
Abstract
Kynurenine aminotransferase-II (KAT-II) is a pyridoxal 5′-phosphate (PLP)–dependent enzyme that acts in the tryptophan metabolic pathway by catalyzing the transamination of kynurenine into kynurenic acid (KYNA). It is one of four isoforms in the KAT family, of which it is the primary homologue responsible for KYNA production in the mammalian brain. KAT-II is targeted for inhibition as KYNA is implicated in diseases such as schizophrenia, where it is found in elevated concentrations. Previously, many different approaches have been taken to develop KAT-II inhibitors, and herein fragment-based drug design (FBDD) approaches have been exploited to provide further lead compounds that can be designed into novel inhibitors. Surface plasmon resonance (SPR) was used to screen a fragment library containing 1000 compounds, of which 41 hits were identified. These hits were further evaluated with SPR, and 18 were selected for inhibition studies. From these hits, two fragments, F6037-0164 and F0037-7280, were pursued and determined to have an IC50 of 524.5 (± 25.6) μM and 115.2 (± 4.5) μM, respectively. This strategy shows the viability of using FBDD in gleaning knowledge about KAT-II inhibition and generating leads for the production of KAT-II inhibitors.
Introduction
Fragment-based drug design utilizes low-molecular weight fragments, typically less than 300 Da, as initial leads for the development of drugs. 1 Libraries of fragment molecules are usually orders of magnitude smaller than those used in high-throughput screening, but the total chemical space for fragments is much smaller, enabling it to be sampled more efficiently. 2 Because of their simplicity, fragments have a greater chance of binding to enzymes and can generate a higher rate of “hits” in a screen. 3 Fragment binding represents a foothold at the start of the difficult process of drug discovery, but its potential has been translated with some success with several targets of interest. 4 Fragments offer greater versatility in being able to be optimized due to starting leads being relatively smaller. 3 However, because of their size, they tend to bind with low affinity to their targets, and hence a sensitive biophysical detection method must be used, such as x-ray crystallography, nuclear magnetic resonance (NMR), mass spectrometry, or surface plasmon resonance (SPR). 2
In approaches guided by SPR, a target is immobilized on a sensor chip surface and any changes in the refractive index upon binding of test compounds can be observed and are measured. 5 The SPR approach consumes less protein than other biophysical screening methods, can rapidly provide the kinetics of interactions, and offers flexibility in allowing multiple channels to be available for referencing or running experiments against different targets in parallel. 5 However, some pitfalls with SPR can arise from using solvents, particularly DMSO, which has a high refractive index, as it can cause a bulk shift in response if the solvent concentration in the sample is mismatched with the running buffer, and may introduce false positives. 6
Kynurenine aminotransferase-II (KAT-II) is a pyridoxal 5′-phosphate (PLP)-dependent homodimer and is one of four KAT isoforms in mammals. 7 Each KAT-II subunit contains a large domain, a small domain containing the C-terminus, and an N-terminal arm, and the interface of the two subunits is where the active site and PLP binding sites are hosted.7,8 KAT-II is clearly differentiated structurally from the other isoforms in that it has a relatively larger, less hydrophobic binding pocket, and also the N-terminal arm undergoes domain swapping as it projects from one subunit into the opposite subunit, helping shape the active site. 9
The KAT isoforms catalyze the irreversible transamination of kynurenine to kynurenic acid (KYNA), a step in the tryptophan metabolic pathway. 10 The KAT-II transamination mechanism is thought to occur first, with kynurenine entering into the active site by a conformational fold of N-terminal residues and generating a π-cation interaction with Arg-20 ( Suppl. Fig. S1 ). 10 The carboxyl of kynurenine acts as a salt bridge and interacts with Arg-399, while the amine group also forms hydrogen bonds with Asn-202 and Gly-39, and together these residues assist in anchoring the substrate in the correct conformation for transamination within the active site. PLP is covalently attached to Lys-263 by an internal aldimine bond that is later broken during the enzymatic reaction. 10
The product of this reaction, KYNA, is an N-methyl-
In this study, we aimed to screen a 1000-fragment library against KAT-II using SPR to find potential binding hits. These hits were validated by evaluating them in different concentrations to minimize the potential for false positives, and any fragment hits advancing further were evaluated for their inhibitory ability using a high-performance liquid chromatography (HPLC) inhibition assay. The possible binding mechanisms of the fragments hits are discussed with a view to aiding the design process of KAT-II inhibitors, and also using these hits as the basis of new leads for new inhibitors.
Materials and Methods
General Procedures
Commercially available reagents were used without additional purification unless otherwise stated. The fragment library (1000 wells, each containing 1 mg of a fragment compound) was purchased from Life Chemicals (Ontario, Canada). Phosphate-buffered saline (PBST; pH 7.4 with 0.05% Tween 20) was purchased from Sigma-Aldrich (Sydney, Australia). SPR reagents (40 mM EDC, 10 mM sulfo-NHS, 10 mM sodium acetate, 1 M ethanolamine HCl, pH 8.5) were purchased from Bio-Rad Laboratories (Gladesville, Australia). Methanol and DMSO were purchased from Thermo Fisher Scientific (Scoresby, Australia).
