
Abstract
Poly(ADP-ribose) polymerase 3 (PARP3) is an important member of the PARP family and shares high structural similarities with both PARP1 and PARP2. The biological roles of PARP3 are currently under investigation; however, several key reports indicate the integral roles of PARP3 in DNA damage repair, and thus it has been investigated as a novel target in oncology. It is clear that the identification of selective PARP3 inhibitors would further advance the understanding of the biological roles of PARP3. Herein, we describe a modified PARP3 screening assay using biotinylated NAD+ as the specialized substrate. This method relies on the activity of PARP3 to transfer the biotinylated NAD+ onto a histone protein to form ADP-ribosylated histone. The biotin label on this histone protein is then detected and quantifies the intrinsic enzymatic activity of PARP3. We optimized the assay with respect to the histone, NAD+/biotinylated NAD+ mixture, DNA, and PARP3. Our developed screening system was then validated with a reported selective PARP3 inhibitor, ME0328, as well as utilizing five other clinically available PARP1/2 inhibitors. We demonstrated that our assay system was sensitive, efficient, and economical, and we reason that it could be useful for the development of highly selective PARP3 inhibitors in the future.
Keywords
Introduction
The poly(ADP-ribose) polymerase (PARP) superfamily contains 17 members that transfer ADP-ribose groups to target proteins and thereby affect various nuclear and cytoplasmic processes. 1 PARP3, also called ADP-ribosyltransferase 3 (ARTD3), is a newly identified member of the family.2,3 The structure of PARP3 is highly conserved across the family, most notably with PARP1 and PARP2; however, the biochemical function of PARP3 remains unclear. PARP3 acts as a mono-ADP-ribosylase4–6 that is substantially activated in the presence of nicked oligonucleotide substrates. PARP3 not only ADP-ribosylates itself and histones H1 and H2B, but also binds to histones H3C and H2BE. 7 PARP3 can interact with and activate PARP1 in the absence of DNA, stabilize the mitotic spindle, and maintain telomere integrity via regulation of the mitotic component’s nuclear mitotic apparatus protein (NuMA) and tankyrase 1.8,9 It is also known to cooperate in complex DNA repair, including the recruitment of mediators of classical nonhomologous end joining (c-NHEJ). It has also been demonstrated through knockout studies that PARP3 delays the repair of double-strand breaks. 10 Additionally, it has been reported that PARP3 is a promoter of chromosomal rearrangements and regulates the formation of G quadruplex DNA in response to DNA damage. 11 Moreover, PARP3 has been implicated in the development of various cancers, such as glioblastoma and breast cancer.12–14 Recently, it has been shown that small-molecule inhibition of PARP3 suppressed tumor growth in vivo and conferred cell radioresistance in glioblastoma. Thus, PARP3 is considered a promising therapeutic target in oncology; however, the selective inhibition of PARP3 is required to deconvolute the biological role of PARP3. Additionally, distinguishing the therapeutic benefit and comparing the activity for pan-PARP PARP3 inhibitors would be highly beneficial for the research community.
Targeting PARP1 and PARP2 via small-molecule pharmacological intervention has been successfully used in the treatment of certain cancers. For example, olaparib (AZD2281) was the first approved PARP1/2 inhibitor used for the treatment of ovarian cancer. 15 Other PARP1/2 inhibitors, such as niraparib and rucaparib, were subsequently approved for clinical cancer therapy.16,17 Intriguingly, during the development of the next-generation PARP1/2 inhibitor, AZD2461, it was demonstrated that AZD2461 had significant advantages compared with olaparib due to its selective inhibitory effects on PARP3. 18 Considering the important roles of PARP3 in DNA repair and tumorigenesis, it is important to evaluate the potential effect of PARP3 inhibition induced by the most widely used PARP inhibitors in the clinic.
In line with its importance as a biological target, several assays have been established for PARP3 inhibitor screening, such as thermal shift assays, fluorescence polarization, and surface plasmon resonance binding assays.19–23 Although powerful, these assays only observed the interaction between protein and compound but did not have a direct readout detailing enzymatic inhibition. However, recently an elegant enzyme-linked assay via PARP3 auto-ADP-ribosylation was developed and applied in the assessment of small-molecule PARP inhibitors. 24
To develop an economic platform for PARP3 enzymatic detection with high sensitivity and accuracy, we established an enzyme-linked, histone ADP-ribosylation, biotin-labeled NAD+ assay, which was designed and optimized for PARP3 inhibitor screening. Histone was employed as the receptor of the biotin-linked mono-ADP-ribose (MAR) or short poly-ADP-ribose (PAR) derived from the substance NAD+ catalyzed by PARP3. We subsequently evaluated the utility of our assay system through the profiling of known and commercially available PARP1/2 inhibitors against PARP3.
Materials and Methods
Reagents
NAD+ and biotinylated NAD+ were purchased from Roche (Basel, Switzerland) and Trevigen (Gaithersburg, MD), respectively. Horseradish peroxidase (HRP)-streptavidin, sheared DNA (cat. D4522), and 3,3′,5,5′-tetramethylbenzidine (TMB) were obtained from Sigma-Aldrich (St. Louis, MO). Histone (cat. LS002544) is from Worthington Biochemical Corporation (Lakewood, NJ). PARP1/2 inhibitors, including olaparib, veliparib, rucaparib, niraparib, talazoparib, and ME0328, were purchased from Selleck Chemicals (Houston, TX). PARP3 antibody was purchased from Abcam. Nicked 5′-phosphorylated (5′P) DNA (5-pGCTGAGCTTCTGGTGAAGCTCAGCTCGCGGCAGCTGGTGCTGCCGCGA-3) was synthesized by Invitrogen (Waltham, MA).
