
Abstract
Atherogenesis has been recognized as a risk factor for lethal cardiovascular diseases. Plasma low-density lipoprotein levels are correlated to the occurrence of atherosclerosis, and their control is critical for both the prevention and treatment of these diseases. Phospholipid transfer protein (PLTP) is one of the key regulators of lipoprotein metabolism; PLTP-deficient mice exhibit decreased apolipoprotein B (apoB)-containing lipoprotein secretion and atherosclerosis, indicating the validity of PLTP as a promising therapeutic target. Here, we demonstrate a high-throughput screening (HTS) method to identify a novel chemotype of PLTP inhibitors. Instead of using recombinant proteins, we used human plasma as a source of enzymes in the first screening, so as to efficiently exclude promiscuous inhibitors. The selected compounds were further confirmed to target PLTP both biochemically and biophysically and were shown to inhibit apoB secretion from hepatic cells with no apparent toxicity. We believe that our approach is suitable for filtering out nonspecific inhibitors at an earlier stage of screening campaigns and that these compounds should have potential to be developed into drugs to treat dyslipidemia.
Keywords
Introduction
Atherosclerosis is often accompanied by dyslipidemia of diabetes and obesity and has been recognized as a crucial issue to be resolved worldwide. 1 A high level of low-density lipoprotein (LDL) containing apolipoprotein B (apoB), which is involved in the control of the assembly and secretion of very low-density lipoprotein (VLDL), can be a risk factor for atherosclerosis and cardiovascular events.
Phospholipid transfer protein (PLTP) is an 80 kDa hydrophobic glycoprotein that mediates the transfer of surface phospholipids from VLDLs to HDLs in plasma by associating with apoA–I, resulting in the production of larger, less dense HDLs.2,3 PLTP is a member of the lipid transfer/lipopolysaccharide-binding protein gene family, which includes cholesteryl ester transfer protein (CETP). 4 PLTP is ubiquitously expressed in animal cells and tissues with higher expression in macrophages and atherosclerotic lesions, and its expression in the liver accounts for around 25% of all plasma PLTP activity. 5 Its expression can be regulated by transcription factors such as peroxisome proliferator-activated receptor, 6 farnesoid X receptor, 7 liver X receptor,8,9 and sterol regulatory element-binding protein 1c, 10 which are involved in lipid synthesis, lipoprotein production, and reverse cholesterol transport (RCT). In addition, a hyperlipidemia mouse model induced by a high-fat, high-cholesterol diet exhibited a significant increase in PLTP expression and activity. 11 Likewise, plasma PLTP levels were elevated in obese and/or type 2 diabetic patients, 12 and the intima-media thickness, an established atherosclerosis marker, was found to be positively correlated with plasma PLTP activity. 13 Therefore, PLTP is regarded as a potential therapeutic target of dyslipidemia and subsequent atherosclerosis.
In addition to phospholipids, PLTP can transfer diacylglycerol, α-tocopherol, cerebroside, and lipopolysaccharides. 14 Although the precise mechanism remains elusive, PLTP is also involved in the secretion of VLDL from hepatocytes and contributes to the progression of atherogenesis, which was revealed by experimental animal models using PLTP-knockout or PLTP-transgenic mice, such as in an atherogenic apoE-deficient background.15,16 It should be stressed that no distinct lipid accumulation was observed in the liver of PLTP-deficient mice. 15 Furthermore, pharmacological inhibition of PLTP activities has been shown to decrease apoB secretion from human hepatic cell lines. 17 These lines of evidence support the notion that the inhibition of PLTP activity is beneficial to inhibit atherogenesis in humans.