The SPR screening protocol was prepared with the Biacore T200 control software, measurements were performed using the Biacore T200 with a CM5 sensor chip, and the results were analyzed with the Biacore T200 evaluation software (GE Healthcare Life Sciences, Sydney, Australia).
The IC50, reported with standard error, was determined using GraphPad Prism v7.02 software (GraphPad, La Jolla, CA), by developing a nonlinear regression fit of the data collected from the inhibition assays. Images of docked compounds were generated by the PyMOL Molecular Graphics System (Schrodinger LLC, Cambridge, MA).
Protein Preparation
The recombinant KAT-II protein was expressed and purified in our lab as previously described. 28
Initial Fragment Binding SPR Screen against KAT-II
Prior to performing the SPR experiments, the solutions were filter sterilized for 10 min at 25 °C. KAT-II was diluted to a concentration of 320 µg/mL using sodium acetate (pH 4.5). The Biacore T200 compartment temperature was controlled at 25 °C, and the flow rate was set to 10 µL/min. Using a 1:1 mixture of 400 mM EDC and 100 mM sulfo-NHS, two flow cells on a CM5 sensor chip (contains a carboxymethylated dextran surface) were activated. Flow cell 1 contained a blank immobilization (PBST buffer injection), and flow cell 2 had the prepared KAT-II sample injected. The two flow cells were deactivated using an injection of 1 M ethanolamine.
The analytes (fragment library samples) were prepared in running buffer (PBST with 5% DMSO) to a final concentration of 20 μM and loaded into a 96-well plate. The analytes were injected using a flow rate set to 30 μL/min, a contact time of 30 s, and a dissociation time of 30 s, with an extra 50% DMSO wash performed after each injection. Solvent correction was performed at the start and end of the experiment, and also after every 50 cycles, to account for the use of DMSO in the analyte sample preparation and the running buffer. The positive control (20 μM NS-1502) and the blank control (running buffer) were injected every 30 cycles.
Further Evaluation of Hits Using SPR
Analytes were prepared in running buffer (PBST with 5% DMSO) in a concentration series from 6.25 to 100 μM. The analytes were injected onto the CM5 sensor chip using a flow rate of 30 μL/min, a contact time of 90 s, and a dissociation time of 90 s, with an extra 50% DMSO wash performed after each injection. Solvent correction was performed before and after the estradiol analyte injections and also every 50 cycles.
Fragment Lead Evaluation Using HPLC Inhibition Studies and IC50 Determination
The KAT-II inhibition assay was performed as previously described.
19
KAT-II (0.5 µg) was incubated at 37 °C in a 50 µL reaction mixture containing 50 µM PLP, 5 mM α-ketoglutarate, and 5 mM
Computational Docking of Inhibitor Compounds at the Active Site of KAT-I and KAT-II
The crystal structure of human KAT-I (PDB ID: 3FVU 29 ) and KAT-II (PDB ID: 2R2N 30 ) was downloaded from the Protein Data Bank. Docking was performed as follows, both with and without PLP in the active site. The protein was optimized and minimized using the protein preparation wizard in Maestro v10.4.017 (Schrodinger LLC), and water molecules not participating in the reaction were removed. LigPrep (Schrodinger LLC) was used to prepare the fragment and inhibitor ligands, using the OPLS-2005 31 force field. The active site grid was determined by the location of kynurenine, the natural substrate, in the PDB file in KAT-II, and the cocrystallized inhibitor, indole-3-acetic acid, in KAT-I. The ligands were docked with XP (extra precision) docking 32 using Glide (Schrodinger LLC).
Data Availability
The data generated and analyzed during the current study are available in the Figshare repository (https://figshare.com/projects/Fragment_Screening_of_Human_Kynurenine_Aminotransferase-II/28293).
Results and Discussion
Preliminary Fragment Binding Screen on KAT-II Using SPR
SPR was used to identify potential KAT-II-inhibiting lead molecules from a fragment library containing 1000 compounds. The fragment library was synthesized to be diverse and has an average molecular weight of 223 Da (with a range of 84–300 Da). An initial consideration with using fragment molecules in SPR is their inherent lower response. The theoretical response limit is dictated by the equation Rmax = MWan/MWlig*RL*Sm, where Rmax is the analyte binding ability, MWan and MWlig are the molecular weights of the analyte and ligand, RL is the ligand immobilization levels, and Sm is the stoichiometric ratio of the interaction. This necessitates that higher ligand binding on the sensor chip is required in fragment binding than it otherwise would be required in, for example, protein–protein interactions. In our experiment, KAT-II was bound onto a CM5 sensor chip by amine coupling and achieved approximately 20,000 RU, which enabled us to elicit a noticeable response from the fragments. All the fragment molecules and positive control (NS-1502) were injected at a concentration of 20 μM, and the running buffer (PBST with 5% DMSO) was used as a blank control. After correcting for DMSO and subtracting the reference channel, the data were adjusted to show the relative response of the analytes in comparison with the positive and blank controls ( Fig. 1 ).
