
Abstract
DNA-encoded libraries (DELs) have emerged as an efficient and cost-effective drug discovery tool for the exploration and screening of very large chemical space using small-molecule collections of unprecedented size. Herein, we report an integrated automation and informatics system designed to enhance the quality, efficiency, and throughput of the production and affinity selection of these libraries. The platform is governed by software developed according to a database-centric architecture to ensure data consistency, integrity, and availability. Through its versatile protocol management functionalities, this application captures the wide diversity of experimental processes involved with DEL technology, keeps track of working protocols in the database, and uses them to command robotic liquid handlers for the synthesis of libraries. This approach provides full traceability of building-blocks and DNA tags in each split-and-pool cycle. Affinity selection experiments and high-throughput sequencing reads are also captured in the database, and the results are automatically deconvoluted and visualized in customizable representations. Researchers can compare results of different experiments and use machine learning methods to discover patterns in data. As of this writing, the platform has been validated through the generation and affinity selection of various libraries, and it has become the cornerstone of the DEL production effort at Lilly.
Introduction
Most pharmaceutical companies and academic groups in drug discovery are facing the need to accelerate their research programs, optimize R&D investments, and expand the current scope of druggable therapeutic targets. Within this context, DNA-encoded libraries (DELs) represent a novel and robust approach to hit identification 1 that can provide access to a broad set of diverse chemotypes for a significantly lower cost than that of high-throughput screening (HTS). 2
The concept of encoding chemically synthetized compounds with DNA was first proposed more than two decades ago. 3 Since then, a very significant adoption of the DEL technology has occurred. Different methods for the production of DELs have been described in the literature, including both single- 4 and double-strand encoding, 5 DNA-templated synthesis, 6 self-assembly libraries, 7 and dual display. 8 In addition, affinity selection methodologies combined with DNA next-generation sequencing (NGS) have progressed to allow rapid and efficient testing of large pools of libraries using negligible amounts of proteins 9 or cells.10,11 This progress has been reflected by an increasing number of publications on the topic,2,12 –14 positioning the technology as a highly valuable tool for internal research programs, as well as open innovation scientific collaborations. 15
While there is an increasing breadth of scientific knowledge available, groups in industry and academia adopting DEL technology still need to evolve their processes and implement technologies to cope with the high-throughput nature of DEL work. Both the production and affinity selection of DELs involve large numbers of building blocks, DNA tags, and reagents. Experimental processes must follow traceable procedures. NGS results usually generate thousands of records per sample. These complexities may be overcome through manual handling of materials and spreadsheet-based data tracking strategies. However, more sophisticated technology platforms based on laboratory automation, custom-built software, and relational databases result in a more sustainable and scalable approach with significant gains in team productivity, speed of DEL production, and quality and reproducibility of the experimental work.16,17
As part of an effort to implement DEL technology, we have developed the DELTA platform, an automation and informatics solution for the design, production, and affinity selection of DELs. To our knowledge, this is the first report of a platform applied to DELs that builds on the concept of capturing and tracking the experimental information of the library synthesis and affinity selection as it is generated. This information is used to command the laboratory instrumentation, maximizing scientific productivity and enhancing the overall quality of DEL processes. Through this approach, we maximize reproducibility of the work by ensuring that experimental protocols captured by scientists reflect the actual manipulations carried out in the laboratory. In addition, we improve scientific productivity by automating a number of sample and analytical data gathering and processing tasks and enhance the overall quality of our DEL by reducing the number of errors resulting from manual execution of the complex liquid handling routines that are required.
To demonstrate the utility of DELTA in supporting our DEL scientific efforts, and as a model for others considering starting their own DEL program, herein we describe the production and affinity selection of DEL-6, a three-cycle library encoded in double-strand DNA and incorporating a triazine central core. This core is functionalized through nucleophilic aromatic substitution (NAS) reactions in each cycle. The DEL-6 design has been described before in the literature 5 and has been produced in different versions by many groups working on DEL.
Material and Methods
DELTA informatics architecture was designed according to a database-centric approach. 19 The foundation for this architecture is an Oracle database, version 12c, Enterprise Edition (Redwood City, CA). This database contains data regarding library designs, experimental procedures, laboratory protocols, building block and DNA tag management, high-performance liquid chromatography–mass spectrometry (HPLC/MS) analysis, affinity selection, sequencing results, and deconvoluted chemical structures of positive binders.
