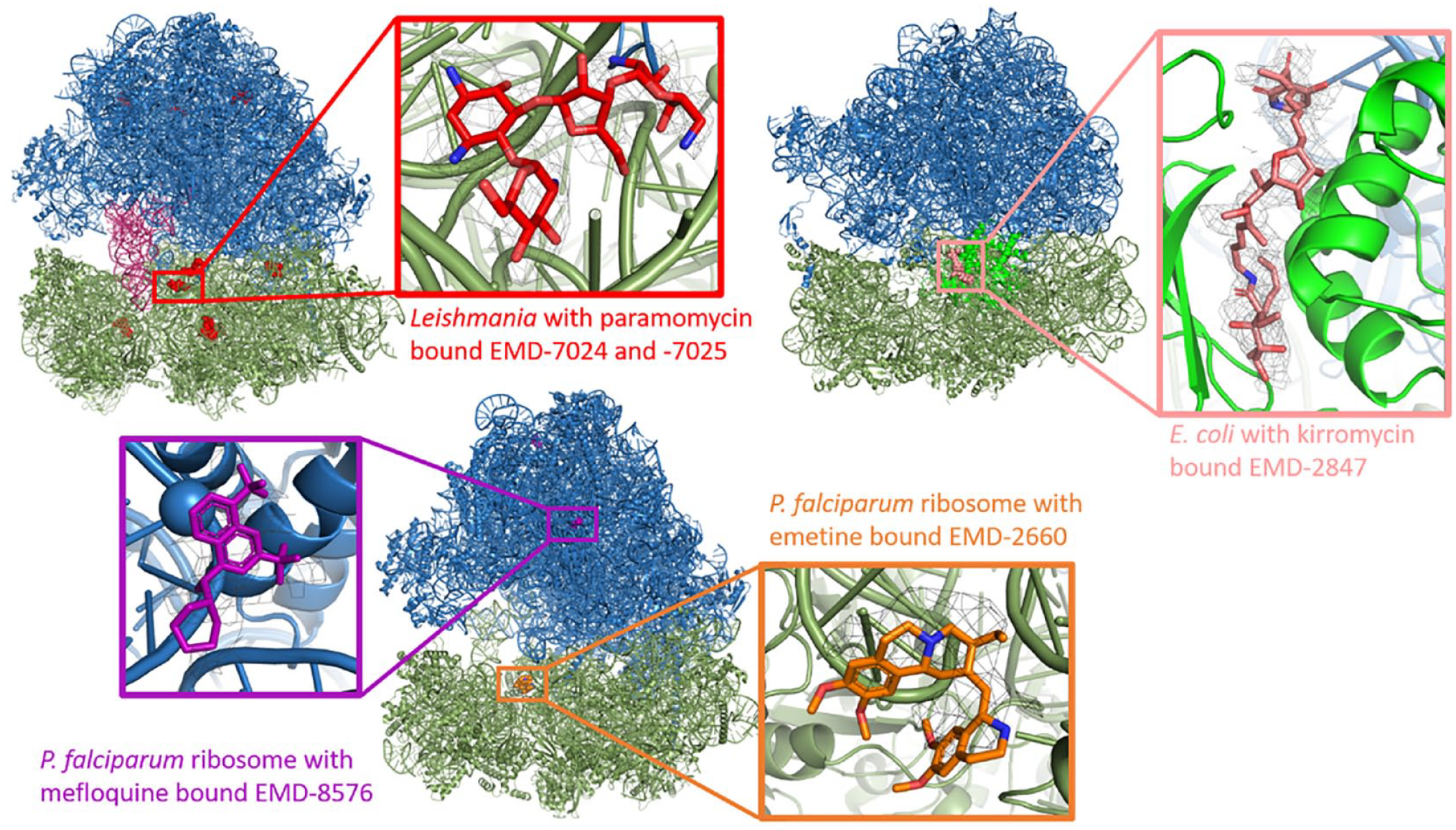
Ligand-bound EM structures available for the ribosome from various species. In all cases, the large subunit is colored blue and the small subunit green. The bound compounds are represented with colored spheres, and then in the inset view the coulomb potential map on which the modeling was based is displayed. Top left: Ribosome from L. donovani with bound paromomycin (red). Bound tRNAs are displayed in dark pink. Fitted models: 6AZ1 and 6AZ3; EM density: EMD-7204. Top right: Ribosome from E. coli complexed with EF-Tu (bright green) and bound kirromycin (salmon pink). Fitted model: 5AFI; EM density: EMD-2847. Bottom: Ribosome from P. falciparum on the left with bound mefloquine (purple). Fitted model: 5UMD; EM density: EMD-8576. The right view is of the emetine-bound structure (orange). Fitted models: 3J79 and 3J7A; EM density: EMD-2660.
