
Abstract
This work presents the validation of a rapid screening procedure for the catalysis of cytochrome P450 on new chemical entities. The assay is tested on the prototypical, catalytically self-sufficient and soluble cytochrome P450 BM3 from Bacillus megaterium that shares a high degree of homology with mammalian counterparts. The so-called alkali assay developed in our laboratory is validated here also by product formation and molecular modeling on a number of derivatives sharing the molecular scaffold of the 1,2,5-oxadiazole ring, a class of molecules very different from the long-chain fatty acids known to be oxidized by cytochrome P450 BM3. The alkali assay reveals the ability of this cytochrome to oxidize NADPH in the presence of nine out of thirteen 1,2,5-oxadiazole derivatives tested. The enzyme shows high affinity and coupling efficiencies when incubated with four 1,2,5-oxadiazole derivatives. The presence of oxidation products deriving from catalysis was also confirmed by high-performance liquid chromatography (HPLC). Molecular docking suggests that a key factor for the 1,2,5-oxadiazole derivatives to enter the active site and induce catalysis is the presence of the –SO2 moiety bridging the 1,2,5-oxadiazole and phenyl rings. These data indicate that the alkali assay is able to quickly and cheaply detect the recognition of new substrates by cytochrome P450. The assay is not intended to substitute HPLC–mass spectrometry analysis, but it is a preliminary screening that allows elimination of obvious nonsubstrates from the start.
Keywords
Introduction
Cytochrome P450 is a class of enzymes important for the oxidation of endobiotics and xenobiotics. 1 The potential persistence of a new chemical entity (NCE) and its metabolites in the human body is often related to the capability of cytochrome P450 to oxidize the molecule prior to its excretion. In some cases, the oxidation by cytochrome P450 leads to the activation of promutagens and procarcinogens,1,2 making knowledge of the interaction between these enzymes and potential new drugs a necessary step in the drug discovery process. Therefore, a rapid high-throughput screening method able to predict if an NCE is metabolized by cytochrome P450 can provide an important tool to test libraries of molecules as potential new drug candidates.
At present, high-throughput screening of drug-P450 interaction is achieved using a competitive fluorescence assay with fluorogenic substrates.3,4 A more precise and thorough, but also more costly and slow approach, is the use of mass spectrometry. 5 One of the major challenges encountered when screening drugs for metabolic studies is that a great variety of chemicals with distinct molecular structures must be analyzed using a generic method. For a method to achieve high-throughput rates, the measurements of the output signal should be preferably compound independent and the data acquisition must be rapid. This has proved to be challenging for methodologies based on mass spectrometry and has sparked the need for investigating alternative approaches more prone to be high throughput. 5 For this reason also, alternative electrochemical screening has been proposed recently.6–8
Here we report the application of the so-called alkali assay (see Di Nardo et al. 9 and Tsotsou et al. 10 for its full description) on a series of 1,2,5-oxadiazole derivatives. Our laboratory has developed and described a method for screening activity of NAD(P)H-dependent oxidoreductases on perspective substrates with an assay that is widely applicable, independently from the substrate considered. 10 The method can be performed in whole Escherichia coli cells and has been adapted to a microplate format.