An ASP.NET web service, implemented in Microsoft C# for .NET framework version 3.5 (Redmond, WA), provides access to this database and manages all user and laboratory processes. This web service represents a centralized implementation of the platform’s core functionality and provides interoperable service for the rest of its components. 20
A Windows desktop application, also implemented in Microsoft C# for .NET framework version 3.5 (Redmond, WA), acts as the platform end-user interface. This application is a client of the aforementioned web service and can generate scripts for automation equipment, as well as input data files for HPLC/MS instruments.
Data generated through the analytical instruments are stored utilizing Agilent OPENLAB Chromatography Data Systems (CDS) technology (Santa Clara, CA).
Laboratory processes are automated through the use of TECAN FREEDOM EVO 200 liquid handlers run by version 2.7 of EVOware controller software (Männedorf, Switzerland). Each liquid handler is equipped with two arms: an Air Liquid Handling arm with eight single tips and a Multi-Channel Arm 96 (MCA96) with gripper. Experiments are carried out in 96-well plate format, and all the aspiration/dispensing steps are done with the MCA96 head. The deck has four shaker-heater-cooler Inheco Thermoshakers (Munich, Germany), with custom-made plate adaptors for optimal temperature transfer. The magnetic bead handling is performed by a MAGNUM EX Universal component (Alpaqua, Beverly, MA).
The HPLC/MS instruments are Agilent 1200 Series run by version B.04.03 of LC/MSD ChemStation software and Agilent Technologies 6530 Accurate-Mass Q-TOF (Quadrupole Time of Flight) LC/MS run by version B.07.00 of MassHunter Workstation Software (Santa Clara, CA).
Target proteins are biotinylated and immobilized on streptavidin beads. These beads are incubated with DELs and unbound molecules are subsequently washed away. Compounds that remain bound to the proteins are eluted by heat denaturation. After three consecutive rounds of affinity selection, the enriched population is prepared for sequencing following the Fusion Method for the Ion Amplicon Library protocol (Life Technologies, Carlsbad, CA). Its quality is assessed by Agilent 2200 TapeStation (Agilent Technologies). Sequencing templates are prepared with the Ion Chef System (Thermo Fisher Scientific, Waltham, MA). Ion PGM and Ion Proton sequencers (Life Technologies) are used to obtain lower and higher sequencing yields, respectively.
Data analysis is carried out in TIBCO Spotfire version 7.5 (Palo Alto, CA). A custom Spotfire application interoperates directly with the database using the native Oracle Data Provider driver. Data processing is carried out by embedded IronPython scripts. Line and plane detection in affinity selection data is carried out by Spotfire’s built-in implementation of UPGMA agglomerative hierarchical clustering based on the Euclidean distances between the sums of counts of all the lines and planes in the chemical space defined by each library.
Affinity selection data processing, deconvolution, and loading of results into DELTA’s database are carried out by scripts written in Python version 2.7 (Python Software Foundation, Beaverton, OR).
Building blocks are delivered in ready-to-use 96-well barcoded plates by our local Sample Management Center (SMC). DELTA is integrated with SMC’s Titian Mosaic database (London, UK) to automatically retrieve plate contents querying by plate barcode. Likewise, DNA tags are purchased in Matrix racks from LGC Biosearch Technologies (Novato, CA), and their sequences and IDs are directly imported into the platform’s database from our supplier’s delivery notes.
Results and Discussion
DEL Production
The DELTA platform has been designed to capture the production of DELs according to the classical split-and-pool approach. Specifically, library synthesis starts with a DNA headpiece that consists of a short DNA double strand linked to a polyethylene glycol (PEG)–based spacer functionalized with an amino group. 5 This headpiece is successively derivatized along two to four cycles that alternate the addition of building blocks and their corresponding DNA tags.
In the DELTA database, this process is captured by means of a linked list data structure 18 ( Fig. 1B ). Starting materials, synthesis steps, and pools are represented as nodes identified by a unique ID. The production is recorded as a sequence of nodes, each of them linked to its predecessor. For example, a library production typically starts by splitting a DNA headpiece into a number of wells. This DNA headpiece is the first node of the sequence and is recorded in the database with a unique Starting Material ID. The ligation of DNA tags to those wells is inserted in the database as the next node in the sequence. This node is identified by an Experiment ID and is linked to the headpiece’s Starting Material ID. The subsequent addition of building blocks represents a new node that receives its own unique Experiment ID and is linked to the ligation’s Experiment ID. Finally, the wells are put together in a pool that is captured as a new node identified with its own Pool ID and linked to the previous Experiment ID. The sequence continues throughout the whole library production as required by the number of cycles in the library design ( Fig. 1B ).