As shown in a previous study, the use of recombinant proteins is a reasonable approach to identify compounds with a PLTP inhibitory activity throughout a screening campaign. However, given that PLTP is a plasma protein, we assume that the use of human plasma as an enzyme source would be favorable to physiologically mimic the conditions under which PLTP functions. Therefore, we chose human plasma as a PLTP enzyme source in the first screens and thereafter validated hit compounds using recombinant PLTPs. Considering that the final hit compounds were confirmed to directly and specifically interact with purified PLTP by a surface plasmon resonance (SPR) assay, there is a high possibility that promiscuous inhibitors were effectively excluded throughout the first screens. Furthermore, we found a novel chemotype of PLTP inhibitors (compounds 1 and 2) and confirmed their inhibitory effect on apoB secretion in HepG2 cells. We consider that it would be meaningful to confirm the validity of PLTP as a possible drug target molecule using other chemotype inhibitors so as to develop new strategies for the prevention and/or treatment of hyperlipidemia and its associated diseases.
Materials and Methods
Preparation of Human Plasma and HDL3
Human blood was obtained from healthy normolipidemic volunteers and was collected in tubes containing heparin (Terumo, Tokyo, Japan). Plasma was isolated by centrifugation at 1500g for 20 min at 4 °C, aliquoted, and then stored at −80 °C. HDL3 fractions were isolated from human blood as described previously. 18
Cloning, Expression, and Purification of Human PLTP
PLTP
Human PLTP was cloned into pcDNA3.1 (Thermo Fisher Scientific, Waltham, MA) with an N-terminal FLAG tag. This expression vector was transfected into HEK293F using 293fectin Reagent (Thermo Fisher Scientific). The expressed PLTP was purified from the culture medium using Anti-FLAG M2 affinity gel (Sigma-Aldrich, St. Louis, MO) and then eluted by competing with FLAG peptides (Sigma-Aldrich). The eluted fractions were pooled, dialyzed with a dialysis buffer (25 mM Tris-HCl [pH 7.4], 137 mM NaCl, and 26.8 mM KCl), and stored at −80 °C.
Biotinylated PLTP
Human PLTP was cloned into pcDNA3.1 with an N-terminal FLAG tag and a C-terminal biotin acceptor protein tag. This expression vector was cotransfected into HEK293F cells with an expression vector encoding biotin ligase BirA for site-specific biotinylation. Biotinylated PLTP, as well as nonbiotinylated PLTP, was purified as described above.