Cytochrome P450 BM3 from Bacillus megaterium is used as a prototype enzyme to validate the alkali assay screening. This is a long-chain (C12–C20) fatty acid monooxygenase, 11 but it has also been shown to be able to catalyze the oxidation of molecules structurally different from the linear amphiphilic known substrates, including drugs commonly metabolized by different families of human enzymes. 9 This enzyme offers the advantage of a high expression in E. coli, as well as the solubility and the fusion of the catalytic P450 domain with its reductase, conferring catalytic self-sufficiency and high coupling efficiency. It is therefore a good starting point to test our screening procedure. Although it has to be taken into account that for this enzyme, N-dealkylation reactions also have been described, giving further evidence on its resemblance to the human enzymes, 9 its general reaction can be written as
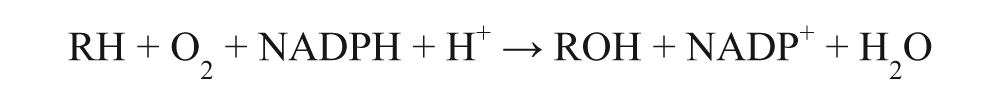
In previous works,9,10 the alkali assay was developed and then used to screen the ability of the bacterial P450 BM3 to turn over commercially available drugs. Here we use the assay to screen NCEs sharing a molecular scaffold, the 1,2,5-oxadiazole, also known as furazan, and the 1,2,5-oxadiazole 2-oxide, known as furoxan. The metabolism of this class of compounds by cytochrome P450 has not been reported before. When the substituents on the ring are different, furoxan derivatives can exist as a pair of position isomers. They are able to release nitric oxide (NO) when incubated in physiological solution in the presence of thiols, 12 and their potential to function as NO prodrugs for the treatment of cardiovascular diseases has been widely demonstrated as they have been shown to have vasodilating properties and to function against platelet aggregation. As the release of NO by furoxans can be modulated by changing the nature of the substituents on the ring, furoxans also have been extensively used for the design of NO-donor molecular hybrids in many therapeutic areas. On the other hand, the corresponding furazans display similar physicochemical properties but are unable to release NO, representing useful tools in comparative studies.
Materials and Methods
Reagents
Arachidonic acid, sodium, and lauric acid were purchased from Sigma-Aldrich (St. Louis, MO). In all experiments, the tetrasodium salt of NADPH (Fluka, Buchs, Switzerland) was used.
Expression and Purification of P450 BM3
Expression of P450 BM3 was carried out in E. coli BL21 DE3 cells transformed with pT7Bm3Z plasmid, carrying the gene of P450 BM3. The purification of P450 BM3 was carried out as described before. 10
Alkali Assay on E. coli Cells Expressing Cytochrome P450 BM3 and on the Purified Protein
The assay is based on the detection and measurement of NADP+ produced from NADPH during the oxidation of the substrate by E. coli BL21 DE3 cells transformed with pT7Bm3Z plasmid expressing cytochrome P450 BM3.
After destruction of the NADPH not consumed during enzymatic turnover by treatment with 0.3 M HCl, 13 NADP+ is measured. It is known that strong alkali act on nicotinamide ribosides to produce a stable fluorescent product that can be used for the quantitative determination of the pyridinium coenzymes. 14 The fluorescent compound may be a secondary condensation product formed from an intermediate pseudobase resulting from the addition of the hydroxyl ion to the nicotinamide nucleus. The pseudobase rapidly rearranges to a stable, highly fluorescent compound having an absorption maximum at 360 nm. 14
The assay was performed in microplates as previously described. 10 A single colony of E. coli BL21 transformed with the plasmid pT7Bm3Z was inoculated into Luria-Bertani (LB)/ampicillin (100 mg mL−1) medium in a microplate well. The expression of P450 BM3 was induced by the addition of 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). After incubation at 37 °C, the cells were harvested and resuspended in 100 mM potassium phosphate buffer (pH 8.0). Aliquots of 1,2,5-oxadiazole derivatives were added periodically to the cells over 30 min, and measurements of alkali product formation were taken at the beginning and after 30 min.
To start the reaction, 1.5 mM NADPH was added to the samples, and an aliquot (typically 70 µL) was transferred in a new microplate. The same volume of 0.3 M HCl was added to stop the reaction and eliminate the remaining NADPH. The final pH in the mixture was 2, and the aliquots were incubated for at least 10 min at room temperature under this pH condition, to ensure complete elimination of the remaining reduced NADPH. An aliquot (typically 80 µL) of the HCl-treated sample was then removed and transferred in a new microplate well with 270 µL of 9 M NaOH, to increase the pH to 14.8. The alkali product was developed under this condition in the dark for at least 2.5 h at room temperature before recording the fluorescence spectrum.
For the assay performed on the purified protein, P450 BM3 (1 µM) was incubated with the 1,2,5-oxadiazole derivatives for 15 min and then with NADPH for 1 h.
Statistical Analysis
To identify positive hits from the screening assay, we performed a statistical analysis of the results. Four replicates for each reaction and each control were considered and averaged and the standard deviation calculated. A t-test allowed the comparison of the average of the fluorescence signals obtained from the reaction mixtures with that resulting from the corresponding control reactions, performed by adding the same volume of buffer/solvent where the substrate was dissolved. The hits where the signals of the reaction mixtures were found to be statistically different from those of the controls were considered positives.