Cryo-EM structures also exist for the ribosome of Plasmodium falciparum, the major parasite causing malaria. The first structure published was at 3.2 Å resolution with the drug emetine bound (PDB: 3J79 and 3J7A EMD-2660). This compound was an existing antibiotic that was shown to have antimalarial activity by affecting protein synthesis, but the binding site on the ribosome was unknown before the structure was solved. Emetine binds close to the P-site tRNA and affects mRNA translation in a similar mechanism to how paromomycin affects the Leishmania ribosome ( Fig. 4 ). 78 A cryo-EM structure with bound mefloquine was also solved to 3.2 Å resolution (PDB: 5UMD EMD-8576). The mechanism of action for mefloquine was previously unknown but through screening was shown to target the ribosome. The drug-bound structure revealed two sites for mefloquine binding, both on the large subunit ( Fig. 4 ). One site is at the periphery, but the primary site impacts the coordination of protein synthesis by affecting GTPase binding. Also, the structure revealed a number of key residues for mefloquine binding. These are conserved in mefloquine-sensitive parasites such as another malaria-causing Plasmodium species, P. vivax, and closely related parasite species Trypanosoma brucei, but absent in mefloquine-insensitive Toxoplasma gondii. This paper is one of the few to describe cryo-EM structures guiding the optimization of a compound to yield derivatives with increased parasiticidal activity. 79
Cryo-EM of the Protozoan Proteasome
