
Abstract
Interleukin-2-inducible T-cell kinase (ITK) plays an important role in T-cell signaling and is considered a promising drug target. As the ATP binding sites of protein kinases are highly conserved, the design of selective kinase inhibitors remains a challenge. Targeting inactive kinase conformations can address the issue of kinase inhibitor selectivity. It is important for selectivity considerations to identify compounds that stabilize inactive conformations from the primary screen hits. Here we screened a library of 390,000 compounds with an ADP-Glo assay using dephosphorylated ITK. After a surface plasmon resonance (SPR) assay was used to filter out promiscuous inhibitors, 105 hits were confirmed. Next, we used a fluorescent biosensor to enable the detection of conformational changes to identify inactive conformation inhibitors. A single-cysteine-substituted ITK mutant was labeled with acrylodan, and fluorescence emission was monitored. Using a fluorescent biosensor assay, we identified 34 inactive conformation inhibitors from SPR hits. Among them, one compound was bound to a site other than the ATP pocket and exhibited excellent selectivity against a kinase panel. Overall, (1) biochemical screening using dephosphorylated kinase, (2) hit confirmation by SPR assay, and (3) fluorescent biosensor assay that can distinguish inactive compounds provide a useful platform and offer opportunities to identify selective kinase inhibitors.
Keywords
Introduction
Interleukin-2-inducible T-cell kinase (ITK) is involved in the regulation of Th2-mediated immunological diseases as well as the development of Th17 cells and their production of IL17A.1,2 Studies of ITK gene knockout mice have suggested that ITK is a dominant contributor mediating TCR signaling, and its inhibition has potential for treating inflammatory disorders.1,3
The catalytic subunits of many protein kinases have high structural similarity, 4 making it particularly difficult to design selective kinase inhibitors. 5 Most kinases exist in conformational equilibrium between two conformational states, active and inactive, under physiological conditions. 6 Targeting the inactive form of kinase offers advantages for achieving selectivity because of their structural diversification. 7 Traditional screening approaches for kinase inhibitors are commonly based on enzyme activity;8,9 however, they do not provide information on the conformational state of the target. Hence, identifying those compounds from the primary screen hits that stabilize inactive conformations has provided an important way of achieving selectivity. To date, some high-throughput detection methods to discover inhibitors that bind more favorably to inactive enzyme conformations have been described. Probe displacement assays measure the displacement of fluorescent ATP-competitive probe. 10 These competitive binding assays allow for the detection of inactive form binders with the usage of an inactive form of kinase. However, in this method, ATP noncompetitive inhibitors can be missed. Cascade assays utilize an active upstream kinase in combination with a dephosphorylated downstream kinase. 11 They are more complex and require additional triage assay using the activated kinase. The second-harmonic-generation (SHG) assay is based on tethering proteins labeled with dye, and measurement of the signal depends on the net orientation of the dye. We recently reported the discovery of inhibitors that stabilize an inactive conformation of ITK using SHG, and it was shown to be useful for distinguishing inactive and active conformations. 12
Here, we report on the application of a fluorescent biosensor assay using ITK. This type of assay is based on the incorporation of an environmentally sensitive fluorophore that reports on conformational changes induced by the binding ligands.13–16 The measurements are performed easily using fluorescence readers commonly found in many laboratories. In the study reported here, we performed high-throughput screening (HTS) of our compound library for dephosphorylated ITK and then selected hits based on selectivity toward FLT3(D835Y) and specificity determined by surface plasmon resonance (SPR) assay. Furthermore, we adopted a fluorescent biosensor assay of ITK for the selection of inactive conformation inhibitors. The results showed that one compound among the screening hits was an ATP noncompetitive binder. We hope that our method for measuring the inactive conformation of ITK can contribute to the identification of conformational changes of other kinases.
Materials and Methods
Materials
FLT3(D835Y) and the ADP-Glo Kinase Assay kit were purchased from Promega (Madison, WI). Paradigm was obtained from Molecular Devices (Sunnyvale, CA). Biacore T200 and 4000 optical biosensors and NTA sensor chips were obtained from GE Healthcare (Little Chalfont, UK). The fluorophore 6-acryloyl-2-dimethylamino naphthalene (acrylodan) was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). The EnVision microplate reader was obtained from PerkinElmer (Waltham, MA).
Cloning, Expression, and Purification of Proteins
Full-length human ITK (ITK-FL) was cloned into pVL1393 (Thermo Fisher Scientific, Waltham, MA) with an N-terminal FLAG tag. Full-length human YopH was cloned into pVL1393 with an Myc tag. The BaculoGold Transfection Kit (BD Biosciences, San Jose, CA) was used to prepare recombinant baculoviruses. Cells were infected with ITK-FL in combination with YopH. The infected cells were harvested 48 h postinfection and stored at −80 °C.