Substrate Binding and NADPH Consumption Studies on Purified Cytochrome P450 BM3
Substrate binding and kinetic studies were performed on a Hewlett-Packard 8452A diode array spectrophotometer (Hewlett-Packard, Palo Alto, CA) equipped with a temperature controller, as described before. 10
Both assays were performed in a 100-mM potassium phosphate buffer (pH 8.0) at 20 °C with a protein concentration in the micromolar range for the substrate binding studies and in the nanomolar range for NADPH consumption studies.
For the substrate binding studies, the 1,2,5-oxadiazole derivatives absorbed themselves in the UV region interfering with the spectrophotometric titration. For this reason, for every molecule tested, a blank titration was performed in the absence of enzyme, and the dissociation constant was calculated from the spectra derived from the subtraction of the background contribution of the 1,2,5-oxadiazole derivatives. The stocks of all 1,2,5-oxadiazole derivatives studied were freshly prepared in N,N-dimethylformamide. During binding or kinetic studies, the N,N-dimethylformamide concentration added to the protein solution did not exceed 1.5% v/v. The solubility of the 1,2,5-oxadiazole derivatives tested in the assay buffer was low (with the exception of
NADPH consumption studies were carried out by monitoring the decrease of signal at 340 nm due to NADPH oxidation by purified cytochrome P450 BM3. The highest concentration of 1,2,5-oxadiazole derivatives applied was as follows: 310 µM for
Uncoupling Assays and High-Performance Liquid Chromatography Analysis of 1,2,5-Oxadiazole Derivatives Metabolites
Uncoupling from substrate oxidation was determined as described before. 10 Briefly, the ratio of oxygen to NADPH consumed was calculated by measuring the oxygen consumption at 24 °C with a Clark-type electrode calibrated by taking the concentration of dissolved oxygen in air-saturated water as 262.5 µM. The P450 BM3 concentration used was in the nanomolar range, the substrate was in excess with respect to the NADPH concentration, and the reaction was performed in a 100-mM phosphate buffer (pH 8.0).
The uncoupling of the reducing equivalents was also monitored by measuring the hydrogen peroxide production during substrate turnover by the horseradish peroxidase (HRP)–ABTS assay. The reaction was performed in 100 mM potassium phosphate buffer (pH 8.0) with an enzyme concentration in the nanomolar range, 150 µM NADPH, and a substrate concentration in excess in comparison to that of NADPH.
For the chromatographic detection of the 1,2,5-oxadiazole derivatives metabolites, 0.65 µM purified P450 BM3 was incubated with 250 µM compound
Docking of the 1,2,5-Oxadiazole Derivatives into the P450 BM3 Active Site
All molecular modeling studies were performed on a Silicon Graphics Indigo II Impact 10000 workstation (SGI, Fremont, CA). The software programs used were the Insight II 95.0 (Biosym/MSI, San Diego, CA) and AutoDock 3.0.3 (The Scripps Research Institute, La Jolla, CA). The crystal structure of the palmitoleate-bound P450 BM3 15 (PDB ID: 1FAG) was used in all cases.
The steps were as follows: (1) The PDB file records of the palmitoleate-bound P450 BM3 structure (1FAG) were deprived of the coordinates of the substrate. (2) Using the Biopolymer module within the Insight II package (Biosym/MSI), bond orders were adjusted and hydrogen atoms were added to the protein to fill unfilled valences on atoms, according to the consistent-valence force field (CVFF) residue library. Hydrogens were assigned at pH 8.0, simulating the pH at which binding experiments were performed. Atom potential types, plus partial and formal charges, were assigned based on the CVFF. Accepting the previously assigned formal charges, the partial charges and atoms potentials were reassigned using the extensible systematic force field (ESFF). ESFF was chosen because it includes parameters for potentials of transition metals in contrast to all other force fields supported by Insight II. (3) Next, the 3D models of the ligands were generated using the Builder module within Insight II. A 2D sketch of the ligand was first produced, which was then converted to a 3D molecule using the Molbuilder/2D•3D command. Atom potentials and partial and total charges were assigned using the ESFF before the model was refined by minimization using molecular mechanics functions and employing default values for the program parameters (Molbuilder/optimize command). (4) AutoDock within the AutoDock package was then used to assign atomic solvation parameters to the macromolecule. (5) A grid map of van der Waals terms, hydrogen bonding, and electrostatic interactions for each atom of the ligand was generated using the AutoGrid program within AutoDock. The grid dimensions were set to 60 × 60 × 60 grid points (spacing between points 0.375 Å). The grid was centered on the Phe87 phenol ring and was sufficiently extensive to cover the active site of P450 BM3. (6) A Lamarkian genetic algorithm was used to predict the 10 ligand positions into the active site with the lowest energy. A population of 200 individuals (random ligand conformations in random orientations and at random translations) was used, whereas default values were employed for the remaining parameters involved in the docking calculation. The calculation was run using the AutoDock program within the AutoDock package.