Measurement of PLTP Activity Using Fluorescence-Labeled Donor Vesicles
PLTP activity was determined using a Roar PLTP Activity Assay Kit (Roar Biomedical, New York, NY). This assay measures the transfer from donor to acceptor particles of fluorescent phospholipid, which is present in a self-quenched state when associated with the donor (Fig. 1A). Briefly, 5 µL of prediluted plasma and 2 µL of compounds were incubated with donor and acceptor particles in a final volume of 18 µL, resulting in the PLTP-mediated transfer of fluorescent phospholipid. This transfer was determined by the increase in the fluorescence intensity, which was detected by SpectraMax Paradigm (Molecular Devices, San Jose, CA).
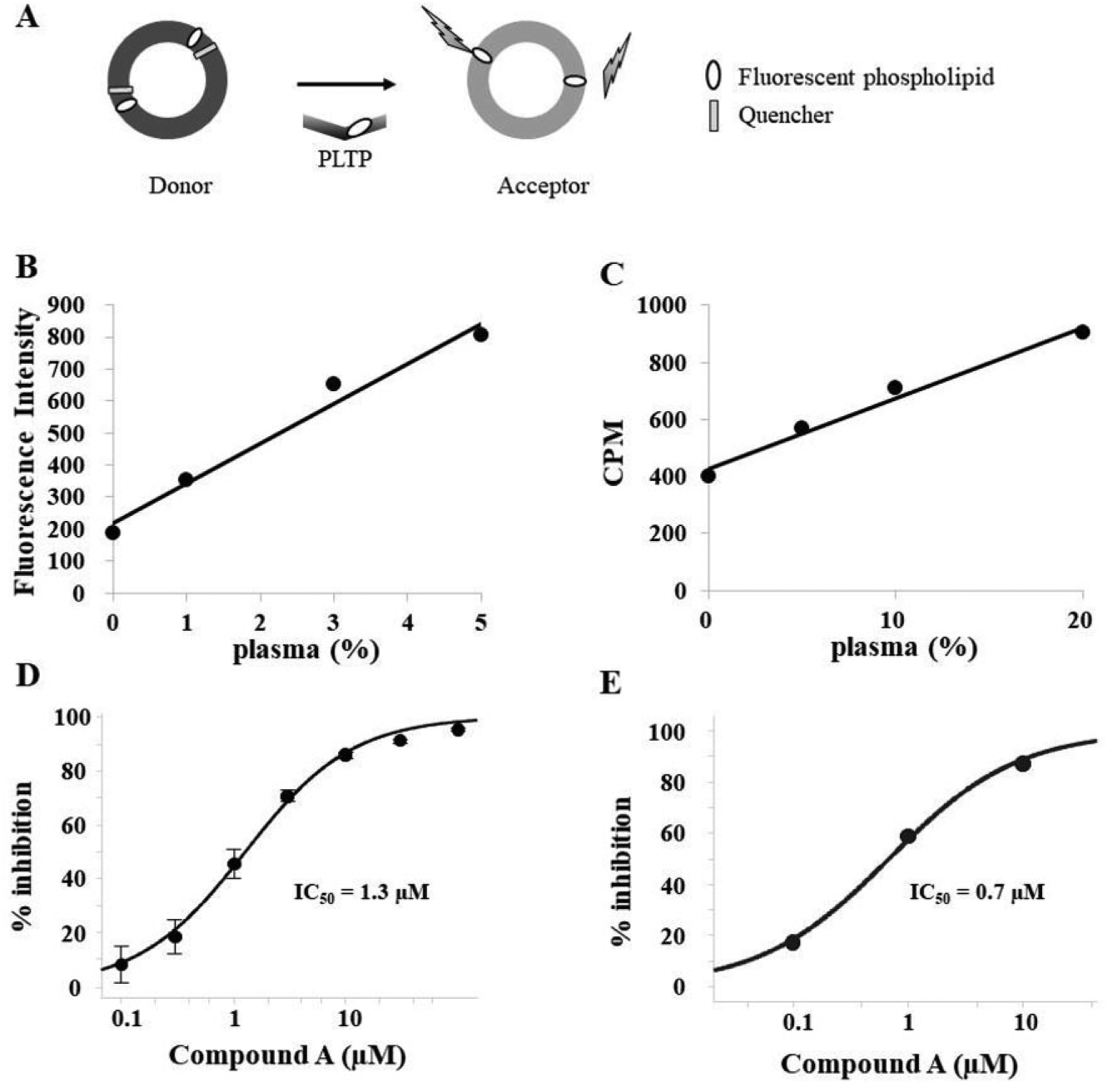
Determination of PLTP activity using human plasma. (
Measurement of PLTP Activity Using Radioisotope (RI)-Labeled Donor Vesicles
PLTP activity was determined by measuring the transfer of radiolabeled phosphatidylcholine (PC) from vesicles to HDL3, as described previously.
17
In brief, 5 µmol of egg PC containing 10 µCi of [
3
H]PC (
Data Analysis for PLTP Inhibition
Concentration–response data were fitted to the following equation using Spotfire (TIBCO, Somerville, MA):
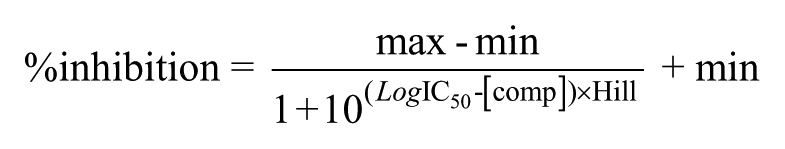
SPR-Based Binding Assay
All experiments were performed using a Biacore 4000 instrument (GE Healthcare, Little Chalfont, UK) at 25 °C. Streptavidin (SA) sensor chips (GE Healthcare) were used to immobilize the biotinylated PLTP. Each SA sensor chip was pretreated with three consecutive injections of 50 mM NaOH in 1 M NaCl to remove any nonspecifically bound contaminants. Biotinylated PLTP (150 µg/mL) in running buffer (25 mM Tris [pH 7.4], 137 mM NaCl, 26.8 mM KCl, and 0.1% Tween 20) was injected for 10 min. Typical immobilization levels ranged from 4500 to 4700 resonance units. The compounds were diluted directly into the running buffer containing 2% DMSO. Serially diluted compounds were injected in a separate cycle for 60 s of association followed by 180 s of dissociation at a flow rate of 30 µL/min at 25 °C. The running buffer was used as a blank buffer and injected both before and after all concentration series of test compounds.