Cell lysates were prepared in a lysis buffer (50 mmol/L Tris-HCl, pH 8.0, 500 mmol/L NaCl, 10% glycerol, and EDTA-free protease inhibitor cocktail). The lysates were centrifuged at 39,000g for 30 min. The supernatants were incubated with Anti-FLAG M2 affinity gel (Sigma-Aldrich, St. Louis, MO), and the protein was eluted by competing with FLAG peptides (Sigma-Aldrich). Eluted fractions were further purified by gel filtration on a Superdex 200 10/300 GL column (GE Healthcare). The final protein buffer contained 50 mM HEPES, pH 8.0, 500 mM NaCl, 10% glycerol, and 1 mM DTT. The proteins were flash-frozen with liquid nitrogen and stored at −80 °C.
Dephosphorylation of ITK-FL by YopH was confirmed by Western blotting using the monoclonal phosphotyrosine antibody clone 4G10 from Merck Millipore (Darmstadt, Germany). The kinase domain of human ITK (ITK-KD) and single-cysteine-mutated ITK-KD (ITK-KD Mutant A) were constructed, expressed, and purified as described previously. 12
A substrate protein, GST-SLP-76, was constructed, expressed, and purified as described previously. 17 The protein was further purified by gel filtration on a HiLoad 26/600 Superdex 200 column (GE Healthcare).
Biochemical Assay
The ADP-Glo Kinase Assay 18 was used to determine compound potency against ITK-FL and FLT3(D835Y) kinase activity. The primary reaction consisted of 2.5 nM ITK-FL, 250 nM GST-SLP-76, 17 and 30 µM ATP in 25 mM HEPES, pH 7.5, 25 mM KCl, 10 mM MgCl2, 1 mM DTT, 0.005% Tween-20, and 0.1% bovine serum albumin (BSA). Three microliters of substrate and 2 µL of enzyme were added to the plate and incubated at room temperature for 1 h. After the incubation, 5 μL of ADP-Glo reagent was added and incubated for 40 min. Then, 10 μL of kinase detection reagent was added and, after an incubation time of 30 min, luminescence was measured on a Paradigm. The assays against FLT3(D835Y) were performed in basically the same way as described for the ITK assay. The FLT3(D835Y) reaction contained 5 nM FLT3(D835Y), 4 µM myelin basic protein, and 54 µM ATP.
SPR Assay
For capture coupling, a flow cell surface was activated for 5 min with a mixture of 0.1 M N-hydroxysuccinimide and 0.4 M N-ethyl-N′-(dimethylaminopropyl) carbodiimide prior to the injection of ITK-KD. ITK-KD (10 µg/mL) was captured in running buffer (25 mM HEPES, pH 7.5, 25 mM KCl, 10 mM MgCl2, 1 mM DTT, and 0.005% Tween-20) for 10 min. Compounds were serially diluted in the running buffer containing 5% DMSO. The compounds were injected at a flow rate of 30 µL/min, and association was measured for 1 min and dissociation for 1–3 min depending on the off-rate of each compound. Competition experiments were carried out in the presence of adenosine diphosphate (ADP) at a concentration of 1 mM diluted in the running buffer.
Biacore T200 or Biacore 4000 evaluation software was used to perform all data analysis. Sensorgram data were solvent-corrected and double-referenced. For kinetic analysis, data were fit to a 1:1 interaction model with a mass transport term. The experimental maximal binding response (Rmax) was determined via kinetic analysis using analytical software. For steady-state analysis, responses at equilibrium were plotted against the compound concentration and fit to a 1:1 Langmuir binding isotherm.
Fluorescent Biosensor Assay Development and Validation
ITK-KD Mutant A and acrylodan were combined in a labeling buffer (50 mM HEPES, pH 7.5, 200 mM NaCl, and 10% glycerol) at a 1:5 ratio and incubated at 4 °C for 5 h. Conjugated protein was subsequently washed three times in a 10-MWCO Amicon Ultra (0.5 mL) centrifugal filter and frozen at −80 °C.
Measurements were performed in a black 384-well plate (PerkinElmer). Labeled ITK-KD Mutant A was diluted to 400 nM in assay buffer (50 mM HEPES, pH 7.5, 25 mM KCl, 10 mM MgCl2, and 0.005% Tween-20). Compounds were prepared in 100% DMSO and prediluted in the assay buffer at double the final desired concentration. Ten microliters of compound was mixed with an equal volume of labeled ITK-KD Mutant A. Plates were incubated for 15 min at room temperature prior to measurement. The excitation wavelength for acrylodan was set at 386 nm, and emission was monitored between 410 and 560 nm.
Screening compounds were added to final concentrations of 2.5, 10, and 40 µM for lower-potency compounds (IC50 ≥ 1 µM) or 0.25, 1, and 4 µM for higher-potency ones (IC50 < 1 µM) and 5% DMSO.