Expression and Purification of hPARP3
The human PARP3 (hPARP3) isoform A (NCBI reference sequence: NM_001003931.3) was amplified by reverse transcription (RT)-PCR with total RNA from A172 cells and cloned into pET-28a. Plasmid pET28a-hPARP3 was transformed into E. coli BL21 (DE3). His-tagged hPARP3 was expressed after induction with 0.5 mM isopropyl-b-
Chemical Quantification of NAD+ Assay
The assay was performed as previously described.25,26 Briefly, PARP3, NAD+, DNA, temperature, and time were titrated to optimize the assay condition for PARP3 activity. First, 30 μL of NAD+ solution (concentrations range from 4 to 256 μM), 5 μL of sheared DNA (concentrations range from 50 to 1600 μg/mL), and 15 μL of hPARP3 (concentrations range from 0.8 to 8 μM) were added to a black 96-well plate to form a 50 μL reaction system. Next, the plate was incubated at different temperatures (ranging from 4 to 45 °C) and different periods (ranging from 10 to 210 min). After adding 20 μL of 2 M KOH and 20 μL of 20% acetophenone, the plate was incubated at 4 °C for 10 min. Finally, 90 μL of 88% formic acid was added and the plate was incubated in an oven at 110 °C for 5 min. The plate was cooled and immediately read on an EnSpire multimode plate reader (PerkinElmer, Waltham, MA) with an excitation of 360 nm and emission of 445 nm (see Fig. 2A–E ).
Enzyme-Linked, Biotin-Labeled NAD+ Assay
The concentration of histone, DNA, biotinylated NAD+, hPARP3, and the ratio of biotinylated NAD+ to total NAD+ were optimized in a concentration-dependent manner. Briefly, a clear flat-bottom 96-well plate was coated with 100 μL of histone in assay buffer (50 mM Tris-HCl, 2 mM MgCl2, pH 8.0) at 4 °C overnight. After washing with phosphate-buffered saline (PBS) containing 0.1% Triton X-100 four times, 30 μL of NAD+ containing biotinylated NAD+ (final concentrations range from 6.25 to 400 μM), 5 μL of sheared DNA (final concentrations range from 0.078 to 5 μg/mL) or nicked 5′P DNA, and 10 μL of hPARP3 (final concentrations range from 8 to 256 nM) were add to each well (total volume 50 μL) and incubated at 25 °C for 1 h. HRP-labeled streptavidin (1:500 dilution) was added to the antibody buffer (0.1 M PBS, 0.2% Tween 20) after four sequential washes and incubated at 25 °C for 30 min. After a further four sequential washes, 50 μL of TMB substrate was added to each well and incubated at 25 °C for 15 min. The stop solution (2 N H2SO4) was then added to each well, and the absorbance was measured at 450 nm.
Determination of IC50 Values of Different Compounds for PARP3
A clear flat-bottom 96-well plate was coated with 100 μL of histone (10 μg/mL) in assay buffer (50 mM Tris-HCl, 2 mM MgCl2, pH 8.0) at 4 °C overnight. After four sequential washes, 30 μL of mixed NAD+ (biotinylated NAD+/total NAD+, 1:5, final concentration of 100 μM), 5 μL of nicked 5′P DNA (400 ng/mL), 5 μL of different concentrations of compound that diluted in assay buffer containing 0.1% DMSO (0.1 nM to 100 μM), and 10 μL of hPARP3 (32 nM) were add to each well (total volume 50 μL) and incubated at 25 °C for 1 h. Antibody buffer was added to HRP-labeled streptavidin after four sequential washes at 25 °C for 30 min. After another four washes, 50 μL of TMB substrate was added to each well and incubated at 25 °C for 15 min. The stop solution was added to each well, and the absorbance was measured at 450 nm.
Determination of IC50 Values of Different Compounds for PARP1 and PARP2
The inhibitory activity of PARP1 and PARP2 was measured essentially as described before. 25 Briefly, 100 μL of histone (10 μg/mL) in assay buffer was coated in a clear flat-bottom 96-well plate at 4 °C overnight. After a washing step, 35 μL of NAD+ (25 pmol NAD+ and 25 pmol biotin-labeled NAD+), 10 μL of PARP1 or PARP2 (0.05 unit), and 5 μL of compound (0.01–100 nM) were added and incubated at 25 °C for 1 h. Then the PAR product was determined as above. IC50 values of compounds were calculated as described below.
Data Analysis
All experiments were performed in triplicate and repeated at least three times independently. The results were analyzed by GraphPad Prism 7 (GraphPad Software, La Jolla, CA).