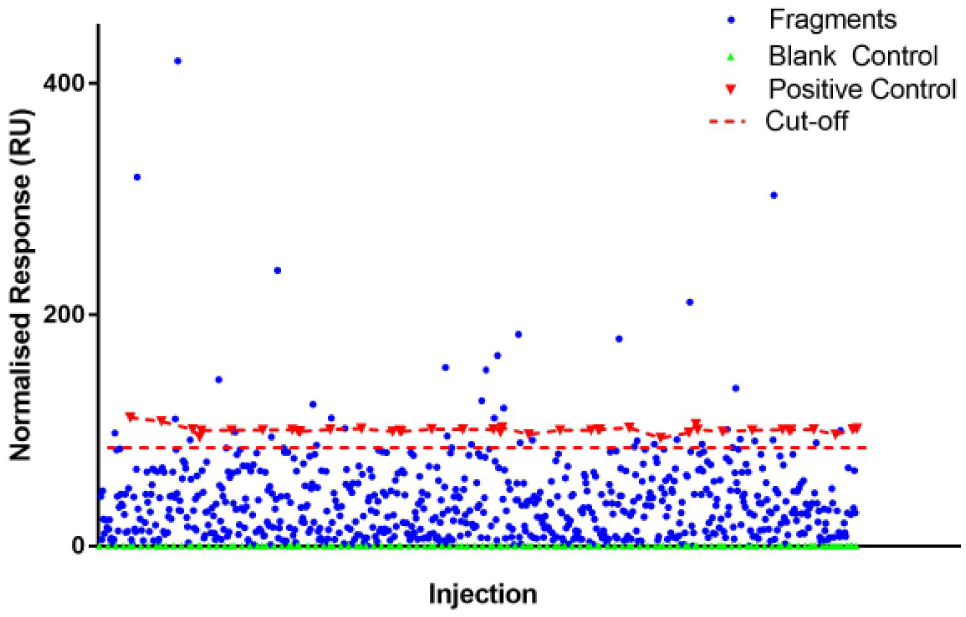
A plot of the SPR response of the initial 1000-fragment screen against KAT-II. Depicted are the blank control (green triangles), positive control (red triangles), fragments (blue circles), and cutoff (red dashed line). The plot has been subtracted with the blank and normalized with the controls. The diagram was produced using GraphPad Prism v7.02.
The screening of the 1000 fragment compounds was done over different days, using samples loaded into a 96-well plate for each run. One negative aspect that was evident from the experiment was the effect the duration of each run (approximately 8–10 h per well plate) had on the quality of the results. Notably, there seemed to be a slight, but increasing, negative shift in response by the blank control as time progressed in some of these runs. Given that between runs the blank control had a normal response at the beginning (before slowly drifting), and given that the positive control seemed to be consistent throughout the runs, the response described is likely not as a result of chip or ligand instability. Instead, it is thought that there was mismatch in the running buffer and the blank control over time. The relative adjustment to the blank control can hence potentially positively skew the response of the fragments in that particular run and produce false positives (a graphical example of this can be seen in Fig. 2 ).
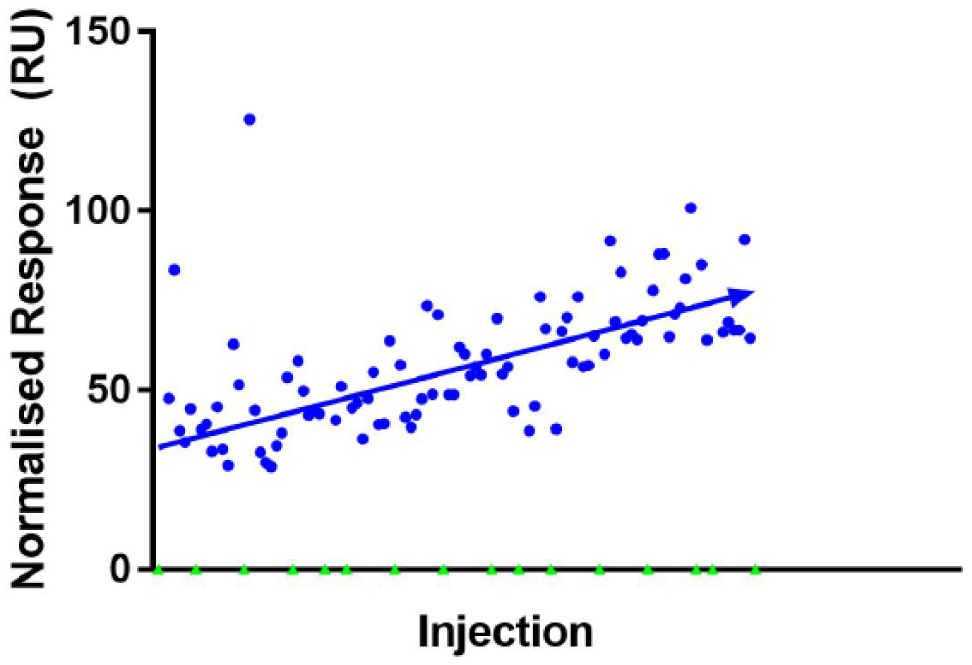
SPR responses of fragments (blue circles) from one run after normalization of the blank buffer control (green triangles). A mismatch in the blank buffer control and running buffer over time appears to skew the response of the fragments in a positive gradient. Diagram was produced using GraphPad Prism v7.02.