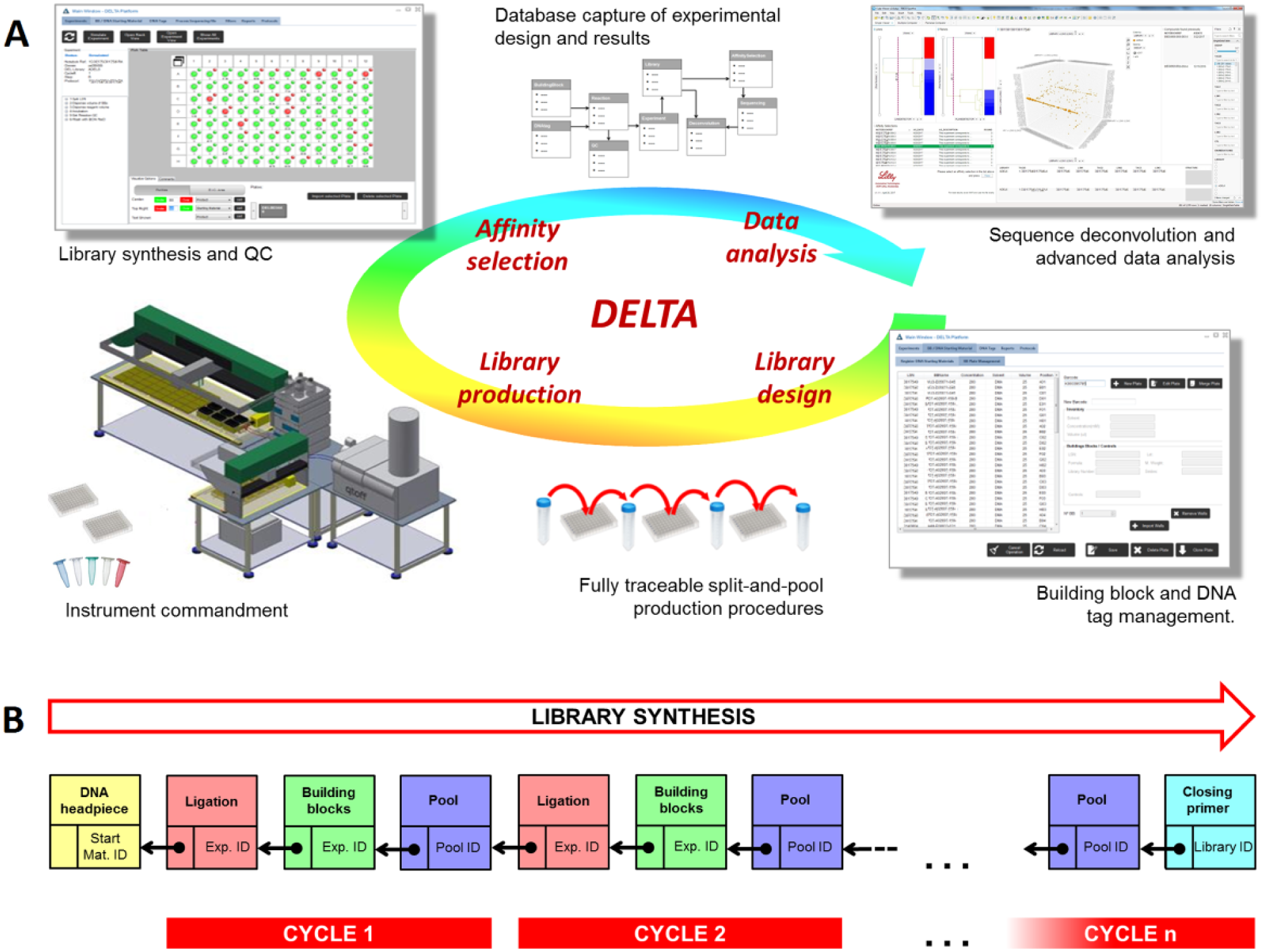
DELTA platform. (
The complexity of a DEL design goes beyond alternating addition of building blocks and ligation of DNA tags. Additional steps in the synthetic route can include functional group deprotections, use of common building blocks as a scaffold, or different chemistries in the same cycle; these steps are stored in the system under their own Experiment IDs and linked to their preceding steps. In order to deal with this complexity, DELTA offers eight different experiment types to capture the library production ( Table 1 ). The ligation of DNA tags corresponds to an experiment Type A, and the addition of building blocks is Type B. In order to illustrate these and other steps and how they can be captured as a sequence of backward-linked experiments, we describe the production of DEL-6, a prototypical, three-cycle, triazine-based, DNA-recorded library that was generally prepared as described in the literature 5 (see DEL-6 flow scheme in Fig. 2B ).