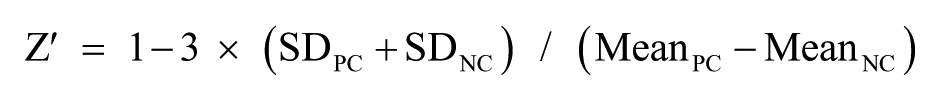
S/B = Mean signal/Mean background
CV (%) = SD/Mean × 100
NAD+ conversion (%) = 100 – (RFUtest – RFUNC)/(RFUPC – RFUNC) × 100
Inhibition (%) = (ODPC – ODtest)/(ODPC – ODNC) × 100
where PC is the positive control, NC the negative control, SD the standard deviation, CV the coefficient of variation, S/B the signal-to-background ratio, RFU the relative fluorescence units, and OD the optical density.
Km values were calculated by Michaelis–Menten. IC50 values were calculated by three-parameter mathematical model of nonlinear regression.
Results
Detection of hPARP3 Enzymatic Activity
Based on our previous work describing the development of a PARP1/2 inhibitor screening assay, 25 we evaluated the enzymatic activity of recombinant hPARP3 via chemical quantification of NAD+. As shown in Figure 1A , NAD+ was converted into a fluorescent compound via a two-step chemical process. In the presence of PARP3, NAD+ was catalytically transferred to nicotinamide and ADP-ribose monomers or polymers without fluorescence, resulting in less NAD+ converted into the fluorescent compound. Thus, the enzymatic activity of PARP3 is directly related to the conversion of NAD+. We first titrated NAD+ concentrations to determine the linear range of the substance ( Fig. 2A ) and experimental Km values ( Fig. 2B ), and we observed that the amount of fluorescent product decreased with increasing PARP3 concentrations ( Fig. 2C ). We also observed that the signal plateaued when the concentration of NAD+ was higher than 50 μM, regardless of the concentration of PARP3 used. Using a fixed NAD+ concentration of 256 μM, NAD+ conversion showed a linear increase in conjunction with PARP3 ( Fig. 2D ). Next, we optimized the concentration of the sensor DNA. The NAD+ conversion began to reach the maximum value with a DNA concentration of 400 μg/mL ( Fig. 2E ). Meanwhile, we observed that nicked 5′P DNA showed a much higher activity in the assay than sheared DNA ( Fig. 2F ). Finally, we explored the optimal reaction temperature and time. After some experimentation, we found that 25 °C was the optimal reaction temperature with a reaction time of 1 h being sufficient for the reaction to reach equilibrium ( Fig. 2G , H ).