Results
HTS and Characterization of Small-Molecule Inhibitors of ITK
To allow compounds to capture the protein in an inactive state, ITK-FL protein was coexpressed with the tyrosine phosphatase, YopH, in baculovirus to fully dephosphorylate them. Western blot analysis using monoclonal phosphotyrosine antibody confirmed that ITK-FL was dephosphorylated (
Suppl. Fig. S1
). To discover ITK inhibitors, we optimized the conditions for the ADP-Glo Kinase Assay. Enzyme kinetic studies of ITK-FL revealed that the apparent Km for ATP was 120 µM (data not shown). To validate the assay, we compared inhibition profiles for three inhibitors against ITK and FLT3(D835Y). Achieving high selectivity of ITK inhibitors over FLT3 might be challenging.19,20 Therefore, we chose to monitor the selectivity for key compounds against FLT3(D835Y), a constitutively active form of FLT3. Known inactive conformation inhibitors for ITK, BMS-509744
17
and Pfizer compound 9,
21
selectively inhibited ITK-FL, while staurosporine potently inhibited both ITK-FL and FLT3(D835Y) (
Suppl. Table S1
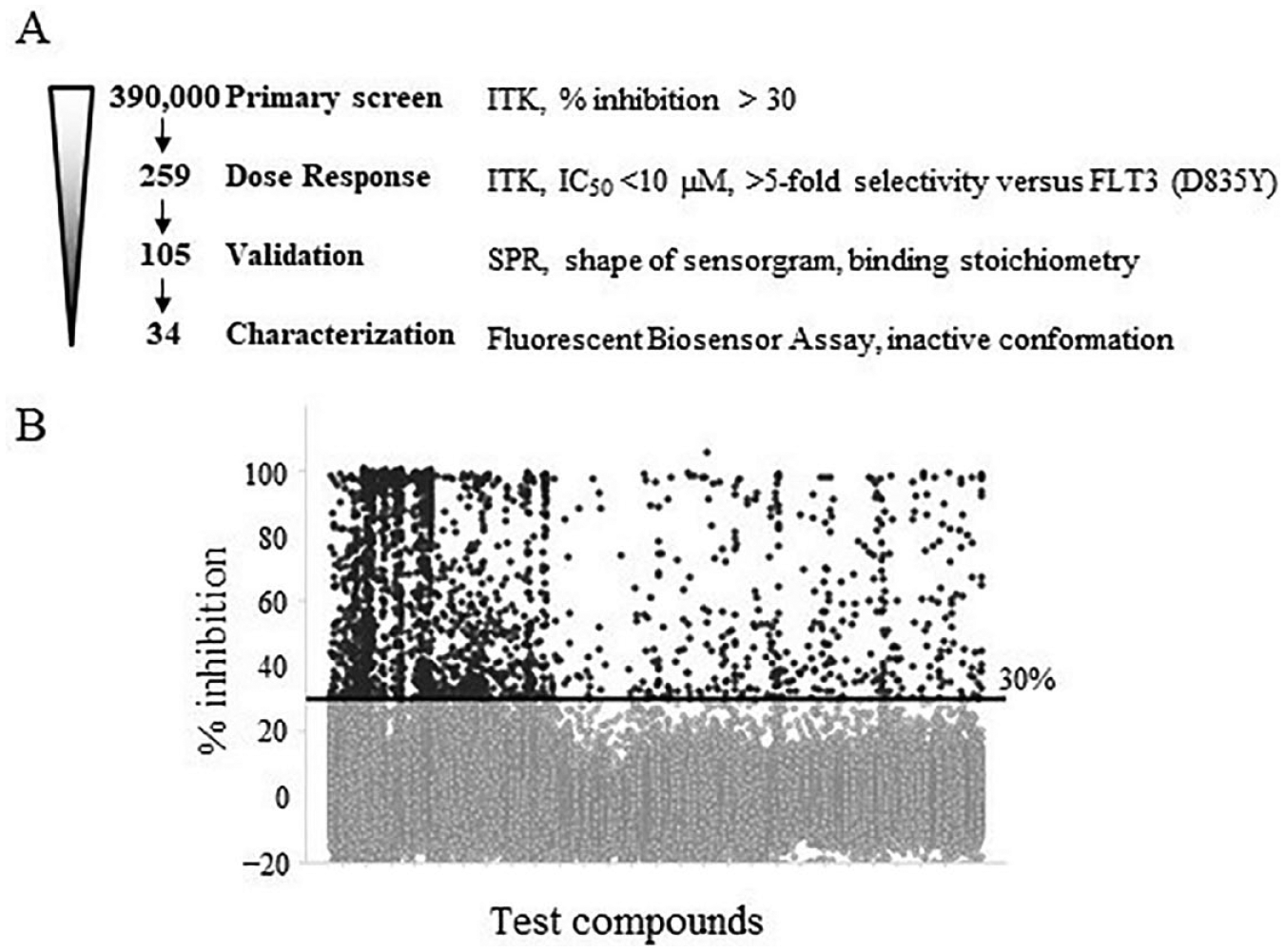
). The ADP-Glo Kinase Assay was used to screen 390,000 compounds against ITK-FL in 384-well plates. The average Z′ factor was 0.88 ± 0.048 for ITK-FL (
Suppl. Fig. S2
). A total of 2754 compounds were selected at more than 30% activity for 10 µM and considered for follow-up assays (
Flowchart and HTS data. (
SPR Hit Confirmation
Typical immobilization levels of ITK-KD ranged from 2900 to 4900 resonance units. ADP was confirmed to bind to the immobilized ITK-KD at a KD of 13 µM ( Suppl. Fig. S3 ), similar to the results of a previous report. 22 We performed precleaning of 259 hits on an ITK-KD-coated surface; 106 compounds were eliminated as being promiscuous according to the criteria of over stoichiometric binding (>3 times the saturation response of positive control) and/or a sensorgram indicating nonequilibrium behavior ( Suppl. Fig. S4 ). 23 Using 25% binding relative to staurosporine as a cutoff, 116 compounds were selected. Finally, we confirmed 105 hits in dose–response experiments.