An inhibitor-bound structure for the P. falciparum 20S proteasome was solved by cryo-EM to a resolution of 3.6 Å ( Fig. 5 ) and revealed that the β2 active site was more open than the human proteasome site (PDB: 5FMG EMD-3231). The inhibitor, WLW-vs, was developed from existing peptide-based inhibitors by studying the different substrate preferences between the human and parasite proteasomes. WLW-vs was later discovered to be a parasite-specific inhibitor. By comparing the human (solved by x-ray crystallography) and Plasmodium proteasome structures, it could be seen that the bulky side chains of WLW-vs could not be accommodated in the human proteasome pocket. 80 The knowledge gained through the comparison of these two structures could be used to further develop parasite-specific inhibitors.
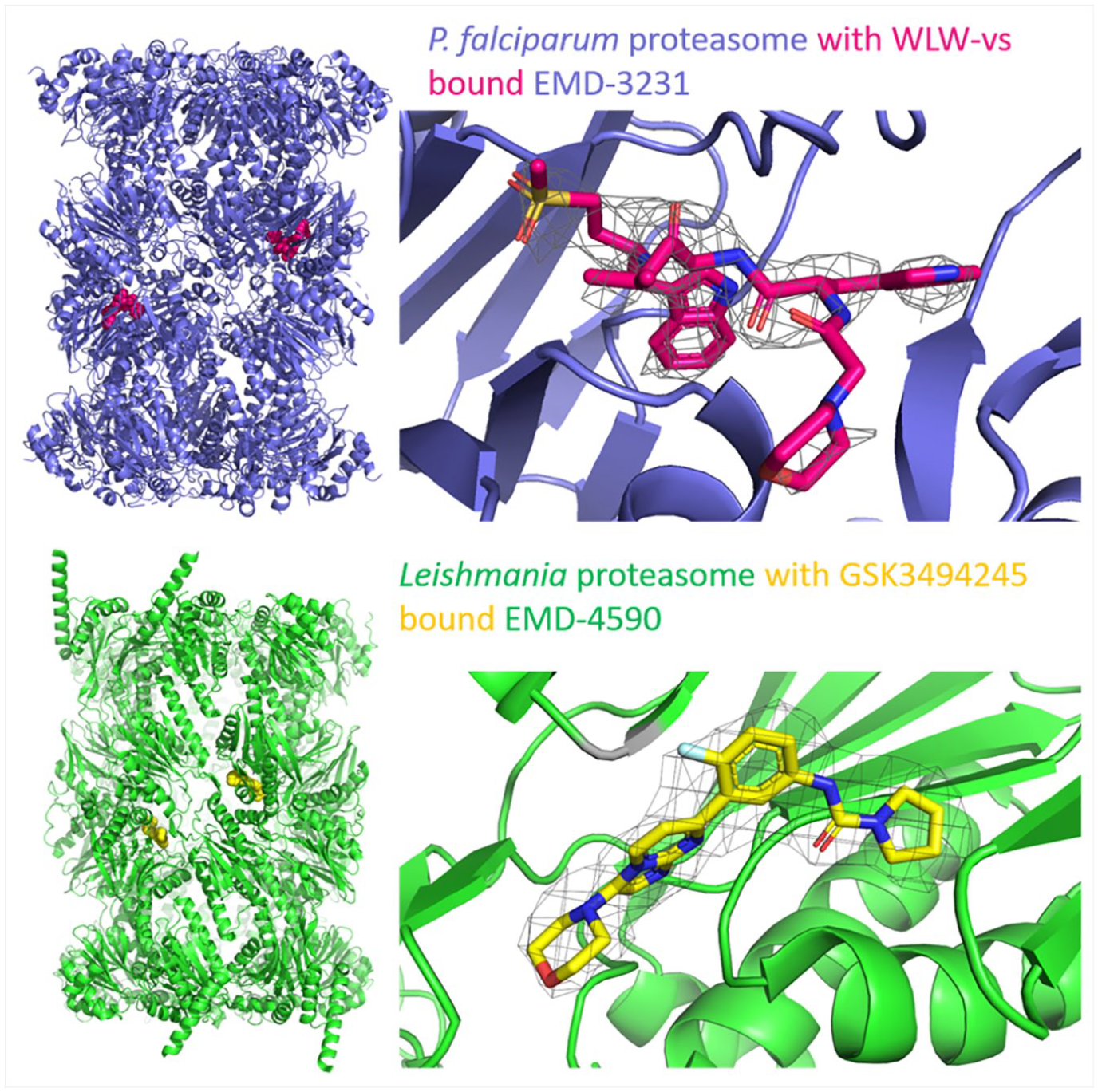
Ligand-bound EM structures available for the proteasome. The fitted models displayed and the ligands are in spheres. The inset views show the modeled compounds and the coulomb potential map on which the modeling was based. Top: P. falciparum proteasome (blue) with bound inhibitor WLW-vs (dark pink). Fitted model: 5FMG; EM density: EMD-3231. Bottom: L. tarantolae proteasome (green) with bound GSK3494245 (yellow). Fitted model: 6QM7; EMD-4590.
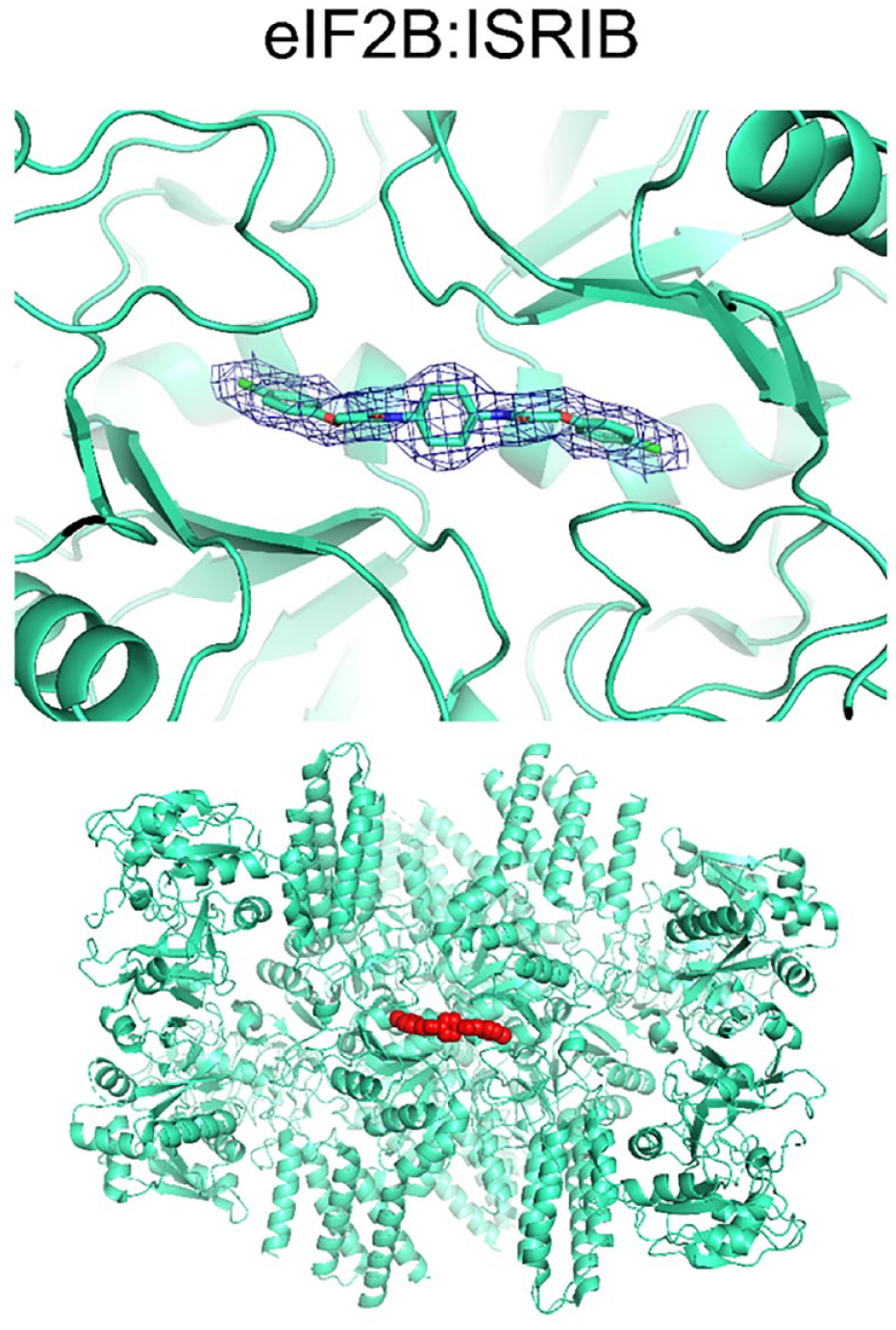
The binding pocket of ISRIB (red) is a cleft at the symmetry interface of the eIF2B decamer (mint). Two alternate conformations could be expected and modeled (only one is shown for clarity). Atomic model: 6CAJ; EM density: EMD-7442.