Data analysis was conducted using Biacore 4000 software as described previously. 19 All sensorgram data were solvent corrected and double-referenced against an intact surface and the average of two blank injections. For kinetic analysis, data were fitted to a 1:1 interaction model with the mass transport term.
ApoB Secretion from HepG2 Cells
HepG2 cells were incubated for 24 h in Dulbecco’s modified Eagle’s medium supplemented with 1% BSA. Then, the cells were incubated in the presence of compounds diluted with 0.4 mM oleic acid/BSA for 24 h. ApoB protein levels in the culture supernatant were measured using an HTRF kit (Cisbio, Codolet, France), as well as cytotoxicity determined by lactate dehydrogenase (LDH) activity in the same medium using an LDH Cytotoxicity Assay Kit (Cayman Chemical, Ann Arbor, MI).
Measurement of CETP Activity Using Fluorescence-Labeled Donor Vesicles
CETP activity was measured using a Roar CETP Activity Assay Kit (Roar Biomedical). Five microliters of prediluted plasma and 2 µL of compounds were incubated with donor and acceptor particles in a final volume of 22 µL, resulting in the CETP-mediated transfer of fluorescent neutral lipid. The samples were incubated for 2 h at 37 °C, and the fluorescent signal intensity was measured by EnVision (PerkinElmer).
Results
Assay Development to Detect PLTP Activity
Prior to high-throughput screening (HTS), we first examined the dynamic range to detect the PLTP activity using human plasma in both fluorescence- and radioisotope (RI)-based assays. We found that the PLTP activity of plasma dose dependently increased in both assays (
HTS and Hit Identification
Based on the results presented above, we performed HTS to discover novel PLTP inhibitors from our original compound library, in accordance with the flowchart shown in