Fluorescent Biosensor Assay Development and Validation
Labeled ITK-KD Mutant A was preincubated with each of three tool compounds to demonstrate that we could distinguish between inactive and active conformation inhibitors. The emission spectra were recorded in the presence and absence of tool compounds. Intrinsic compound autofluorescence was corrected by subtracting fluorescence intensities of the respective compounds, as measured alone in buffer. Inactive conformation inhibitors caused a bathochromic shift of the acrylodan emission spectrum (maximum from 488 to 508 nm) (
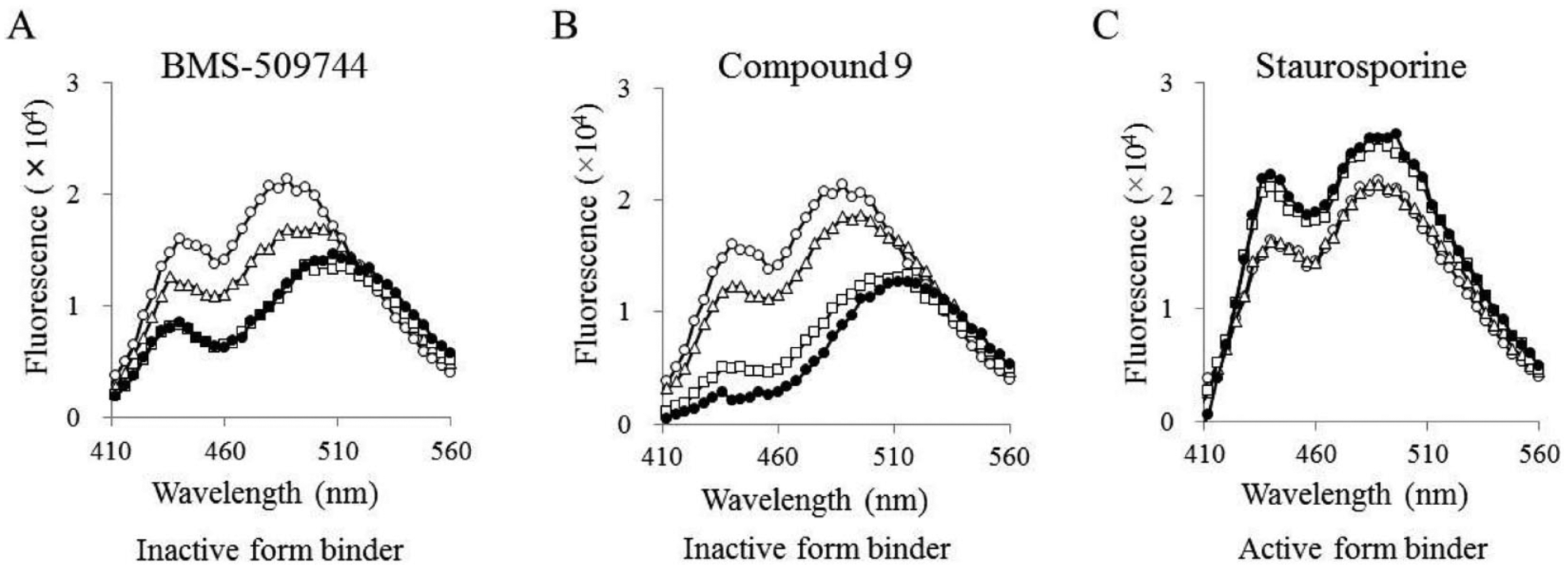
The results of the fluorescent biosensor. Emission spectra recorded in the presence of BMS-509744 (