Results and Discussion
Screening of P450 BM3 Activity toward 1,2,5-Oxadiazole Derivatives
Thirteen 1,2,5-oxadiazole derivatives were screened for P450 BM3 activity, and their molecular structure is shown in Figure 1A . The alkali assay is based on the measurement of the alkali product of NADP+ deriving from the oxidation of NADPH coupled to enzymatic activity. The alkali product of NADP+ is developed when the enzymatic reaction is stopped and the pH is raised to 14.8 by the addition of NaOH. The alkali product absorbs at 360 nm, and upon excitation at this wavelength, it is fluorescent with a maximum at 414 nm, 10 making the presence of NADP+ detectable also at very low concentrations. A control, consisting of cells in phosphate buffer added with the same volume of the solvent used to dissolve the 1,2,5-oxadiazole derivatives but without the NCE, was run for each sample. The results of the alkali assay were statistically analyzed for the identification of positive and negative hits. The signals generated by the enzyme reaction (including standard deviation) and those of the controls were compared.
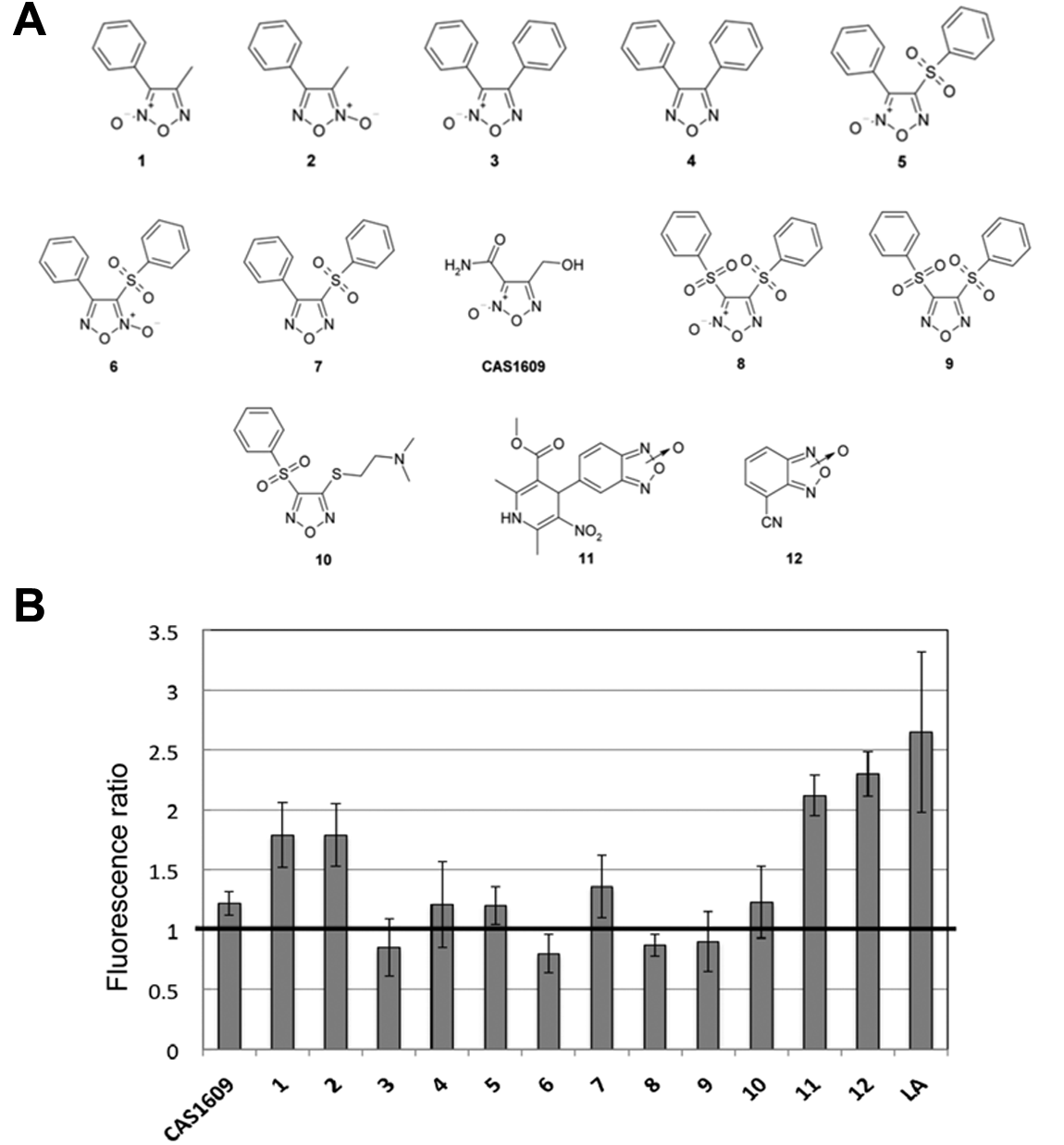
(
For compounds
For compounds