The cutoff used from the initial screen included those fragments that achieved a relative response of 85%+ of the positive control, NS-1502. In total, 41 fragments were selected for more extensive investigation with SPR to provide more accurate information on their binding and hence maximize chances of true positives while ensuring that any false results were eliminated.
Further Evaluation of Hits Using SPR
The 41 fragments were prepared in a concentration series from 6.25 to 100 μM and analyzed using multicycle kinetics ( Suppl. Fig. S2A–C ). NS-1502 was also prepared in the same concentration and used as a positive control.
With increasing concentration, a concomitant increase in response can be expected from each fragment. Hence, some fragments could immediately be discarded as they did not respond in this way, and likely arose from binding nonspecifically or simply as a result of the experimental conditions, such as requiring DMSO, which has a high refractive index and could result in false data if not treated with care. Removing these fragments from further consideration, there were 26 fragments remaining from which we could identify those truly most suitable. These fragments were compared with NS-1502, and 18 fragments were selected for a greater response to KAT-II than the positive control. Eight out of the 18 fragments were observed to have a similar structural moiety, with an aromatic ring fused to a five-membered ring ( Suppl. Fig. S3 ).
HPLC Enzyme Assay Screening and IC50 Determination
Out of the 1000 compounds in the fragment library, the 18 selected fragments were determined to be the best binding to KAT-II, as determined by SPR, but this did not necessarily indicate that they would be inhibitory. An HPLC inhibition assay was required to measure the inhibitory ability of these fragments at one concentration.
As seen in Supplemental Table S1 , 15 of the selected fragments did not exhibit inhibition of KAT-II or did so only weakly. Fragments by nature can be highly indiscriminate in their binding, and it is possible that some of these compounds bound to off-target locations when they were eliciting a response to SPR. However, two fragments (F0037-7280 and F6037-0164) were pursued for further study as evidence showed that they were inhibitors of KAT-II. F3412-0030 was a third fragment that seemed to inhibit KAT-II relatively well; however, given the high level of structural similarity to F0037-7280, this fragment was not followed up initially.
F6037-0164 and F0037-7280 were prepared in a concentration series from 1 to 2000 μM. F6037-0164 was found to have an IC50 of 524.5 (± 25.6) μM ( Fig. 3A ). By varying the concentration of PLP in the reaction mixture, F6037-0164 was found to be reversible (data not shown), like NS-1502. 19 In these same inhibition assay conditions, NS-1502 has an IC50 of 315 μM, so the IC50 determined for F6037-0164 is a promising lead for a reversible inhibitor.
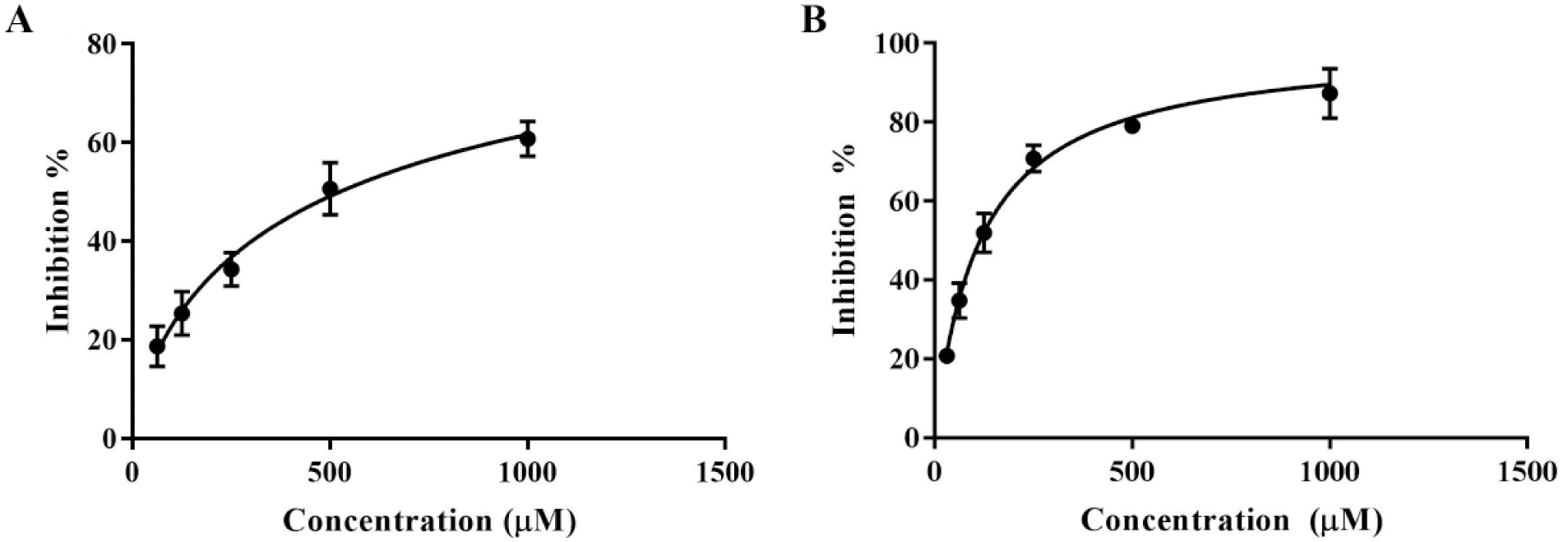
(
F0037-7280 was found to have an IC50 of 115.2 (± 4.5) μM ( Fig. 3B ). Unlike NS-1502 and F6037-0164, changing PLP concentration did not seem to have an effect on F0037-7280 inhibition (data not shown), suggesting that it may act irreversibly or in a noncompetitive manner with respect to the cofactor. However, the primary cause of irreversible inhibition in KAT-II inhibitors is the formation of a covalent adduct via a primary amine in the inhibitor, forming an external aldimine bond with PLP, which F0037-7280 would be unable to achieve.