We previously showed the binding affinity of known ITK inhibitors for ITK-KD.12 The ratiometric fluorescence value (R = FI508/FI456) against two ligand concentrations of various compounds was plotted (
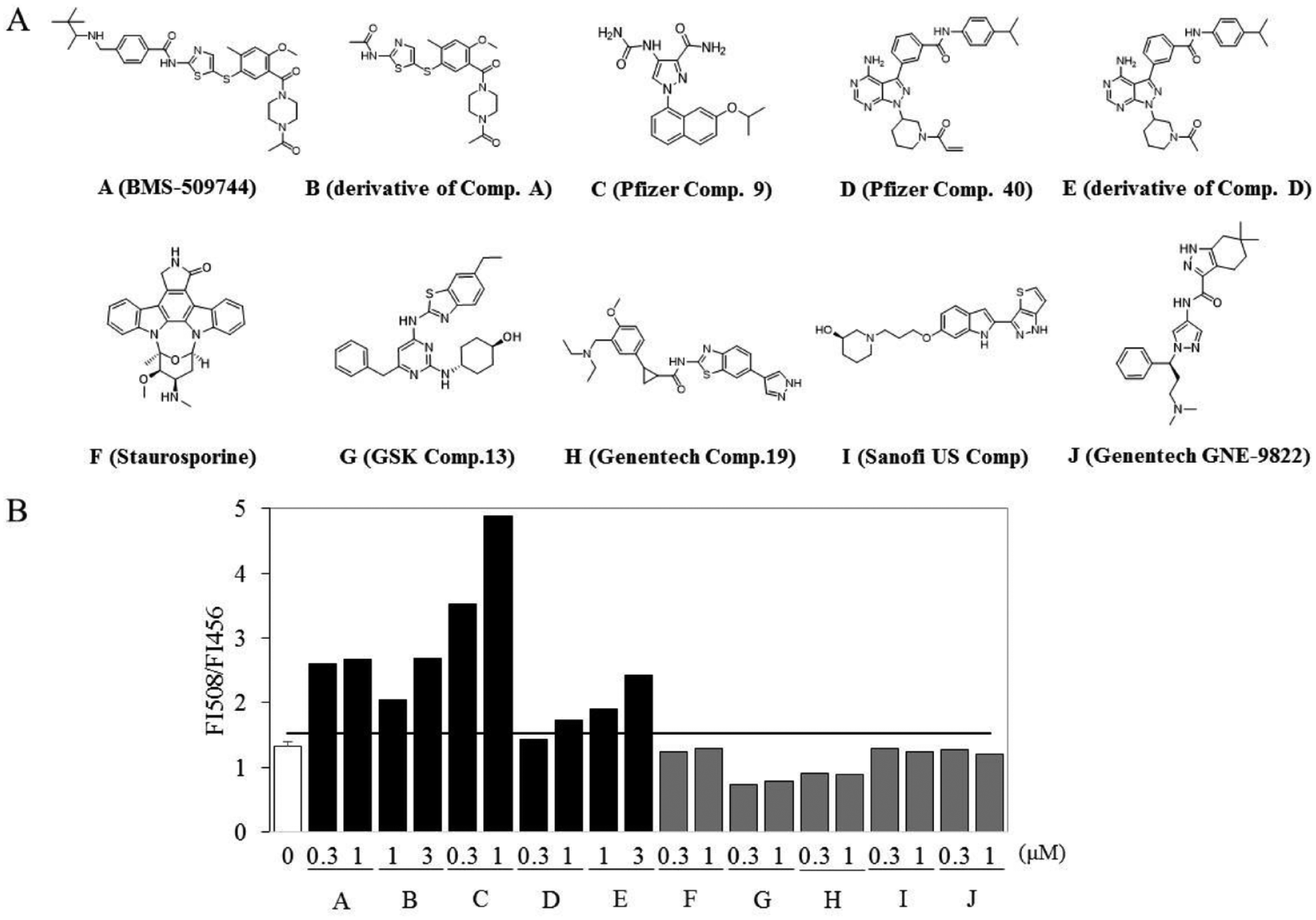
Characterization of inhibitor types. (
Hit Identification
After the fluorescence response of labeled ITK was validated, SPR hits (105 compounds) were screened by a fluorescent biosensor assay. Background plates were used to correct for intrinsic compound fluorescence. The performance of the screen was assessed by monitoring the ratiometric values of the positive control (BMS-509744) and DMSO control of all plates and yielded a calculated Z′ factor of 0.65 ± 