The alkali assay was then repeated on the purified enzyme to avoid false positives due to the potential of these molecules to release NO in E. coli cells and therefore to be metabolized by P450 BM3 after this modification. The results obtained showed an increased signal at 414 nm for the nine derivatives previously selected by the whole-cell assay when compared with the control.
These compounds were therefore selected for further studies in terms of binding and catalysis using the purified enzyme.
Binding of 1,2,5-Oxadiazole Derivatives by P450 BM3 and NADPH Consumption Studies
UV-vis absorbance spectroscopy was used to study the binding of 1,2,5-oxadiazole derivatives to the active site of purified P450 BM3, as the shift of the Soret peak from 418 to 387 nm due to the low-to-high spin transition of heme iron is indicative of substrate binding. This shift is typical of the so-called type I spectra derived from the substrate-induced displacement of the water molecule present as a sixth iron ligand. As compounds
Titrations with increasing amounts of substrates were performed, and the binding constants (KD) were calculated (
Table 1
). The KD values were on the same order of magnitude of those measured for fatty acids (
Table 1
). The fact that furoxans
Binding, Kinetic, and Uncoupling Parameters for P450 BM3 and 1,2,5-Oxadiazole Derivatives
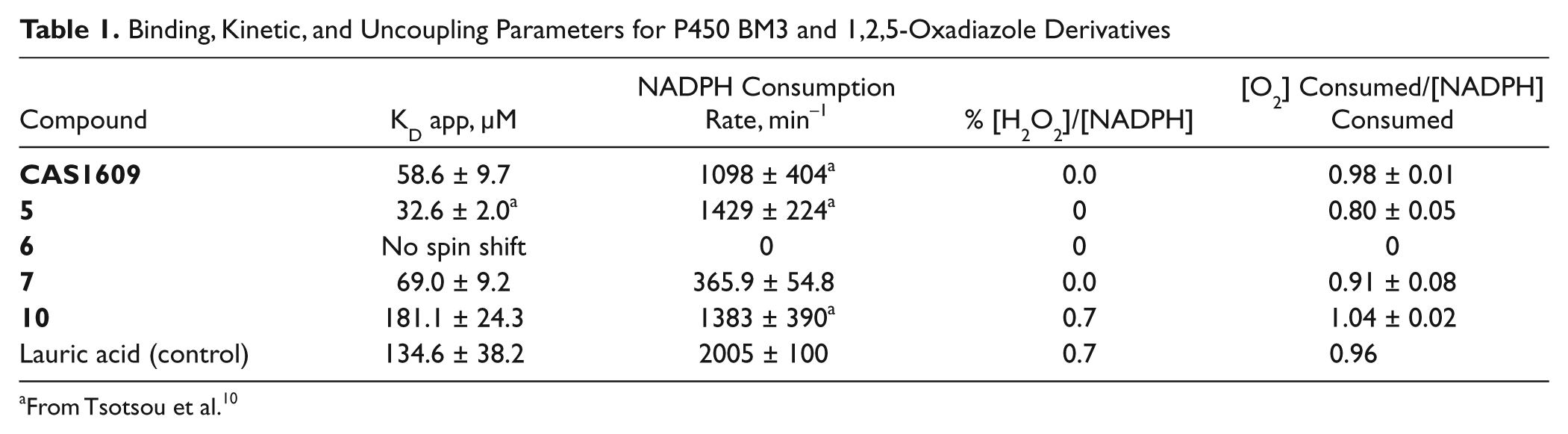
From Tsotsou et al. 10
The rates of NADPH consumption previously determined