Figure 2A
. We adopted the fluorescence-based assay in the first screens because it is a high-throughput homogeneous assay. We evaluated the PLTP inhibitory activities at a single concentration of 10 µM in 1.5% DMSO in the enzyme reaction solutions, since a DMSO tolerance test indicated that DMSO concentrations up to this level hardly affected the PLTP activities (
Fig. 2B
). In order to obtain compounds with an IC50 value of <10 µM, we chose ones whose inhibitory activities exceeded 40%, considering that the level of the coefficient of variation was 10%. The mean Z′-factor value of the first screening was 0.8 (
Fig. 2C
), and 87 compounds were selected (
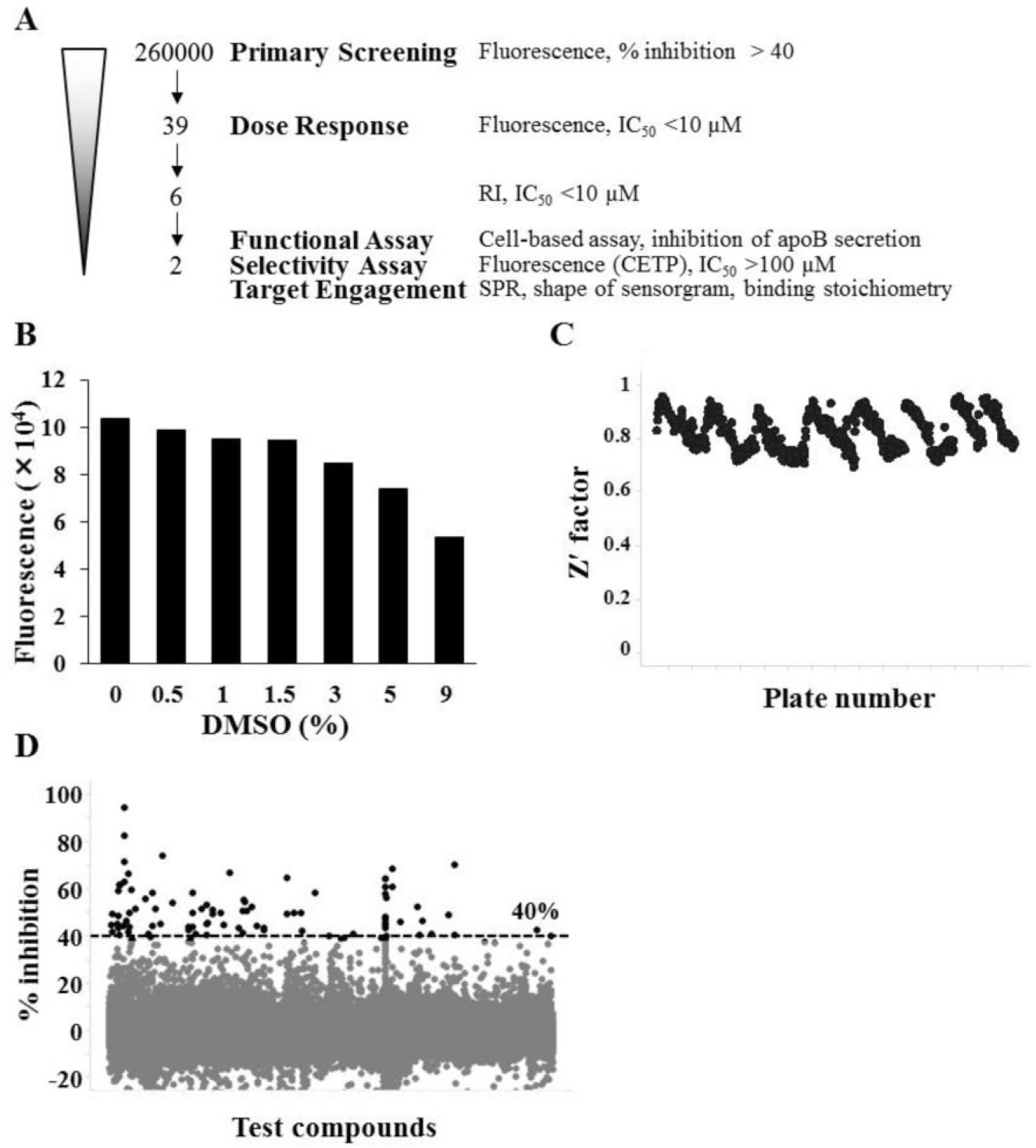
HTS by the fluorescence-based assay. (
In order to filter out the compounds that impede the fluorescent signal rather than the PLTP activity, we performed an RI-based assay. The results revealed the successful identification of two related compounds (compounds 1 and 2) ( Fig. 3A ), which showed similar PLTP activities as determined by both fluorescence- and RI-based assays ( Fig. 3B ). Furthermore, it was found that these compounds were able to inhibit apoB secretion from HepG2 cells as well as compound A ( Fig. 3C ). It should be noted that while compound A exhibited some cytotoxicity at higher concentrations, as revealed by our LDH assay, compounds 1 and 2 did not. In addition, it was revealed that compounds 1 and 2 did not suppress the activity of CETP, the closest PLTP homolog of lipid transfer/lipopolysaccharide protein sharing no redundancy in the function of PLTP ( Fig. 3D ). 20
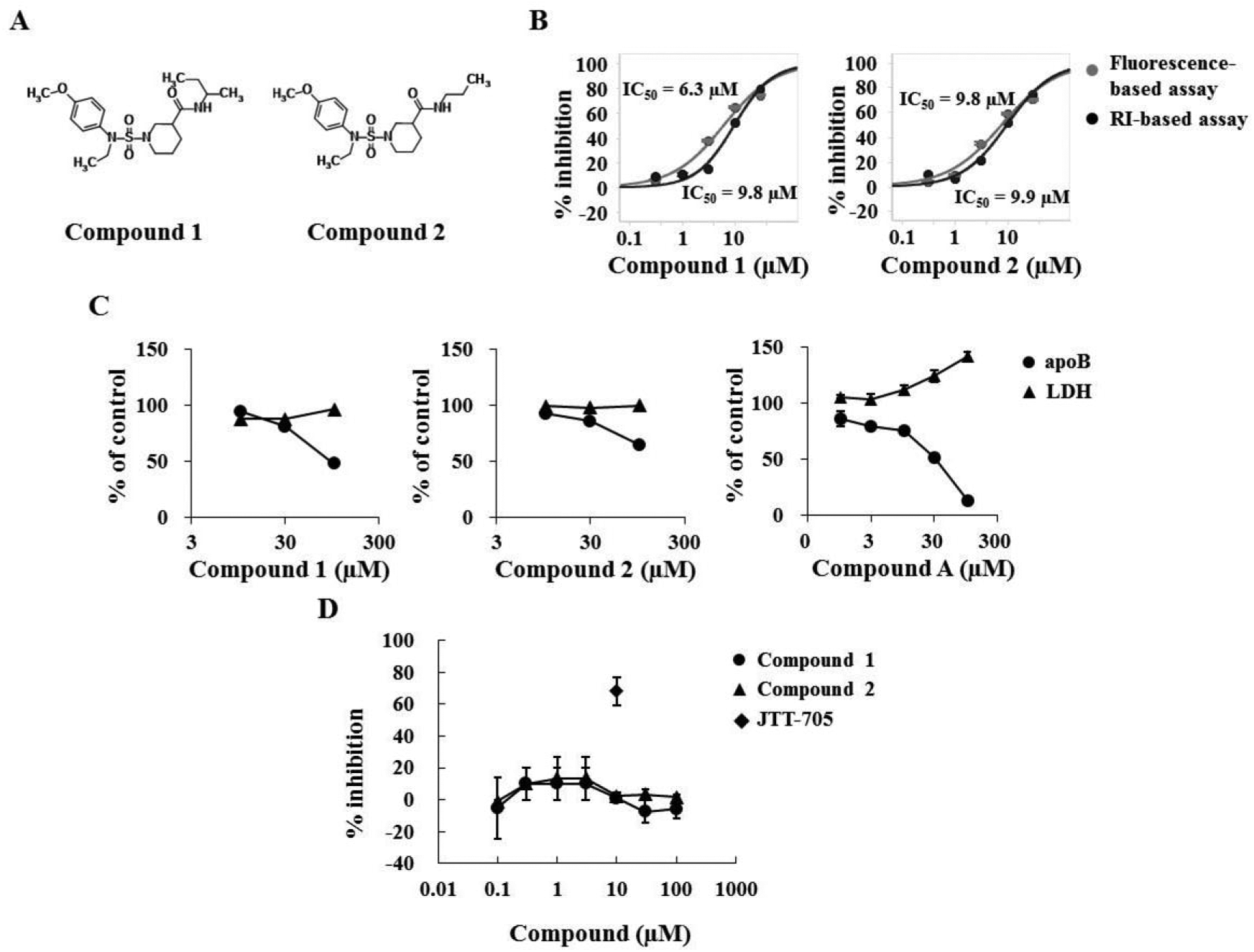
Characterization of hit compounds. (
Target Engagement of Compounds 1 and 2 Using Recombinant Human PLTP