0.089 for the entire screen (Suppl. Fig. S5). Thirty-five compounds that reached the cutoff value defined as the mean plus 3 standard deviations (SD) of the DMSO control (
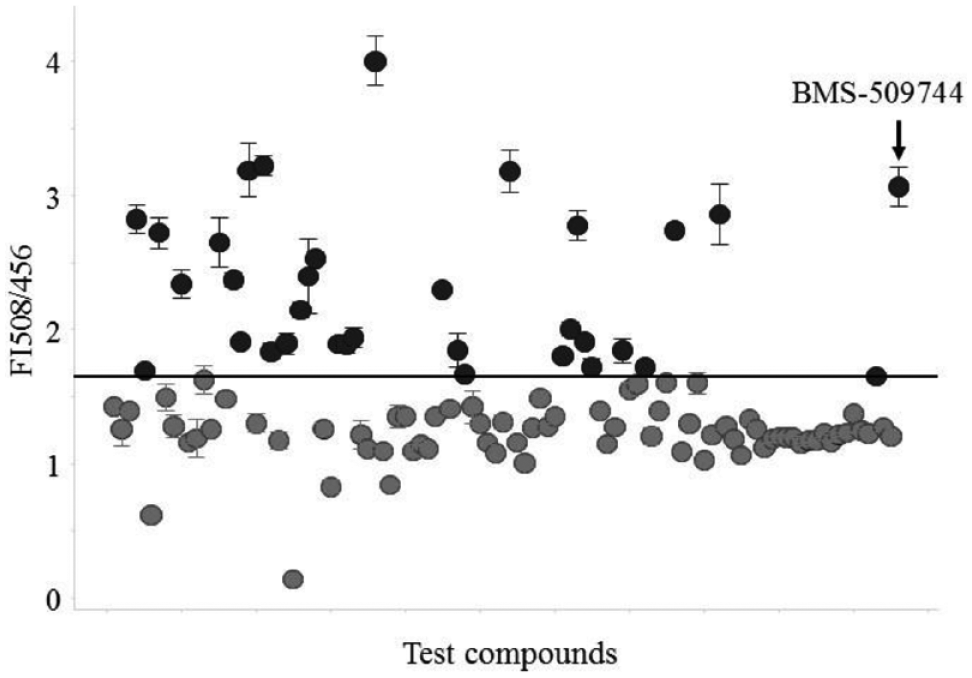
Ratiometric assay readout of ITK-KD Mutant A supplemented with the highest inhibitor concentration (4 or 40 μM). All data are shown as the SD of triplicate measurements. A hit threshold of 1.65 (mean of DMSO control + 3 SD) was chosen.
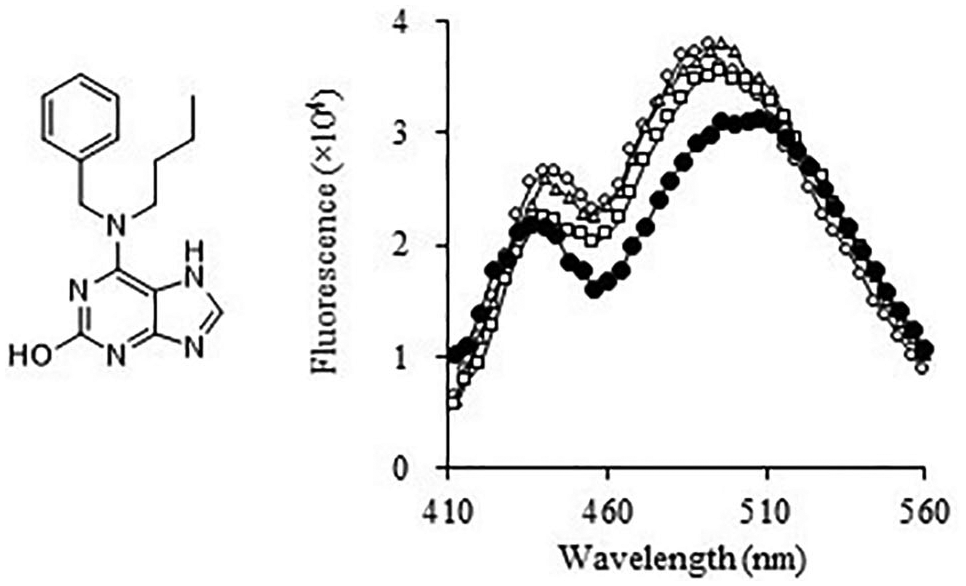
Chemical structure and emission spectra for J-13. Emission spectra recorded in the presence of J-13 at varying concentrations (0 μM [open circles], 2.5 μM [open triangles], 10 μM [open squares], and 40 μM [filled circles]).