The structure of the Leishmania proteasome has also been solved by cryo-EM to 3.3 Å resolution (PDB: 6QM8 EMD-4591). This structure was reported in a paper from GSK, along with the development of compound-8, GSK3494245/DDD01305143. Compound-8 was developed from a hit identified when initially targeting Trypanosoma cruzi but has been repurposed. The binding site of compound-8 was identified to be at the β4-β5 interface ( Fig. 5 ) through determination of the inhibitor-bound structure to an overall 2.8 Å resolution (PDB: 6QM7 EMD-4590) and reveals how mutations lead to resistance and how selectivity arises. A nonpathogenic strain, Leishmania tarantolae, was used for both structures, but the potency of the drug was similar with both species. 81 Novartis have also been working on targeting the Leishmania proteasome with compound GNF6702, which is structurally related to compound-8. 82 Structural information about the binding mode of GNF6702 has not yet been collected, 82 but compound-8 does not bind in the proposed GNF6702 binding site. 81 By mutating various residues in the protein constructs, similar biochemical results were obtained with both compounds, suggesting that the binding sites may partially overlap. Comparison of the Leishmania and human structures reveals that the human binding site is more open and many of the residues that compound-8 interact with are not present, hence explaining parasite selectivity. Compound-8 is currently in preclinical development and is expected to progress into human trials. 81 It is hoped that it will become a treatment for this neglected tropical disease and reduce the likelihood of fatality.