Furthermore, to shed light on the possibility that other related or unknown proteins might inhibit the transfer of PC from donor vesicles to HDL3 acceptors, we performed a RI-based assay using recombinant human PLTP, instead of human plasma, as an enzyme source. We preliminarily confirmed the enzyme dose-dependent signal increase of PLTP activity ( Fig. 4A ) and the inhibition by compound A with an IC50 value of 0.24 µM ( Fig. 4B ). The results showed that compounds 1 and 2 inhibited PLTP activity with IC50 values of 4.2 µM (compound 1) and 4.3 µM (compound 2) ( Fig. 4C ), which are similar to those obtained when utilizing human plasma, indicating that the transfer of PC was mediated by PLTP. In order to further confirm the specific binding of compounds 1 and 2 to PLTP, we conducted an SPR analysis. For this, biotinylated PLTP was captured on an SA sensor chip, and compound A was used as a positive control to evaluate and optimize the assay conditions. The sensorgram showed an excellent fitting curve at a stoichiometric ratio of 1:1 ( Fig. 4D ), and the kinetics was fitted to a 1:1 binding model, giving an association rate constant ka = 1.7 × 105 M−1 s−1, a dissociation rate constant kd = 4.2 × 10−3 s−1, and an equilibrium dissociation constant KD = 0.024 µM. The KD value is roughly consistent with the IC50 value (0.24 µM) ( Fig. 4B ) and indicates the validity of the assay. Under the same assay conditions, the kinetics was fitted to a 1:1 binding model, yielding kinetic constants ka = 1.8 × 104 M−1 s−1, kd = 5.3 × 10−2 s−1, and KD = 3.0 µM for compound 1 and ka = 1.3 × 104 M−1 s−1, kd = 3.5 × 10−2 s−1, and KD = 2.7 µM for compound 2 ( Fig. 4D ). The KD values are also similar to each IC50 value. Overall, they showed specific binding interactions to PLTP like compound A. These results clearly indicate that compounds 1 and 2 are novel chemotypes of specific PLTP inhibitors.