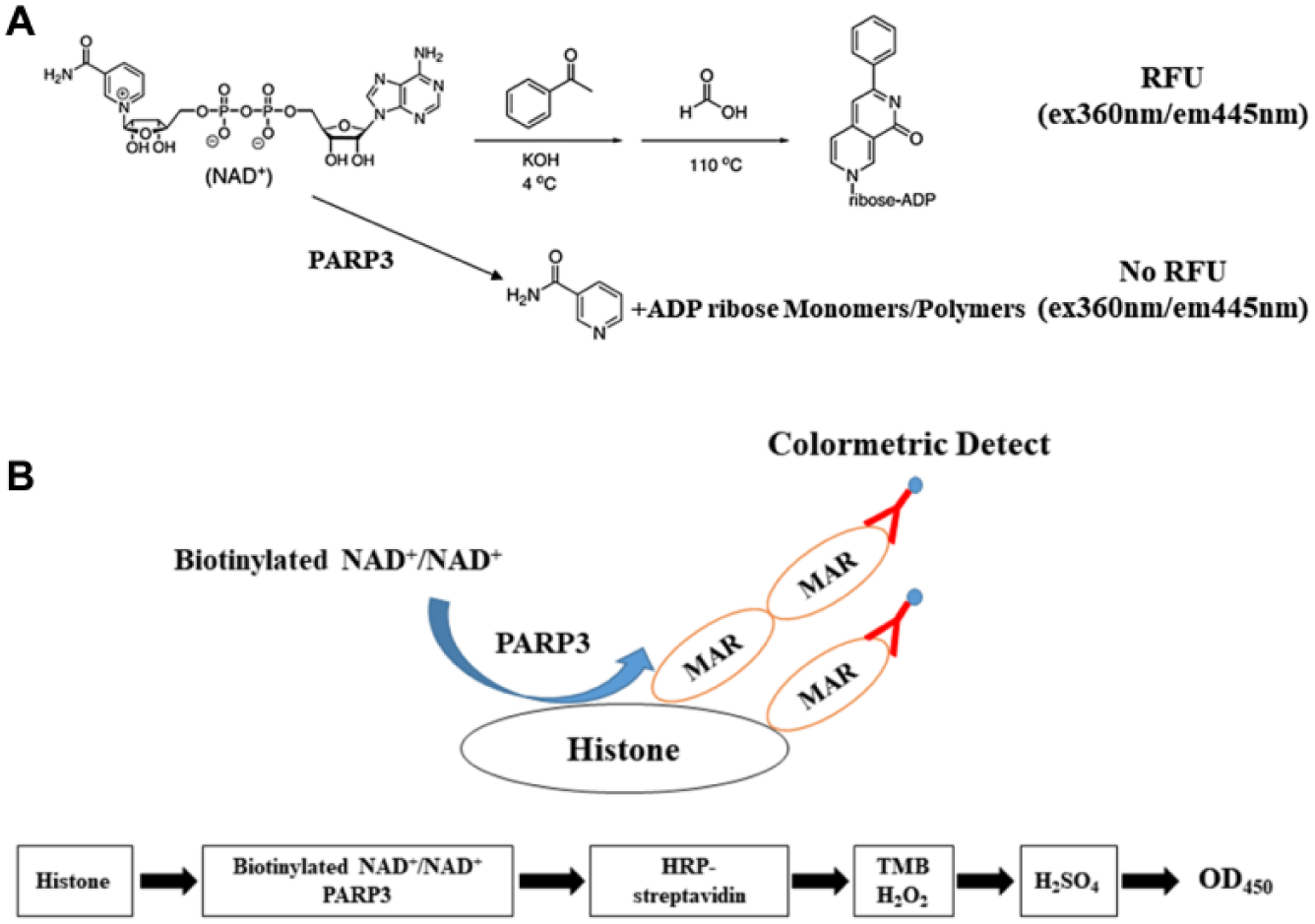
Flowchart of platform for PARP3 inhibition. (
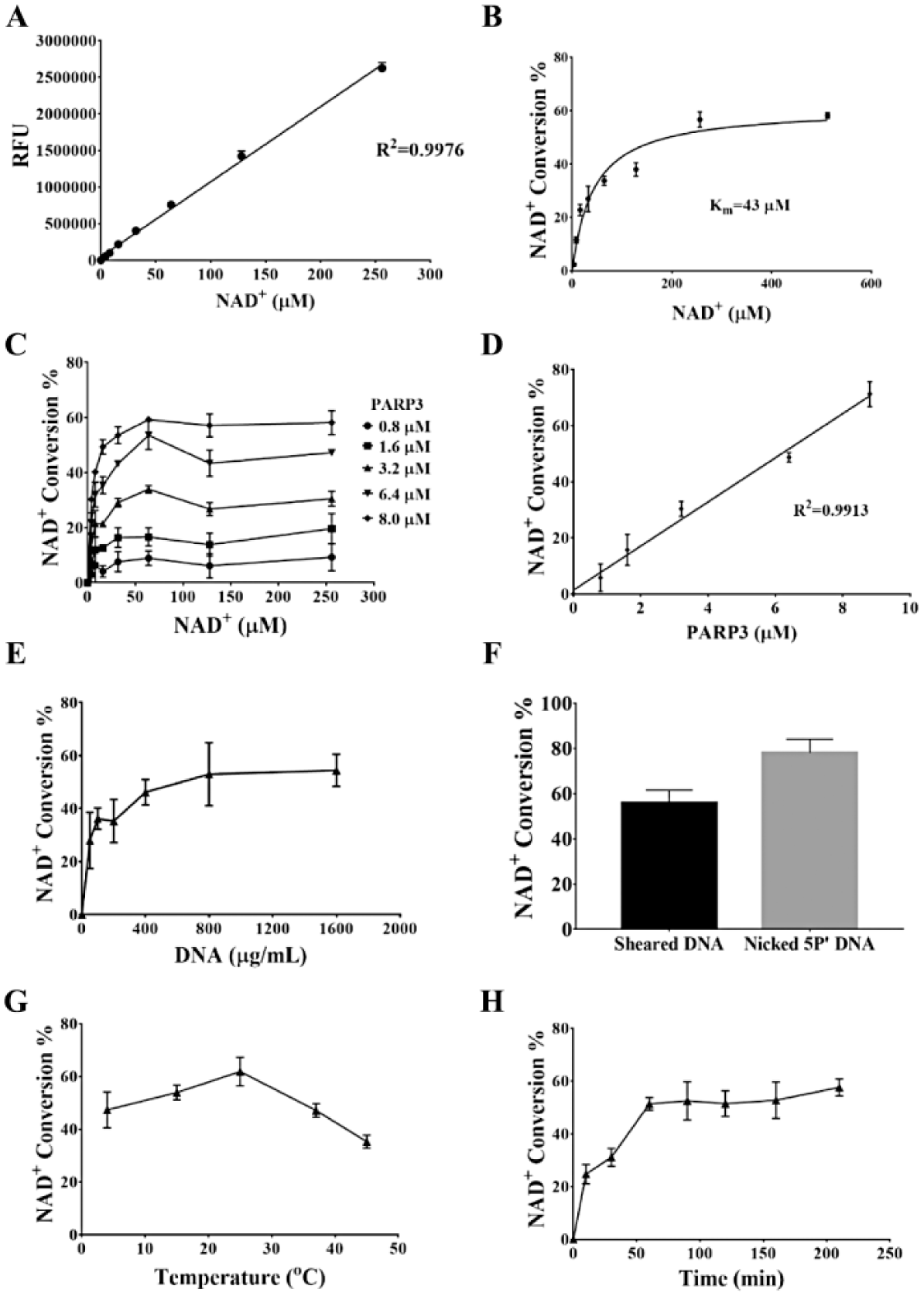
Detection and optimization of PARP3 enzymatic activity assay via chemical quantification of NAD+. (
Development of hPARP3 ELISA
Although the aforementioned chemical assay showed robust and sensitive detection and quantification of PARP3 activity, and therefore could potentially be used for PARP3 inhibitor screening, the assay required large amounts of recombinant PARP3 and sheared DNA, which was impractical for large-scale screening. Therefore, we established and optimized an hPARP3 enzyme-linked immunosorbent assay (ELISA) using an enzyme-linked, biotin-labeled NAD+ histone ADP-ribosylation quantification method ( Fig. 1B ). We first detected the Km value for NAD+ in this assay ( Fig. 3A ) and subsequently determined the optimal concentrations for histone (10 μg/mL), sheared DNA (2 μg/mL), and biotinylated NAD+ (100 μM) using a fixed concentration of 3.2 nM hPARP3 ( Fig. 3B–D ). The signal reached a steady plateau at 100 μM NAD+, which also ensured that there was sufficient quantity of NAD+ in the assay. Subsequently, to ensure that the assay was as economical as possible, we optimized the ratio of biotinylated NAD+ to total (biotinylated and nonbiotinylated) NAD+. Our data suggested that when the total concentration of NAD+ was set at 100 μM, the ideal ratio of biotinylated NAD+ to total NAD+ was 1:5 ( Fig. 3E ). Furthermore, we compared the activator nicked 5′P DNA with sheared DNA. As shown in Figure 3F , 400 ng/mL of nicked 5′P DNA activated the reaction more easily than sheared DNA at a concentration of 2 μg/mL.