Both of these compounds structures have a resemblance to a phthalimide core, with F0037-7280 differing in having the aromatic ring two carbon lengths distant from the succinimide-like moiety, and with F6037-0164 having an imidazole ring instead of a succinimide. Both fragments extend toward an aromatic ring but differ in the length of the molecule, with F6037-0164 being significantly bulkier. Based on the features of these compounds, and knowledge of the specific atomic interactions kynurenine has in the active site, it may be possible to predict the manner in which these fragments bind.
Binding Conformation of the Fragments
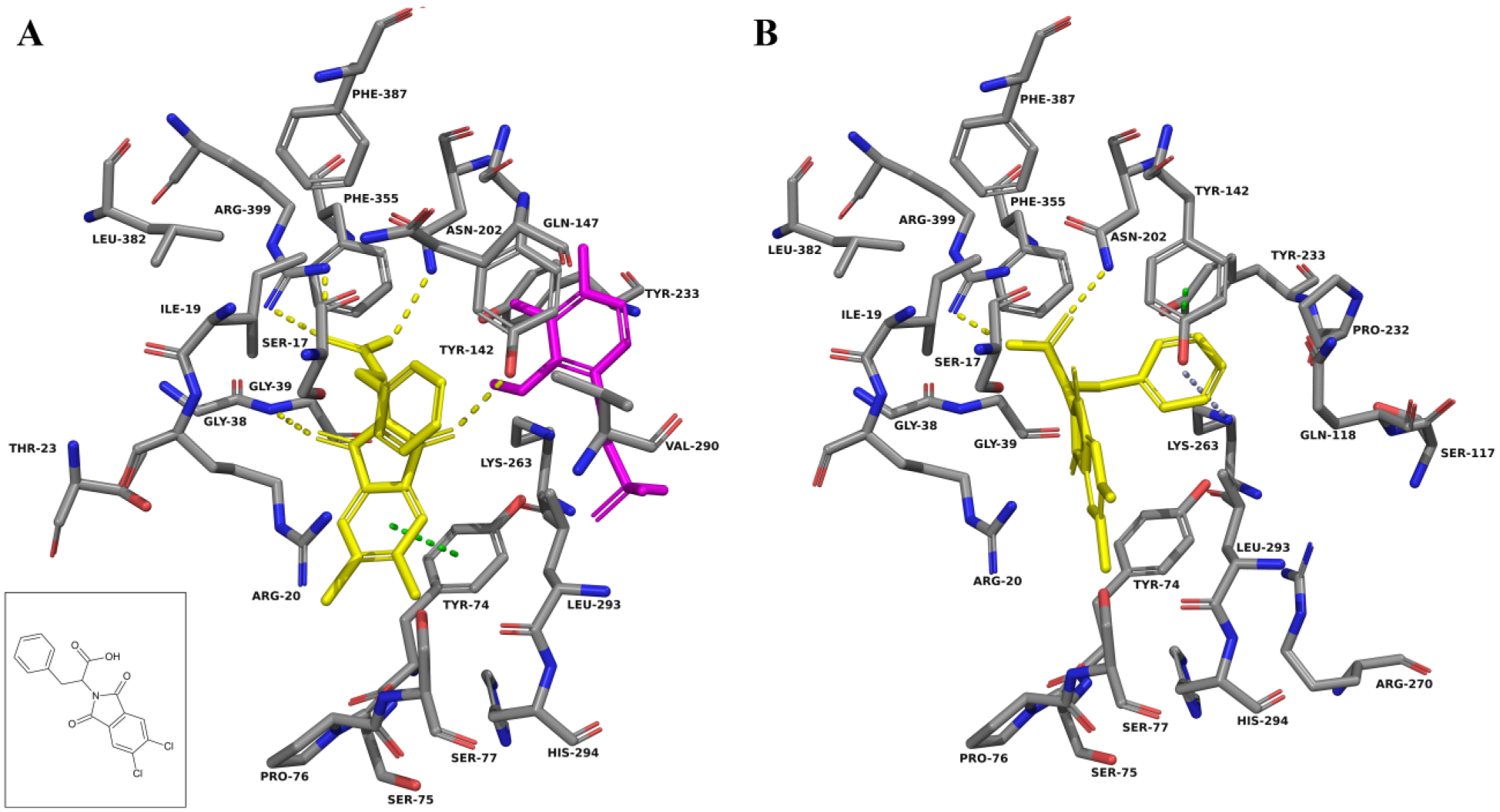
In both the three-dimensional structure and the docking in KAT-II with PLP, kynurenine is observed to favor the aromatic ring forming π-cation interactions with Arg-20, and the carboxyl group forming a salt bridge with Arg-399 ( Suppl. Fig. S1 ). The amine group also forms hydrogen bonds with Asn-202 and Gly-39, which, in addition to the carboxyl, helps situate it in the correct conformation within the active site. On the basis of modeling, a similar conformation is expected with F0037-7280, with the benzene moiety situated adjacent to Arg-20 and the succinimide moiety forming hydrogen bonds with Arg-399, Asn-202, and Gly-39 ( Fig. 4A ). F6037-0164 is similar, with the benzimidazole core near Arg-20 and the carbonyl hydrogen bonding to Arg-399 and Asn-202 ( Fig. 5A ). As F6037-0164 is bulkier, the remainder of the molecule curves back around in the binding pocket to avoid clashing with PLP. F3412-0030 is also a potential hit that was not explored and docks correspondingly to the structurally similar F0037-7280, but with fewer notable interactions ( Suppl. Fig. S4 ).