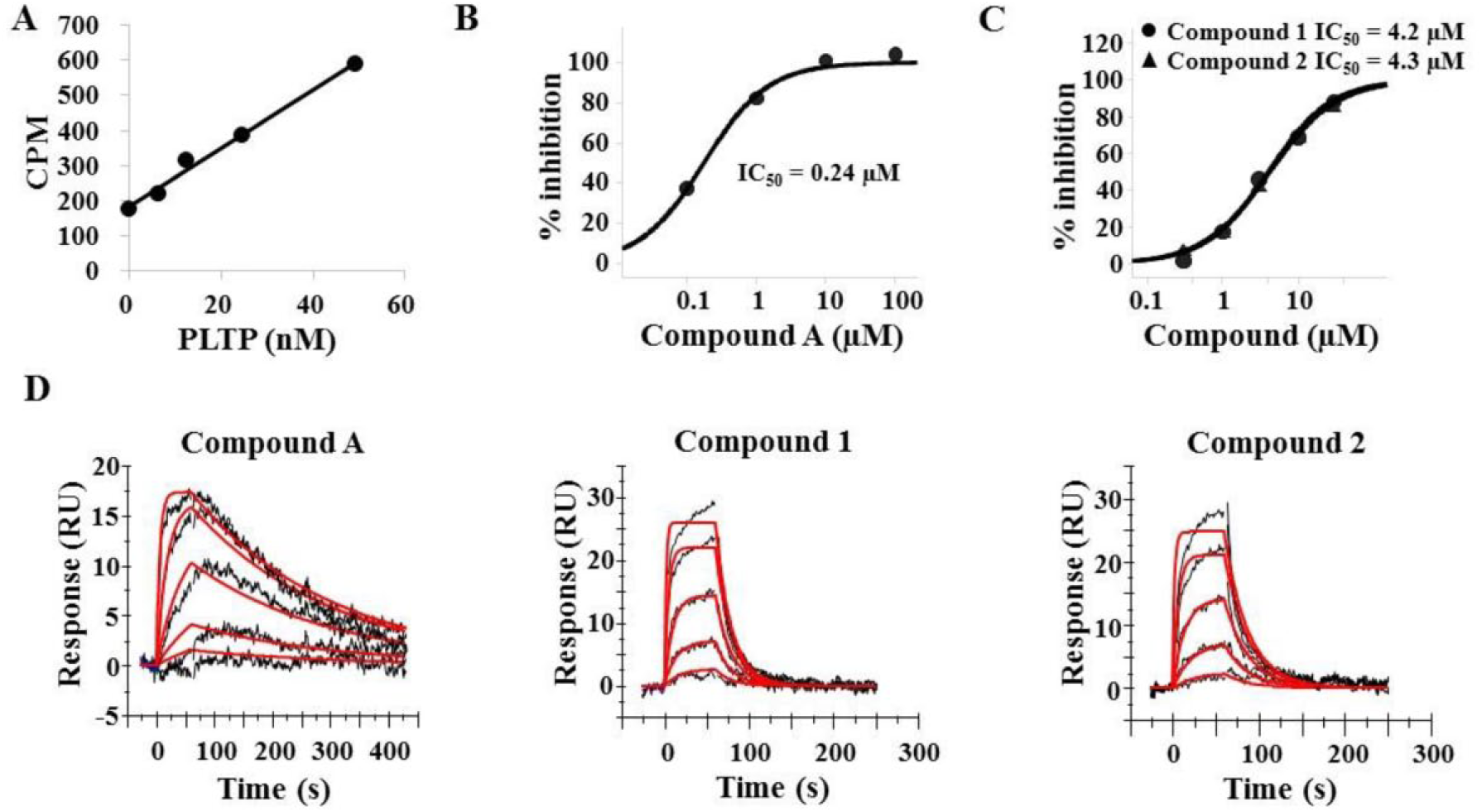
Target engagement of compounds 1 and 2 using recombinant human PLTP. (
Discussion
In this study, we carried out HTS to identify novel PLTP inhibitors by utilizing fluorescence-labeled PC as a donor vesicle and human plasma as a PLTP enzyme source. Following the screening scheme to narrow down hit compounds as depicted in
Figure 2A
, we successfully discovered compounds 1 and 2, which belong to the same chemotype of novel PLTP inhibitors. We found that they were able to inhibit PLTP activity as well as apoB secretion from HepG2 cells and specifically bind to the purified protein, as revealed by the SPR analysis that we established (
We believe that the use of human plasma in the first screens contributed to conditions that strongly resembled the physiological environment in which PLTP functions; this effectively eliminated the promiscuous compounds because plasma contains a huge number of proteins other than PLTP. In other words, this type of approach resembles phenotypic screening in terms of the fact that compounds were screened in crude mixtures and subsequent target confirmation was performed. Hence, it is plausible that a similar screening strategy is worth adopting if the majority of target proteins and their inhibition by small compounds are expected in the plasma.
On the other hand, the effect of PLTP inhibition on RCT is currently controversial. 5 It has been suggested that just increasing the total amounts of HDL would not directly lead to the treatment of dyslipidemia and cardiovascular diseases.22,23 Therefore, we postulate that investigation using PLTP inhibitors with a higher affinity and inhibitory activity may resolve the issue of whether