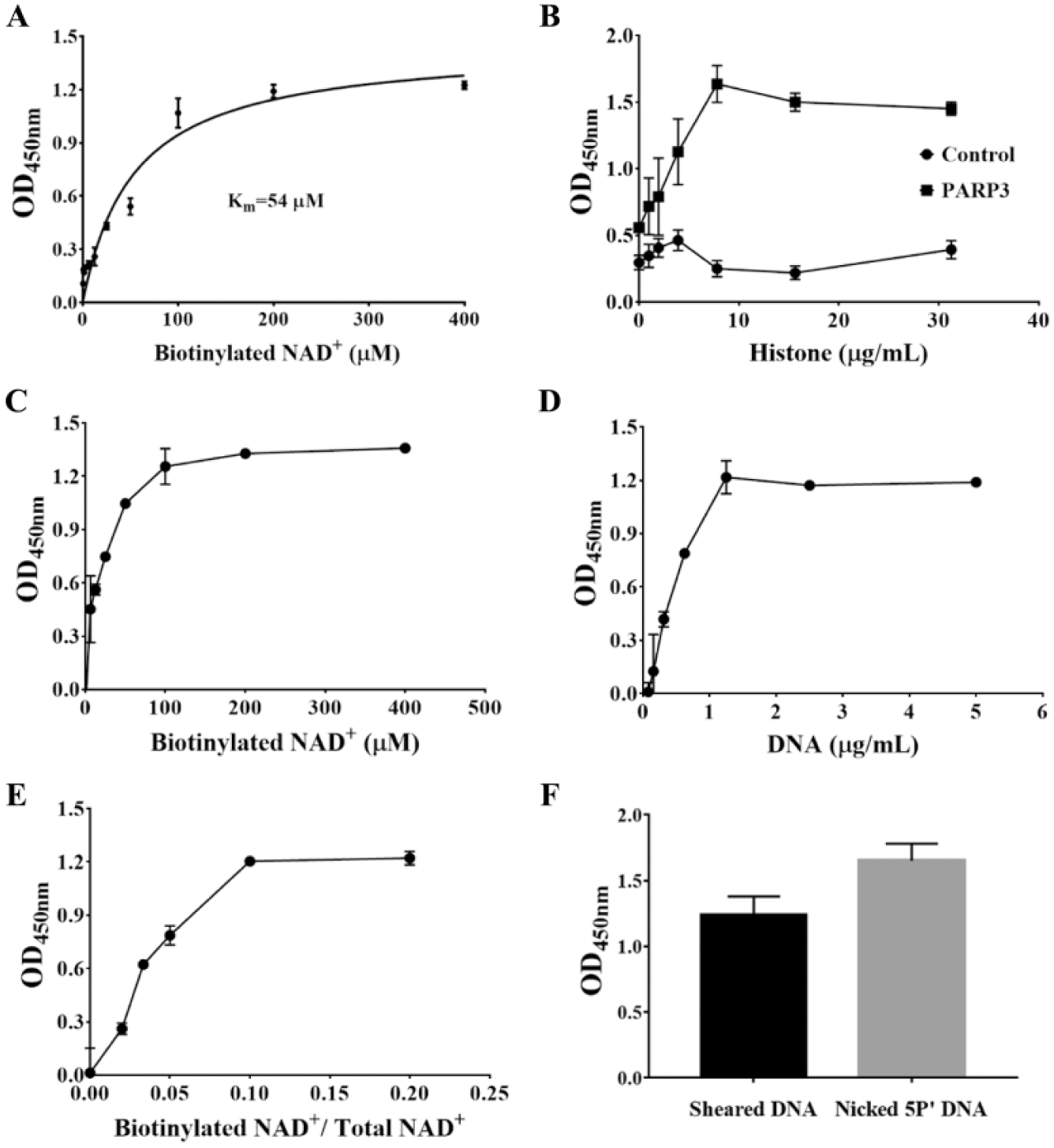
Development of ELISA for PARP3 inhibition. The assay was performed at 25 °C with 32 nM hPARP3 in a 50 μL volume. (
Finally, we aimed to optimize the concentration of hPARP3, with our data showing that the OD value reached the maximum plateau beyond the linear range of the reaction ( Fig. 4A ), and we therefore chose a concentration of 3.2 nM hPARP3 for the assay. Additionally, based on the Z′ factor, the S/B, and the CV of the assay, we can confidently say that our assay system is suitable for high-quality PARP3 inhibitor screening ( Fig. 4B ).