Biophysical Characterization of the Binding Site
To characterize the J-13 binding site, we performed the Biacore competition assay with the compound and ADP. J-13 bound to ITK in the absence and presence of ADP (
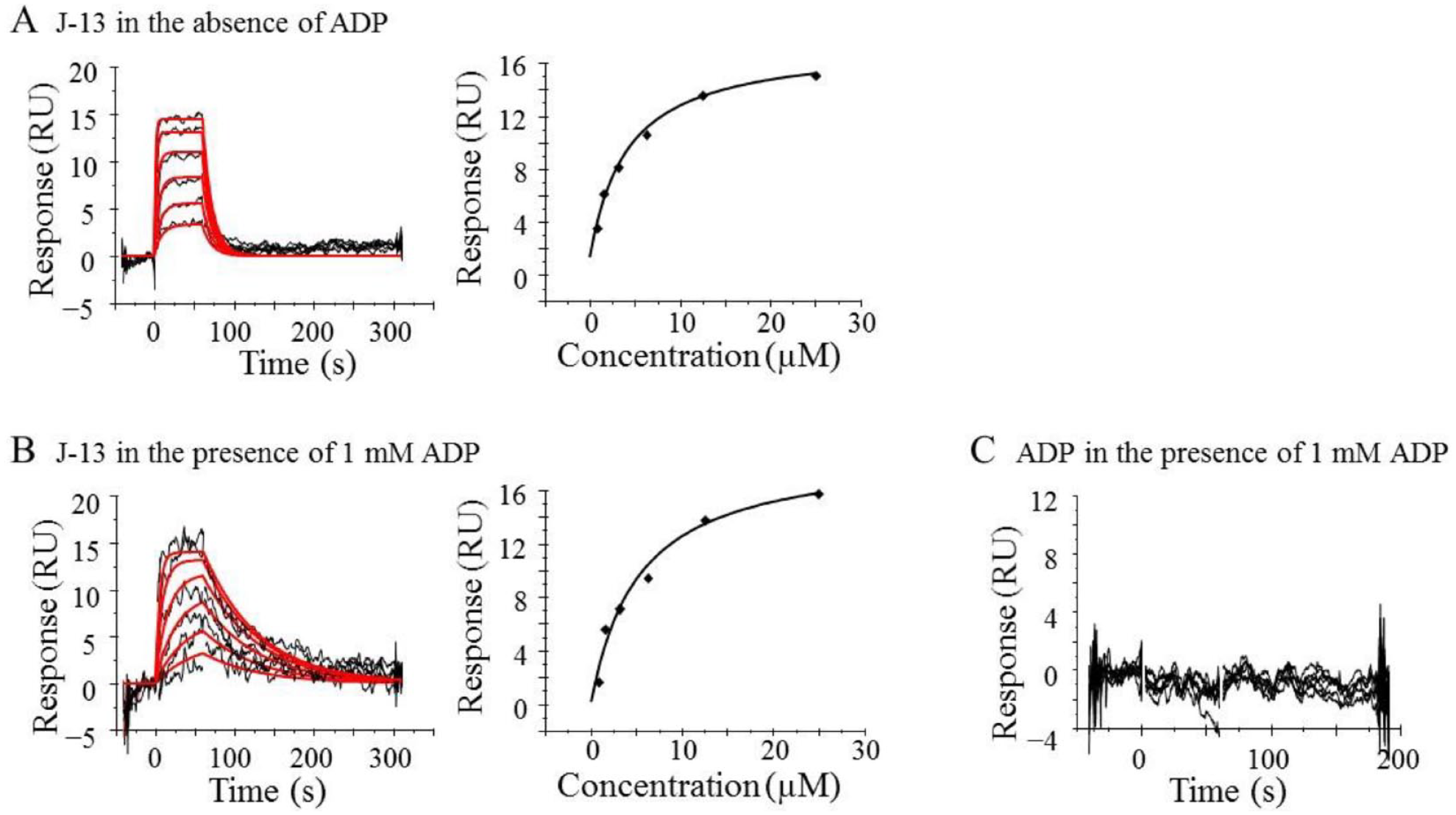
SPR analysis of J-13. (
Kinase Selectivity of ITK Inhibitor
The kinase selectivity of the ITK inhibitor was evaluated by profiling it in the KINOMEscan (Eurofins, San Diego, CA) competitive binding assay. J-13 inhibited only one kinase (NEK3) >65% ( Suppl. Table S2 ), indicating good selectivity.
Discussion
The inactive forms of kinases are attractive targets for designing selective kinase inhibitors. In this study, high-throughput biochemical screening was conducted with a library screen of 390,000 compounds using dephosphorylated ITK-FL, which is activated by autophosphorylation during the assay. We identified 259 compounds with <10 µM against ITK-FL and showing >5-fold selectivity versus FLT3(D835Y). Then, the binding of 105 compounds to dephosphorylated ITK-KD was confirmed by SPR assay. Furthermore, we developed a fluorescent biosensor assay and identified 34 compounds that stabilize inactive conformations of ITK-KD. J-13 was selected as an example that bound to a site other than the ATP pocket and was a highly selective ITK inhibitor against the kinase panel.