a change of HDL metabolism by PLTP inhibition would contribute to the treatment of the above-mentioned diseases. There are also seemingly contradictory reports about inflammatory responses mediated by PLTP,24–27 so the use of specific inhibitors might be appropriate to clarify this issue. In this context, the SPR assay that we developed should accelerate research on structure–activity relationships to obtain compounds with a higher affinity to PLTP by tracking KD values. However, it should be noted that lipoprotein metabolism differs between humans and mice; for example, there are high levels of HDL in mice, probably owing to their lack of CETP activity. 28 Taking these factors into consideration, it would be crucial not only to carefully choose experimental animal models, but also to delicately analyze the data obtained from them.
In summary, we identified a novel chemotype of PLTP inhibitors through an HTS campaign using human plasma as an enzyme source. We further examined their specific binding to PLTP by SPR analysis and confirmed that they are authentic PLTP inhibitors. They exhibited characteristics distinct from those of another PLTP inhibitor, compound A, that was developed by Pfizer Inc. 17 We hope that our study will lead to a better understanding of lipoprotein metabolism and the pathogenesis of dyslipidemia and eventually to the treatment of related diseases.
Footnotes
Acknowledgements
We thank Drs. Jun Nishihata and Atsuhito Yoshida for helpful discussions. We also thank Mr. Kenji Yamanaka for technical support.
Authors’ Note
Yu Takahashi is currently affiliated with the Department of Applied Biological Chemistry, Graduate School of Agricultural and Life Sciences, The University of Tokyo, Tokyo, Japan.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.