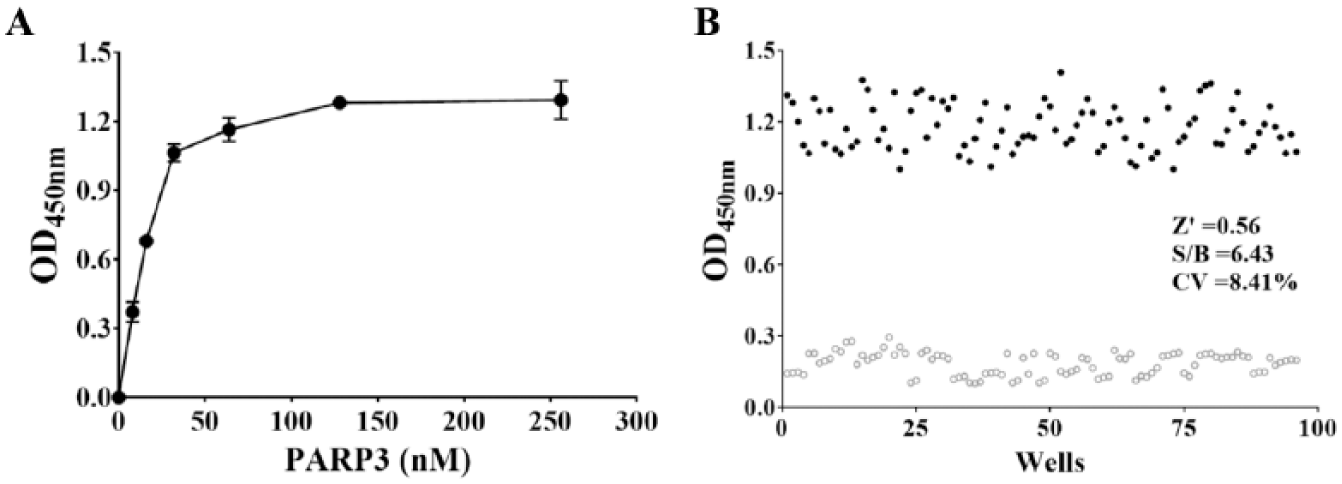
Optimization of hPARP3 concentration. (
Validation of hPARP3 ELISA
In order to validate our screening platform, we initially tested the known PARP3 inhibitor, ME0328, in a dose-dependent manner. Our data confirmed that ME0328 was an efficient PARP3 inhibitor with an IC50 value of 180 nM ( Fig. 5 ), which was lower than the previously reported value (0.89 μM), 20 suggesting that the assay was highly sensitive toward PARP3 inhibition. The inhibition of PARP1 and PARP2 by ME0328 was also detected and quantified via a similar assay with IC50 values of ME0328 on PARP1 and PARP2 of 1.25 and 1.26 μM, respectively.