Cryo-EM of E3 Ligase Complex Components
Another strategy involving the proteasome is in targeting specific proteins for degradation. Small molecules can be designed to exploit the UPS, recruiting nonnative substrates to an E3 ligase complex that are then ubiquitinated and targeted for degradation. This method opens opportunities to degrade proteins that were previously thought to be “undruggable.” An example of this substrate recognition modulation occurs with the drug thalidomide. Thalidomide was initially given to pregnant women for relief of morning sickness but was later withdrawn as the children were born with various deformities. Thalidomide does have a positive effect against leprosy and multiple myeloma, though, and this was later revealed to be because it changes the substrate recognition of cereblon, part of the CUL4 E3 ligase complex. 83 Cryo-EM, due to its tolerance of flexibility and heterogeneity, may be helpful in determining the structure of ternary complexes consisting of a substrate receptor protein such as cereblon, a small molecule, and a neosubstrate. Two papers described structure determination for E3 ligase components DDB1 (adaptor/scaffold protein), DDA1 (accessory protein), and DCAF15 (substrate recognition protein) bound to small molecules and recruiting part of neosubstrate RBM39. One such small molecule was the anticancer drug indisulam in a structure solved to 3.5 Å resolution by cryo-EM (PDB: 6SJ7 EMD-10213), 84 and a second was the related compound E7820 in a structure solved to 4.4 Å overall resolution (EMD-20553). 85 These structures may now guide optimization of the small molecules to enhance binding affinity or variations that could be made to recruit other substrates. Bifunctional molecules are also being developed with binding sites for the E3 ligase complex and the target protein connected by a linker region. Any structures that can be solved for these complexes will help in understanding the selectivity toward the widened target scope enabled with these substrate recognition modulators.
eIF2B and the ISR
In eukaryotic cells, protein synthesis is finely tuned via a common pathway in response to various stress signals, through modulation of translation initiation. Responsible for this is the integrated stress response (ISR)86,87 that provides high-level regulation and has downstream effects on a variety of functions, such as development, metabolism, immunity, and even memory. Certain pathological circumstances, most notably neurological disease settings, call for weakened signaling and attenuation of the pathway. A small-molecule inhibitor of the ISR (called ISRIB) has been identified 88 and shown to improve outcomes of neurodegeneration and brain injury in mouse models,89,90 though other areas of interest have also been proposed. 91
The key player of this pathway is eIF2. eIF2 is a multimeric complex that, in its active and nonphosphorylated forms, carries the initiator methionyl transfer RNA to the ribosomes, as part of an even larger preinitiation complex. 87 eIF2 is a GTP-dependent carrier and is activated by its specific guanine nucleotide exchange factor eIF2B. 91 eIF2 activity is inhibited by several kinases that phosphorylate it in response to external stressors. 92 The inactive phosphorylated form in turn inhibits eIF2B by forming an unproductive complex and therefore ultimately limiting the supply of Met-tRNA to the ribosomes. 93
Cryo-EM of eIF2B
Both eIF2 and eIF2B are heteromultimeric proteins. While several crystal and solution structures of isolated subunits and the Schizosaccharomyces pombe eIF2B complex structure have been available, the real breakthrough in our understanding of the human eIF2 and eIF2B modulation at a molecular level was provided by a series of cryo-EM structures in recent years. These groundbreaking EM studies followed two main avenues.
First, several groups succeeded in determining the structure of the heterodecameric eIF2B in complex with ISRIB, with resolutions reaching 2.8 Å overall.94,95 Human eIF2B consists of two sets of five subunits (ɑ to ε), of which ɑ, β, and γ form two stacking heterotrimers at the center of the complex that sandwich between two sets of δ and ε units at the periphery. The C-terminal domain of eIF2Bε has been shown to carry most of the catalytic activity, whereas the central hexamer of ɑ2β2γ2 is involved in the recognition of the substrate eIF2. The catalytic C-terminal HEAT domains of the two ε subunits are not well resolved in uncomplexed structures of eIF2B, suggesting that binding of the substrate protein eIF2 is required for this domain to adopt a well-defined position.
ISRIB, the small-molecule inhibitor of ISR, binds in the central cavity of the eIF2B. The symmetrical ISRIB molecule is located exactly on the symmetry axis of the complex, with its cyclohexyl ring occupying a central position and the two distal chlorophenyl rings extending into deep pockets forming β and δ subunits on either side ( Fig. 6 ).
Visualization of the ISRIB-eIF2B complex now enables us to explain the SAR of ISRIB analogs and guide future chemistry efforts toward therapeutic agents. For instance, compounds lacking the ether linker cannot form H-bonds to the protein and therefore appear less potent. On the mode of action level, mutagenesis, ultracentrifugation, and structural studies demonstrated 94 that ISRIB promotes the formation of the β2γ2δ2ε2 subcomplex by stabilizing this hetero-octamer by acting as a molecular staple. The β2γ2δ2ε2 subcomplex in turn has an enhanced affinity to binding ɑ2 and forming the decameric holoenzyme.