10
and the value measured in this work for compound
Docking of 1,2,5-Oxadiazole Derivatives into P450 BM3 Active Site
AutoDock was used to study the binding of the thirteen 1,2,5-oxadiazole derivatives to the P450 BM3 active site (
Fig. 2
). Palmitoleic acid was used as a positive control, and the position of the complex was consistent with the crystal structure data published.
14
All the compounds positive in the alkali assay, such as compound
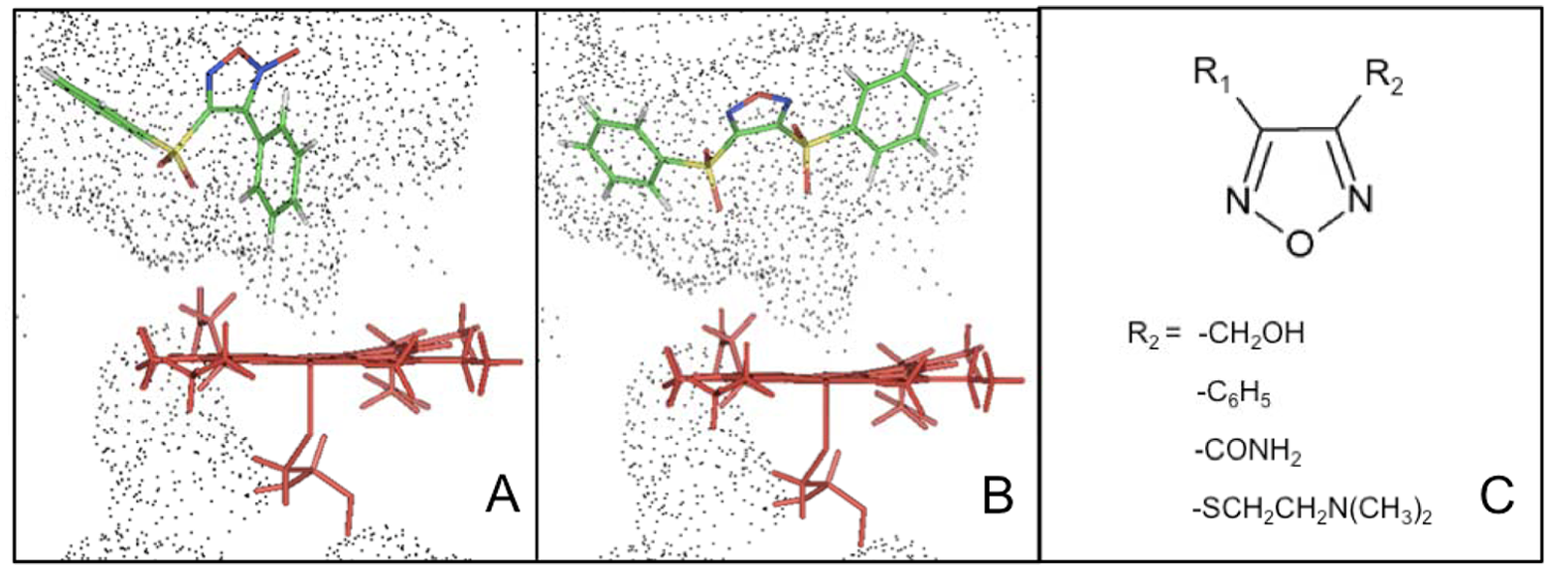
Rigid docking of (
Analysis of the docking data suggests that the substituents at the 1,2,5-oxadiazole ring, rather than the heterocycle itself, could represent a pattern for substrate recognition by P450 BM3. In particular, the simultaneous presence of two –SO2 moieties bridging the heterocycle ring with the phenyl rings (compounds
Measurement of Uncoupling
The possibility that NADPH is oxidized by uncoupled reactions was then checked by measuring the hydrogen peroxide formed during the turnover and by calculating the ratio between the oxygen consumed and the NADPH oxidized. Hydrogen peroxide formation and oxygen consumption, in the presence of the sub-stoichiometric concentration of NADPH relative to the 1,2,5-oxadiazole derivative substrates, were determined for the positive ones with purified P450 BM3.