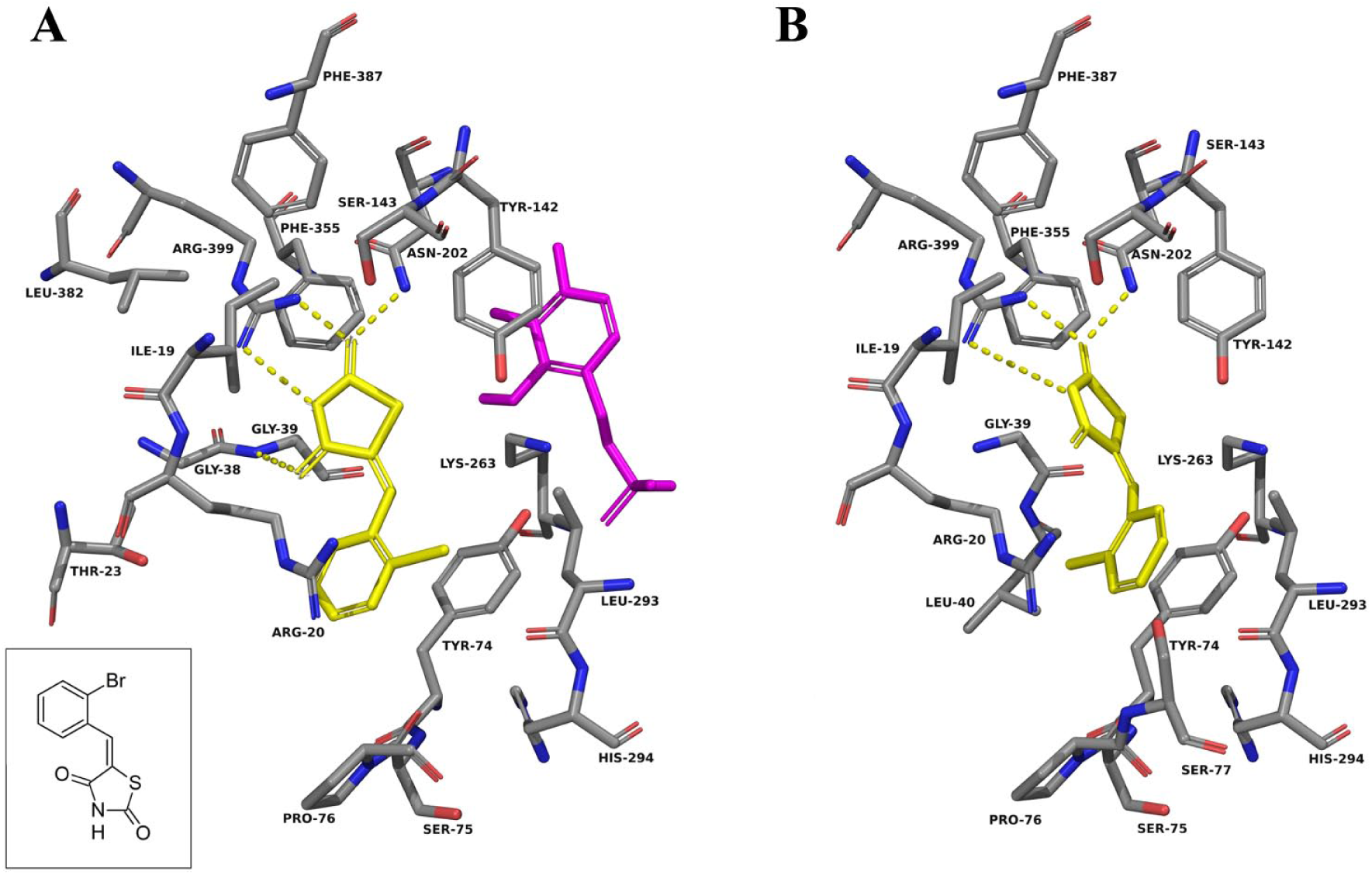
F0037-7280 docked into the active site of KAT-II. The amino acids with an atom within 5.0 Å of F0037-7280 (yellow) were chosen for display. (
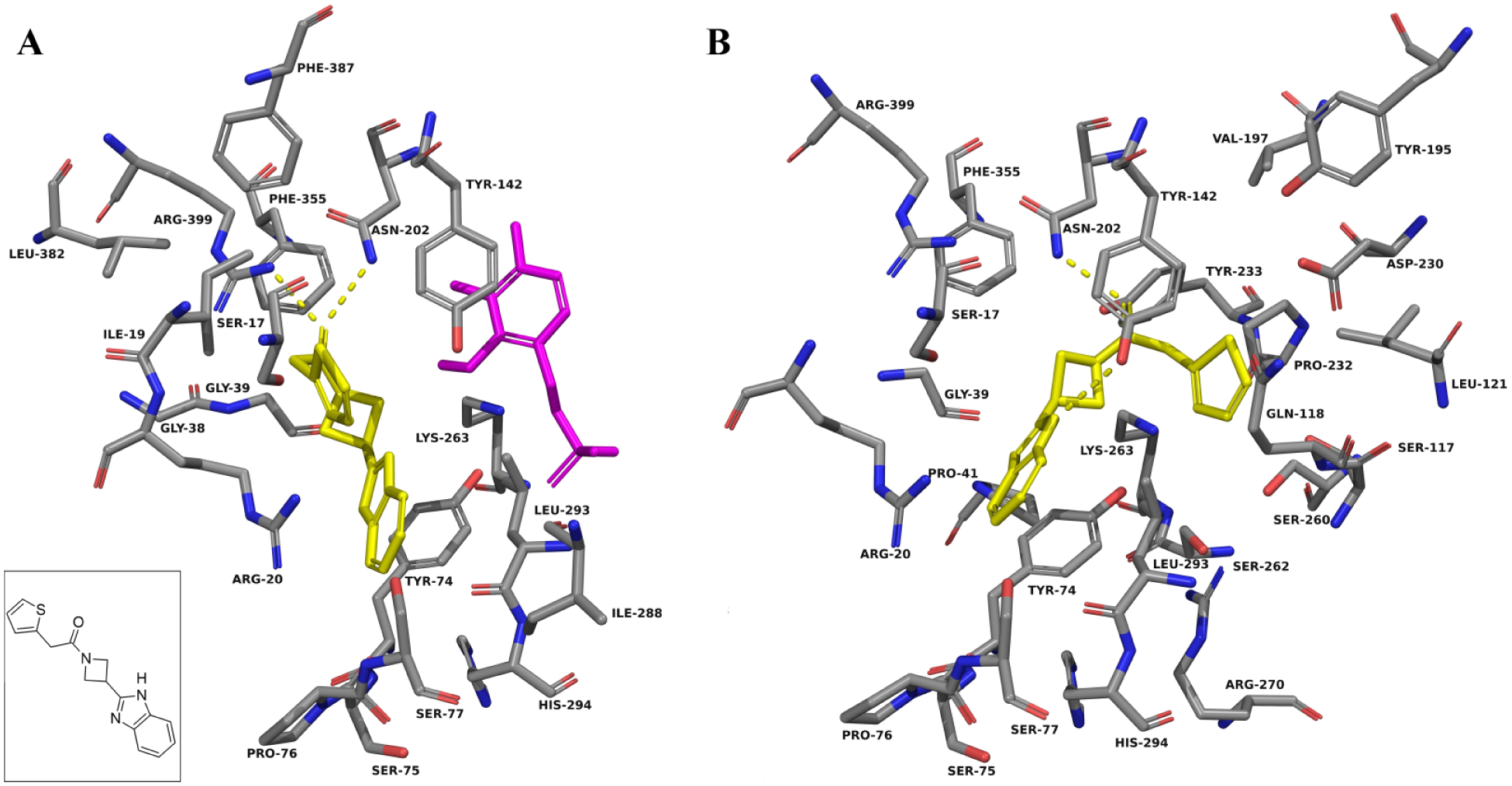
F6037-0164 docked into the active site of KAT-II. The amino acids with an atom within 5.0 Å of F6037-0164 (yellow) were chosen for display. (
F0037-7280 docking calculations were also performed in KAT-II with PLP removed, and no significant change in the docking pose was observed ( Fig. 4B ). This is consistent with the F0037-7280 binding being unaffected by the presence of PLP, as was observed in the HPLC assay when a range of PLP concentrations in the reaction mixture were used but inhibition was very similar. These observations contrast with those of F6037-0164 ( Fig. 5B ) and NS-1502 ( Fig. 6A , B ), which have a different proposed pose and dock near and interact with Lys-263, the residue normally involved in covalently binding to PLP, in the absence of the cofactor. Unlike F0037-7280, the inhibitory ability of these compounds decreases with increasing PLP concentrations, and hence they are in competition with it and may take up a less favorable conformation within the active site or get displaced completely when PLP levels are increased. Estrogen disulfate, another relatively strong inhibitor of the KAT enzymes, 33 also has been reported to compete with PLP, and it has been argued that its inhibitory action occurs by competing for the binding site with the apoenzyme form of KAT. 34 A similar mechanism of inhibition is conceivable for F6037-0164 and NS-1502.