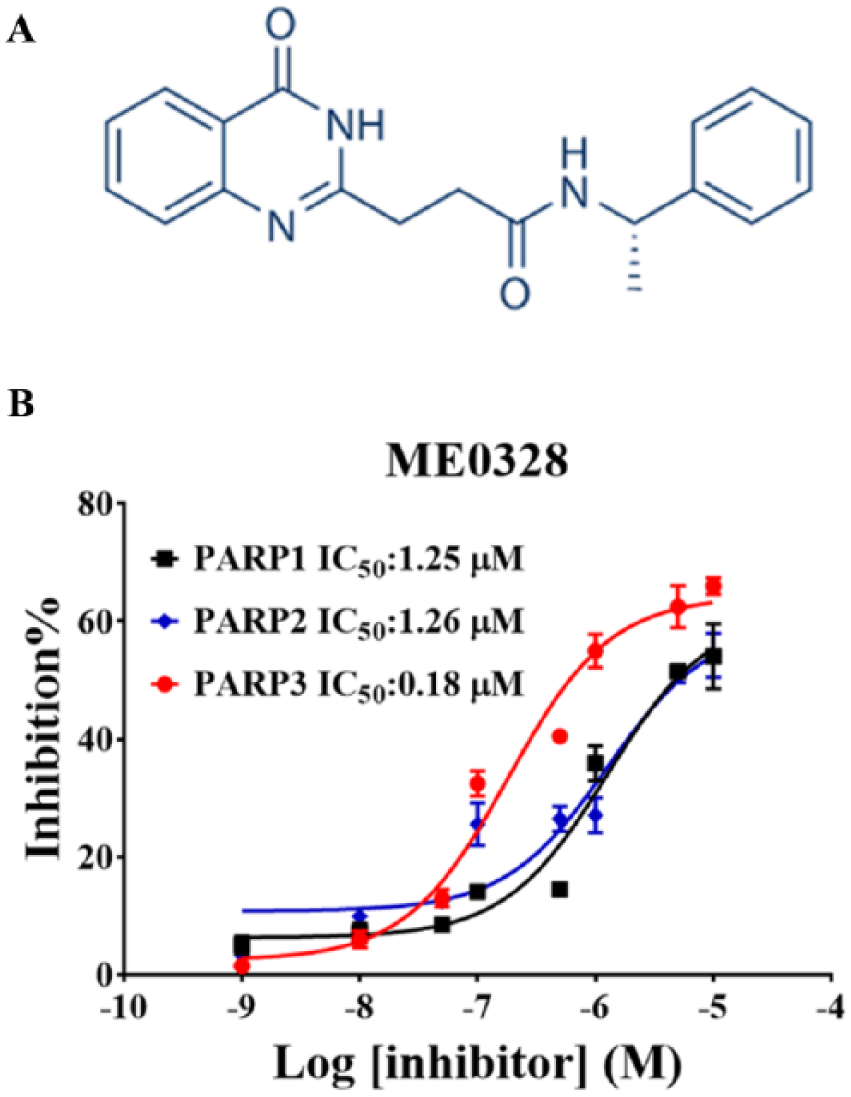
Validation of ELISA-based assay for PARP3 inhibition. (
Compound Screening
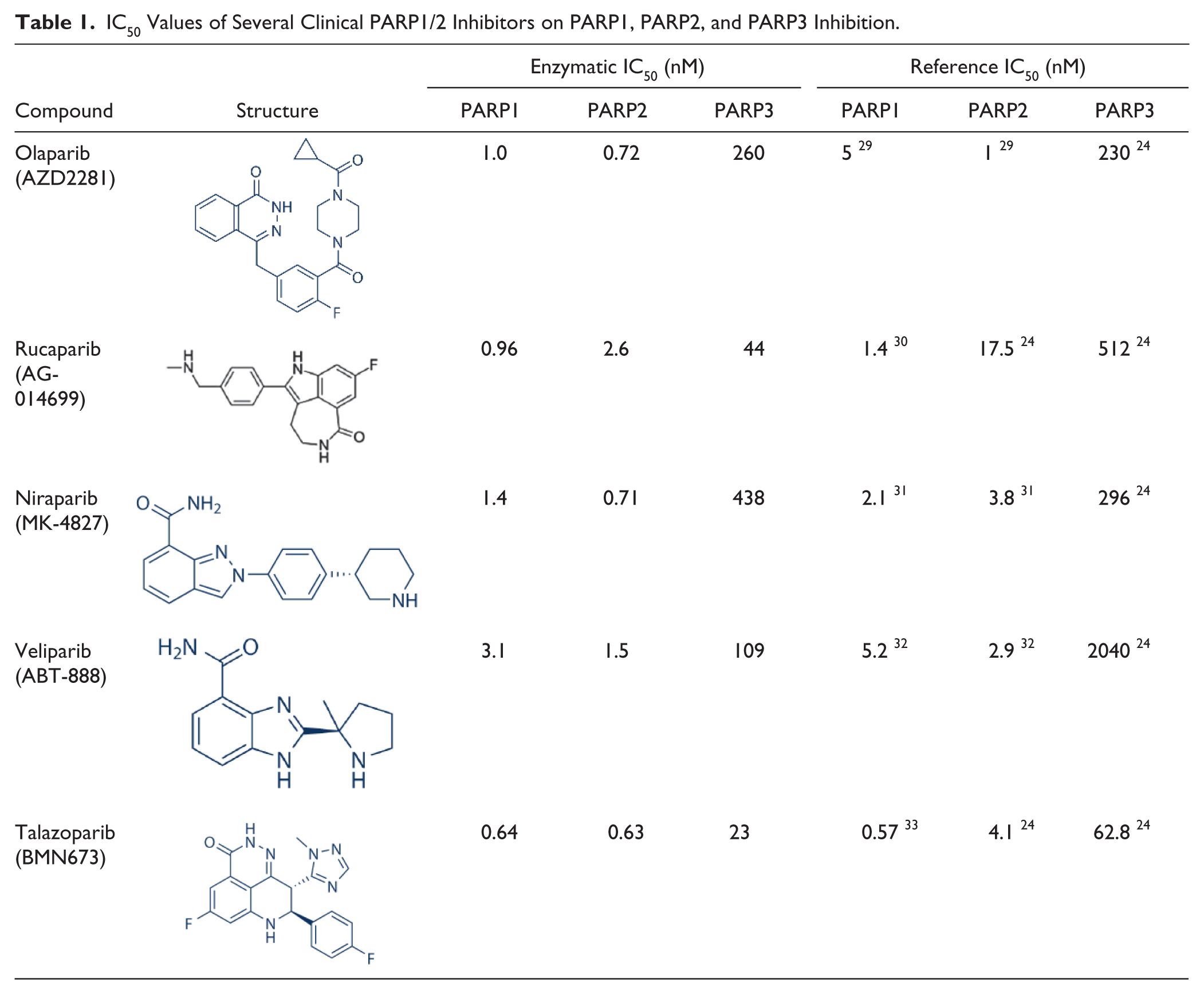
Thorsell et al. recently reported their investigation on the levels of PARP3 inhibition by clinically available PARP1/2 inhibitors.
24
We therefore decided to benchmark our assay against these results with the aim to validate our developed assay with the experimentally observed IC50 values of these compounds shown in
Table 1
and
IC50 Values of Several Clinical PARP1/2 Inhibitors on PARP1, PARP2, and PARP3 Inhibition.