The rate of oxygen consumption in the samples containing the 1,2,5-oxadiazole derivatives varied from 28.8 to 4.4 µM oxygen consumed/min/µM P450 BM3 for
The presence of a reaction product was also checked for compound
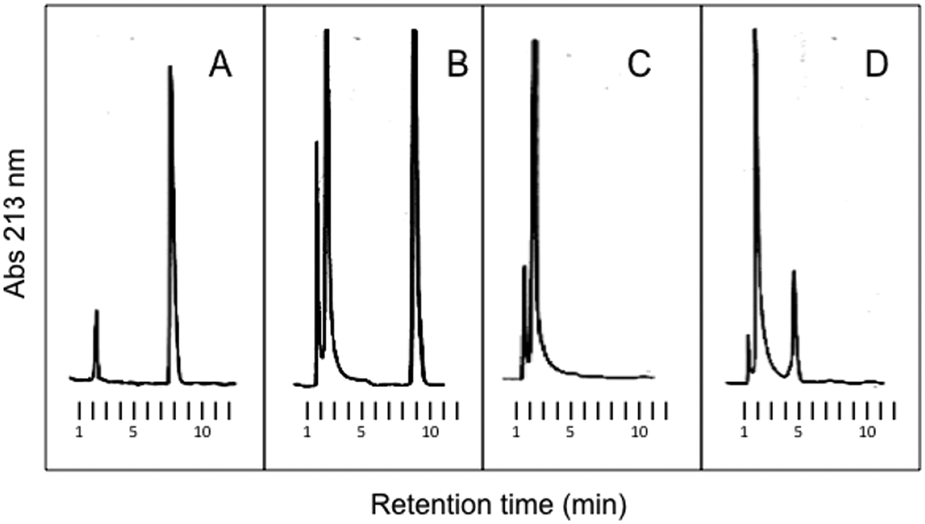
Reverse-phase high-performance liquid chromatography (HPLC) separation of metabolites formed during incubation of P450 BM3 (0.65 µM) with 12 (500 µM) and NADPH (700 µM). Reaction buffer, 100 mM KPi, pH 8.0; reaction temperature, 25 °C; incubation time, 3.5 min. In
In conclusion, this study demonstrates that it is possible, out of a series of similar compounds that might have the same effectiveness from a pharmacological point of view, to identify the ones that can be metabolized by cytochrome P450 by using a fast screening method to rapidly identify the positive ones. According to the statistical analysis performed, in a small library of chemically similar compounds, two false negatives were found, representing 15.4% of the compounds tested. Computational methods can also offer a tool to establish patterns for the optimization of a new drug design.
The alkali assay presents the advantages of low cost and time required, and it is independent of the nature of the substrate. It is not intended to substitute HPLC–mass spectrometry (MS) analysis, but it is a preliminary screening that could eliminate obvious nonsubstrates from the start. Once performed, the presence of metabolites from the molecules positive to the assay can be checked by liquid chromatography (LC) or LC-MS analysis since false positive may arise from uncoupling. Here the alkali assay indicates P450 BM3 activity toward molecules sharing the 1,2,5-oxadiazole ring. The results indicate that the assay is able to predict the interaction between the enzyme and the 1,2,5-oxadiazole derivatives, regardless of the presence or the position in the ring of the N-oxide moiety.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.