NS-1502 docked into the active site of KAT-II. The amino acids with an atom within 5.0 Å of NS-1502 (yellow) were chosen for display. (
The ability of F0037-7280 and F6037-0164 to inhibit KAT-II can be seen to be a consequence of the interactions they could have with key amino acid residues within the active site. Of particular importance seems to be the presence of an aromatic ring near the entry point of the active site, situated adjacent to Arg-20, whether alone (seen in F0037-7280) or as part of a system of rings (seen in F6037-0164). Moieties that can exploit hydrogen bonding interactions with Arg-399, Asn-202, and Gly-39, such as the carbonyl groups in F0037-7280 and F6037-0164, are also very important for binding.
The unique features of KAT-II, in particular its wider active site and flexible N-terminal arm, can be exploited in the development of selective inhibitors. The bulkiness of a ligand has been observed to be a significant determinant of selectivity as larger compounds can be more readily accommodated in the KAT-II active site. In contrast, there is more steric hindrance in the narrower, rigid active sites of the other KAT isoforms.22,35 The docking of F6037-0164 showed that in KAT-II, even as an elongated compound, it can be enclosed in the active site; however, in KAT-I this fragment directs more toward the surface of the enzyme ( Suppl. Fig. S5 ). This suggests that KAT-II selectivity may be invoked by incorporating more bulk and rigidity into the structure, as the whole molecule may not be contained within the active site of KAT-I. F0037-7280 is a much smaller molecule and has more potential to be optimized in the future with similar considerations.
The design of selective KAT-II inhibitors remains an important endeavor. KYNA, thought to be predominantly produced by KAT-II in the brain, is a downstream product of tryptophan degradation and is implicated in normal cognitive function; however, it is also found to be elevated in patients with schizophrenia. KAT-II inhibitors, which reduce KYNA levels, notably have been shown to have positive effects on cognitive function in animal models. To aid in the improvement of existing inhibitors and in the development of new KAT-II inhibitors, in this study we screened a library of fragments compounds using SPR. From the screening and subsequence inhibition assay, we have found two fragments, F0037-7280 and F6037-0164, to pursue as lead KAT-II inhibitor compounds.
The most important protocol design consideration that became evident during this work was ensuring that the solvent concentration was controlled in the samples and the running buffer for use in SPR. As DMSO has a high refractive index, a mismatch during its use can cause a large bulk shift in response and confound the data. This situation is likely more problematic in a long screening run, as we found that for extended runs the mismatch between the two grew. To overcome this, it would be more advisable to split the runs into smaller ones with freshly made samples and running buffer. In the work presented here, the false positives were eliminated by running the fragment hits in a concentration series and reevaluating the SPR curves generated. This method, however, adds an additional step to the protocol; hence, an initial minimization of the effect of DMSO would be more beneficial in terms of time.
The fragment-based screening found 18 hits from the SPR experiments. Despite many of these hits being poor inhibitors, they can potentially be developed into more potent leads as they are proven KAT-II binders. Evidence suggests that three of the hits are already strongly inhibitory, with F6037-0164 and F0037-7280 actively pursued and F3412-0030 in consideration for future research. Analyzing the interactions with which F0037-7280 and F6037-0164 may bind, it appears that the important features in KAT-II binding and inhibition include interactions with Arg-20, Arg-399, Asn-202, and Gly-39 for reversible inhibition. It also appears that a smaller compound, such as F0037-7280, may be noncompetitive with respect to the cofactor PLP, whereas a larger compound, such as F6037-0164, may be partially competitive with PLP (along with the substrate kynurenine) in binding to the apoenzyme.
Supplemental Material
DS_DISC764620 – Supplemental material for Fragment Screening of Human Kynurenine Aminotransferase-II
Supplemental material, DS_DISC764620 for Fragment Screening of Human Kynurenine Aminotransferase-II by Gayan S. Jayawickrama, Alireza Nematollahi, Guanchen Sun, W. Bret Church in SLAS Discovery
Footnotes
Acknowledgements
The authors acknowledge support from the Australian Postgraduate Research Awards and University of Sydney (G.S.J., A.N.) to conduct this study. The authors wish to acknowledge the Sydney Computational Drug Discovery Group for software access. The authors thank members of their research group for a supportive and collegial environment. We also wish to acknowledge the support received from the Bosch Institute’s Molecular Biology Facility and the expert help of facility staff, especially Dr. Donna Lai and Dr. Sheng Hua, on using the Biacore T200 system.
Author Contributions
Conceived and designed the experiments: W.B.C., G.S.J., G.S., A.N. Performed experiments: G.S.J. Analyzed the data: G.S.J., A.N., G.S. Wrote the paper: G.S.J., W.B.C.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Author W.B.C. discloses receipt of the financial support for part of the research of this article from The Rebecca L. Cooper Medical Research Foundation.
Supplementary material is available online with this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.