Discussion
Recently, it has been shown that small-molecule inhibition of PARP3 was responsible for the lower tolerability and enhanced toxicity associated with certain PARP1/2 inhibitors, most evidently observed in a myelosuppression mouse model. 18 Contrary to this finding, knockdown of PARP3 suppressed tumor growth in vivo, and it was subsequently considered a therapeutic target for cancer by the pharmaceutical industry. Much further work is required to determine whether the inhibition of PARP3 is beneficial in the treatment of various cancers. Therefore, the development of a robust and sensitive screening platform may help in the development of novel PARP3 inhibitors, which have the potential to further our understanding of the role of this understudied PARP member’s biological systems, as well as facilitating in the development of novel PARP family-specific inhibitors.
Although several PARP3 activity assays have been established, such as thermal shift assays, fluorescence polarization, and surface plasmon resonance binding assays, these assays focused on the interaction between PARP3 and compounds and not PARP3 enzymatic activity.19–23 Enzyme-linked screening assays for PARP1 and PARP2 activities have also been widely applied for the discovery of novel small-molecule inhibitors.25,27,28 However, these assays, although highly useful, were not suitable for PARP3 inhibitor screening. This is due to the fact that PARP3 is a mono-ADP-ribosylase, and it catalyzes NAD+ to produce MAR or short PAR. However, there is no commercially available MAR antibody, and the PAR antibody cannot recognize the short form of PAR. To overcome this issue, biotin-labeled NAD+ was successfully applied and HRP-streptavidin was used to detect the NAD+, with several commercial and published assays having been developed via this solution.24,28 These assays utilized hexahistidine-tagged PARP3 immobilized on Ni2+-chelating plates and detected PARP3 activity via PARP automodification. However, one downside of this technology was that it required a large amount of the PARP3 enzyme. It is therefore imperative to develop an economical and sensitive assay for selective PARP3 inhibitor screening, which can not only uncover the biological functions of PARP3 but also assess PARP3 inhibitory effects for compounds.
In this study, we first performed a regular PARP3 enzymatic assay to quantify NAD+, which can react with acetophenone to form a fluorescent product. However, this assay is not amenable for high-throughput screening due to the low efficiency and the large amount of recombinant hPARP3 required to successfully run the screen. Thus, we aimed to establish and optimize an assay via an enzyme-linked, biotin-labeled NAD+. It was reported that PARP3 could monoribosylate histones H1 and H2B, 5 and we therefore selected mixed histones to immobilize the plate as the receptor of MAR or short PAR. Meanwhile, biotin-labeled NAD+ was applied to reduce the consumption of biotinylated NAD+, and we analyzed different ratios of biotinylated NAD+ to total NAD+. At a ratio of 1:5 (100 μM total concentration of NAD+), the absorbance at 450 nm reached the upper plateaus. In addition, it has been reported that nicked 5′P DNA could strongly activate the reaction, 5 and we therefore applied a nicked 5′P DNA to replace the sheared DNA in our system. Indeed, compared with sheared DNA, nicked 5′P DNA showed a much higher activity in the assay. Furthermore, we validated our system’s sensitivity by testing the available PARP3 inhibitor, ME0328, and the IC50 value was demonstrated to be lower than previously reported, 20 suggesting that our assay is very sensitive. We also analyzed the enzymatic inhibition of ME0328 on PARP1 and PARP2 with IC50 values of 1.25 and 1.26 μM, which is consistent with published data (IC50 of PARP1, 6.3 μM).
Additionally, we tested the PARP3 inhibitory activity of five clinical PARP1/2 inhibitors. Our data indicated that talazoparib and rucaparib showed the strongest PARP3 inhibitory activity among the five PARP1/2 inhibitors tested. However, the IC50 values of rucaparib and veliparib are much higher than reported data. 24 This may be attributed to the discrepancy of enzymatic activity of PARP3. Based on our data, only niraparib exerted appreciable PARP1/2 selectivity.
In summary, our study provides a modified ELISA for the characterization of PARP3 inhibitors using an enzyme-linked, biotin-labeled NAD+ histone ADP-ribosylation quantitation method. This assay is both highly sensitive and economical. Use of this platform to screen our in-house compound library and discover novel PARP3 inhibitors is currently underway.
Supplemental Material
DS_DISC767843 – Supplemental material for The Development of a Biotinylated NAD+-Applied Human Poly(ADP-Ribose) Polymerase 3 (PARP3) Enzymatic Assay
Supplemental material, DS_DISC767843 for The Development of a Biotinylated NAD+-Applied Human Poly(ADP-Ribose) Polymerase 3 (PARP3) Enzymatic Assay by Ming Ji, Liyuan Wang, Nina Xue, Fangfang Lai, Sen Zhang, Jing Jin and Xiaoguang Chen in SLAS Discovery
Footnotes
Acknowledgements
We thank the Beijing Key Laboratory of Non-Clinical Drug Metabolism and PK/PD studies for support. We greatly appreciate Dr. Shuan Rao and Dr. Shane Cronin for their advice on scientific discussion and writing.
Supplemental material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by the National Natural Science Foundation of China (NSFC No. 81402957), CAMS Innovation Fund for Medical Sciences (2016-12M-3-009), and PUMC Graduate Innovation Fund.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.