While ISRIB-eIF2B complex structures revealed the molecular mechanism of ISRIB on the target level, high-resolution structures of the eIF2-eIF2B complex gave insight into the ISR regulation on the pathway level. Again, several groups published eIF2B in complex with both phosphorylated and unmodified eIF2, using human and yeast variants of the proteins.
The eIF2 molecule is a heterotrimeric complex consisting of ɑ, β, and γ subunits. The active, nonphosphorylated form of eIF2 binds with its γ subunit sandwiched between the main body of eIF2Bε and its HEAT domain. The latter is well ordered in this substrate-bound form, and its atomic model could be built unambiguously. In the eIF2-eIF2B complex, catalytic residues of eIF2Bε form close interactions with eIF2γ, suggesting that this form of the assembly positions its components so that they are ready for the catalysis of nucleotide exchange. 96 Comparison of structures of eIF2-eIF2B with 1:1 and 2:1 stoichiometry reveals that eIF2 binds identically to eIF2B in both cases, without allosteric modulation. 96
On the other hand, eIF2 phosphorylated on Ser51 of its ɑ subunit, and hence the inactive form binds to eIF2B in a different fashion. Phosphorylated eIF2ɑ bridges the ɑ and γ subunits of eIF2B and the phosphorylated loop extends into the central cavity. In this arrangement, eIF2γ can no longer engage with eIF2Bε; thus, eIF2 and eIF2B remain trapped in an unproductive complex. Furthermore, nucleotide exchange is sterically blocked at the second eIF2Bε subunit. 97
These recent structural studies of proteins of the ISR highlight the tremendous speed with which cryo-EM drives our understanding of molecular pathways and their small molecular modulators. We expect further ISRIB derivatives to be designed, synthesized, and characterized in the future, building on the structural information discussed above.
Fragment Screening Using Cryo-EM
A recent study by Saur et al. 98 explores the viability of fragment screening and fragment-based drug discovery using cryo-EM as the primary screening method. While this work does not offer groundbreaking novelty in terms of our understanding of the investigated enzymes, it probes the boundaries of the use of cryo-EM in SBDD in a previously unprecedented direction: to fragment-sized small molecules. The structural biology community very much welcomed cryo-EM as a high-resolution alternative to crystallography for large and difficult-to-crystallize systems; however, its use in a high-throughput fashion and on smaller ligands has been met with skepticism. Saur et al. demonstrate that under favorable circumstances, such as reproducible sample preparation, using new rapid data acquisition and streamlined data processing techniques, fragment-sized molecules indeed can be unambiguously detected and liganded structures delivered on timescales that meet the requirements of SBDD projects.
In this work, two enzymes were studied: Escherichia coli β-galactosidase and human PKM2. Over the recent years, β-galactosidase has become the guinea pig of cryo-EM for its reproducibility and rigidity, leading to remarkably high-resolution structures being reported. PKM2 is also precedented, though, prior to this series of EM structures, only crystals structures had been deposited (e.g., Wang 99 ). Both proteins form homotetramers, that is, have a fourfold internal symmetry that can be exploited in 3D reconstructions and can reduce the amount of data required to achieve suitably high resolution.
Structure solution and map interpretation were aided by automated data processing and ligand fitting. Saur et al. developed Webcryo, a computational platform to streamline steps of data processing from micrographs to the final map, in order to reduce human intervention and for bookkeeping in larger-scale projects. Webcryo also includes techniques borrowed from crystallography pipelines, such as molecular replacement into EM maps and ligand fitting.
The β-gal studies paved the way to the fragment screening experiments of PKM2. This protein is a known oncology target, as it is upregulated in multiple tumor cells. Unlike β-gal, this protein does not form entirely rigid particles, but has flexible domains, though the ligand binding site is located in the more rigid core. Despite the structural flexibility, overall resolutions of 2.5–3.2 Å could be achieved in the reported structures. Fragment-sized ligands were screened both individually and in cocktails of four. The sample preparation appears to follow that of crystallographic fragment screening; for both single fragments and cocktails, the compounds were first solubilized in d6-DMSO and added to the protein solution at concentrations of 5–25 mM. This treatment was overall well tolerated.
The authors report example structures resulting from both single-fragment and cocktail experiments. In all cases, the ligands could be fitted to the map unambiguously, demonstrating that resolutions (3.2 Å in this example) that historically would have been labeled mediocre can still lead to a successful fragment screening campaign.