Several selective inhibitors preferentially bind to the inactive conformation of kinase, as exemplified by the PERK inhibitor GSK-2656157, 24 CSF1R inhibitor GW-2580, 25 and BTK inhibitor CGI-1746. 26 There are some high-throughput methods for monitoring conformational change. The SHG assay measures the dye-labeled protein’s physical orientation relative to the surface. We previously reported the development of the SHG assay as a method for discriminating inactive and active conformation inhibitors. The SHG assay requires the protein to be immobilized. We selected a fluorescent biosensor approach, allowing the evaluation of the protein–compound interactions in solution, to identify inhibitors that stabilize the inactive conformation of ITK. Our fluorescent biosensor assay was implemented for HTS in the 384-well format, demonstrating robust performance with acceptable Z′ scores. The fluorescent biosensor assay could be applied to a large compound library to identify more novel chemical scaffolds. A limitation of the fluorescent biosensor is the interference from compound fluorescence. Of the 70 compounds that did not reach the set cutoff on the fluorescent biosensor screen, 10 compounds had a higher intrinsic fluorescence intensity than that of the fluorophore. The shift of emission maximum of these compounds might be masked by intrinsic fluorescence. Usage of a red-shifted fluorophore would reduce compound interference. 27
Two types of potent ITK inhibitor have been reported in the literature. The first is the so-called type I inhibitors, which bind to the ATP pocket in the active conformation of ITK. The other class of inhibitors target the inactive conformation of ITK. Examples of this latter type include a selective ITK inhibitor, BMS-509744, and the dual binder at the ATP site and at an allosteric pocket reported by the Pfizer group. To our knowledge, no type II inhibitors that target Asp-Phe-Gly-out (DFG-out) conformation of ITK are known. 28 A comparison of the reported ITK crystal structures of inactive conformation (Protein Data Bank ID codes 3MJ2, 29 4M14, 21 and 4HCU 30 ) with those of active conformation (Protein Data Bank ID codes 4L7S, 31 4MF1, 32 and 4PQN 33 ) revealed that an αC-helix of inactive ITK rotates away from the catalytic center. We focused on movements of the αC-helix as an important determinant of inhibitor selectivity. By labeling the αC-helix of ITK with acrylodan, we were able to develop a kinase biosensor that can detect the binding of inactive conformation inhibitors. ITK-KD Mutant A was also used previously in our SHG assay. In the SHG assay, BMS-509744, 29 Pfizer compound 9, 21 and Pfizer compound 40 30 showed low signal intensities, whereas staurosporine, GSK compound 13, 31 Genentech compound 19, 32 Sanofi US compound, 34 and Genentech GNE-9822 33 showed high signal intensities. These results suggest that these active form inhibiters induced αC-helix-in conformation and oriented the SHG dye on αC-helix parallel to the z axis. In the fluorescent biosensor assay, BMS-509744, Pfizer compound 9, and Pfizer compound 40 shifted the emission maximum in the red direction (bathochromic shift). The fluorescent biosensor assay distinguished between the inactive form and active form inhibitors, as well as the SHG assay. The bathochromic shift in the emission by J-13 was similar to that by known inactive conformation inhibitors. These shifts indicated increased exposure to the polar solvent and are consistent with the rotation of the αC-helix. The fluorescent biosensor methods for reporting conformational changes in proteins should be applicable to other kinases, 14 phosphatases, 35 nuclear receptors, 36 and G-protein-coupled receptors (GPCRs). 15
Hit confirmation and characterization by SPR assay revealed that J-13 undergoes noncompetitive binding with ADP. We believe that J-13 is a specific allosteric inhibitor of ITK because the binding agreed well with the 1:1 binding model, and the experimental Rmax did not exceed the theoretical Rmax (36 RU). Preliminary selectivity assessment results in a broad kinase panel indicated that the compound has good selectivity and a unique chemical structure. Thus, it represents an interesting potential starting point for future medicinal chemistry exploration and further drug discovery. Structural studies of ITK complexed with the compound will facilitate an understanding of the binding mechanism of the compound to the target.
In conclusion, using the combined platform of the biochemical assay, the SPR assay, and the fluorescent biosensor assay with dephosphorylated kinases, we successfully identified inhibitors that stabilize the inactive conformation of ITK. The platform that we present in this paper could potentially provide a powerful tool to identify selective inhibitors of kinases.
Supplemental Material
Supplemental_Material_for_Identification_of_a_New_by_Hantani_et_al – Supplemental material for Identification of a New Inhibitor That Stabilizes Interleukin-2-Inducible T-Cell Kinase in Its Inactive Conformation
Supplemental material, Supplemental_Material_for_Identification_of_a_New_by_Hantani_et_al for Identification of a New Inhibitor That Stabilizes Interleukin-2-Inducible T-Cell Kinase in Its Inactive Conformation by Rie Hantani, Saya Hanawa, Shohei Oie, Kayo Umetani, Toshihiro Sato and Yoshiji Hantani in SLAS Discovery
Footnotes
Acknowledgements
We thank Dr. Gentaroh Suzuki for helpful discussions. We also thank Mr. Kenji Yamanaka and Ms. Aoi Horiike for help with the SPR assay.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.