This study clearly validated cryo-EM as a viable technique in terms of resolution, detectable ligand size, and general feasibility in the context of fragment screening as part of SBDD. This has likely been aided by the selection of target proteins with internal symmetry, a rigid core, and previously known binders, though there are no apparent theoretical limitations. Ambiguous ligand placement cases could be resolved using additional (e.g., protein–ligand interaction) terms as part of the overall ligand-fitting scoring mechanism, which can prove particularly powerful at 3+ Å resolution and in the case of symmetrical ligands.
Conclusion
Apart from limitations in resolution, the most criticized aspect of cryo-EM in the pharmaceutical community has undoubtedly been its mediocre throughput, when compared with crystallography as a screening platform. Industrial structural biology groups have begun to augment this by developing computational pipelines and information management systems that bring the timeline of the sample preparation to the fitted ligand process into a feasible range. Webcryo, described by Saur et al., integrates data processing, molecular replacement into reconstructed maps, and model building into a single platform. At the time of writing, none of the other publicly available pipelines provide this level of integration, though many cover individual parts of the process. For instance, Genentech has built GP2S, 100 a multiuser laboratory information management system to track projects in high-throughput settings, ranging from protein samples to models, which is publicly available. The main advantage of such in-house tools over off-the-shelf solutions is that they are bespoke to the hardware and software environment of the group and are intimately integrated into the larger organization’s various other workflows and databases, such as protein production, small-molecule compound synthesis, and assays.
The typical resolution of cryo-EM maps, though rapidly improving, still often necessitates the use of a large pool of prior knowledge when fitting small-molecule ligands into cryo-EM maps. Software developers have offered valuable tools, built on decades of computational chemistry and high-resolution structural biology observations, for instance, the pipeline GemSpot, which produces ligand poses based on quantum mechanics energy calculations and on the coulomb potential maps. 101
Further developments in data acquisition, such as faster detectors, queueable data acquisition, and on-the-fly data processing, are expected to make cryo-EM a suitable platform to support the fast turnaround required to support medicinal chemistry design cycles. We predict that even fragment screening will become a viable hit-finding technique for real-life targets of pharmaceutical interest, especially for large and difficult to crystallize proteins. New hardware improvements, specifically improved monochromators, spherical aberration correction, and energy filters, are being reported that are pushing the resolutions obtained for favorable samples to the level at which single atoms can be resolved 7 and are delivering better than 2 Å resolution structures. 57
At present, the cases discussed above do not yet give a full picture of the extent of cryo-EM-facilitated SBDD. Publications of drug design stories, irrespective of the structure solution technique used, are slow to emerge, and typically several years pass between experiments and the medicinal chemistry report to appear in print. Cryo-EM-based compound design is very much expected to gain traction in medicinal chemistry journals in the coming years, featuring multiple compound-bound structures of the same protein, reflecting the workflow of iterative design in a high-throughput context. On the other hand, one should not underestimate the amount of rational compound design that can be derived from a single high-resolution structure of a previously unprecedented and intractable protein target, such as membrane proteins and large, difficult-to-purify multiprotein complexes. In such cases, compound docking and modeling of smaller conformational changes within the protein can become orders of magnitudes more accurate compared with working with homology models or orthologs.
In summary, it is now clear that pharmaceutical companies are fast establishing cryo-EM capabilities internally within their research groups. These investments are driven by the expansion of target space for which high-resolution structural biology insights can impact. Success relies on the timely delivery of these structures within the project timeline, and the advances in the speed of data acquisition and, critically, in data processing and the streamlining of EM data analysis are beginning to enable SBDD projects. Further developments of these processes are desired; however, the field is advancing rapidly. The diverse examples discussed in this review demonstrate the broad impact across discovery portfolios that cryo-EM can have and underline what is currently achievable. All the examples are underpinned by significant sample preparation expertise and demonstrate that strong protein chemistry is a prerequisite to the successful implementation of cryo-EM within industrial research teams. The speed with which impactful insights can be delivered is perhaps best exemplified by the rapid determination of the SARS-CoV-2 target:ligand complex structures. 102 In short, the resolution revolution is live.
Footnotes
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: All authors are employed by AstraZeneca, and their research and authorship of this article was completed within the scope of their employment with AstraZeneca.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: All authors are employees of AstraZeneca.