Experiment Types Currently Available in DELTA.
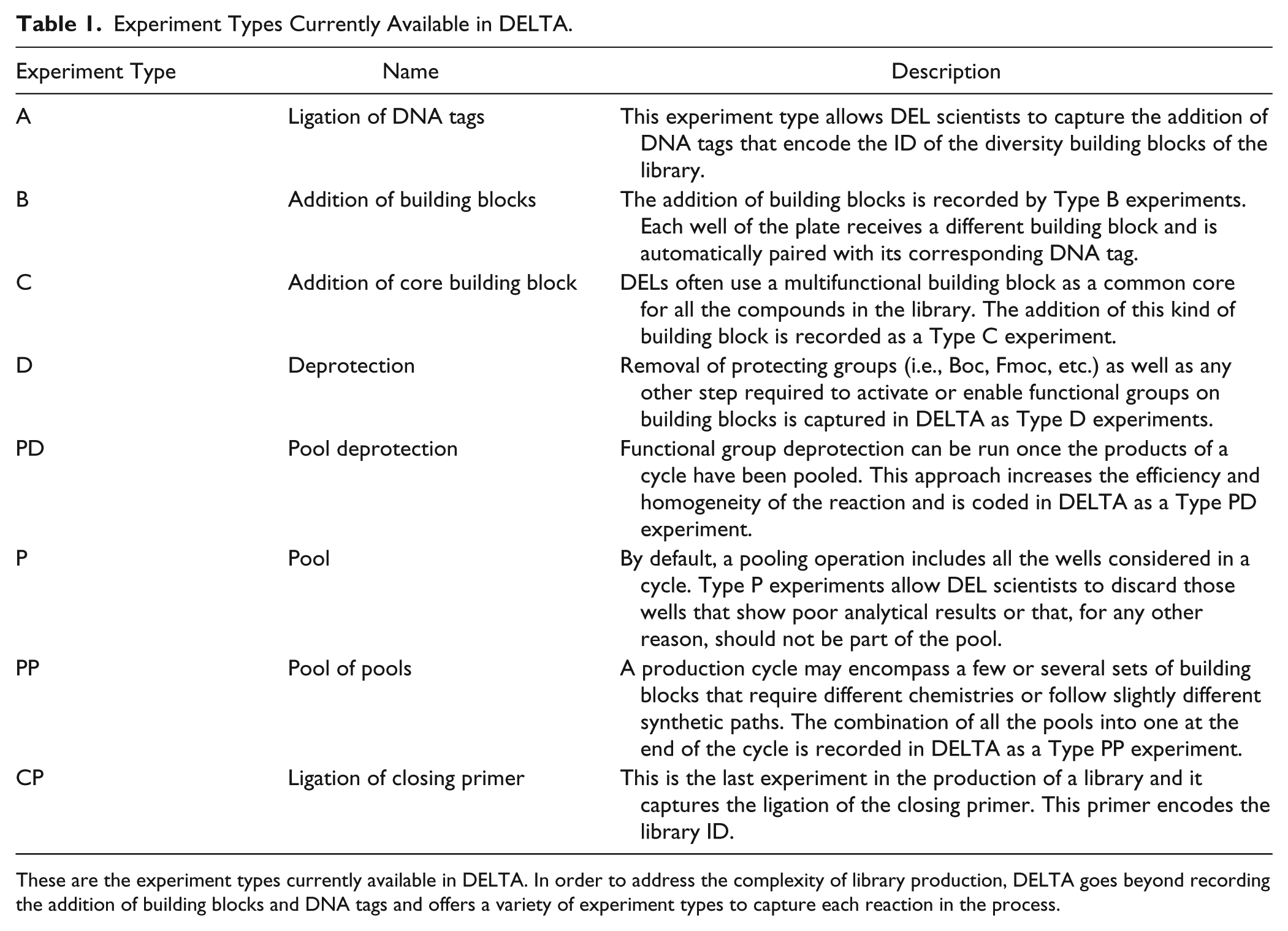
These are the experiment types currently available in DELTA. In order to address the complexity of library production, DELTA goes beyond recording the addition of building blocks and DNA tags and offers a variety of experiment types to capture each reaction in the process.
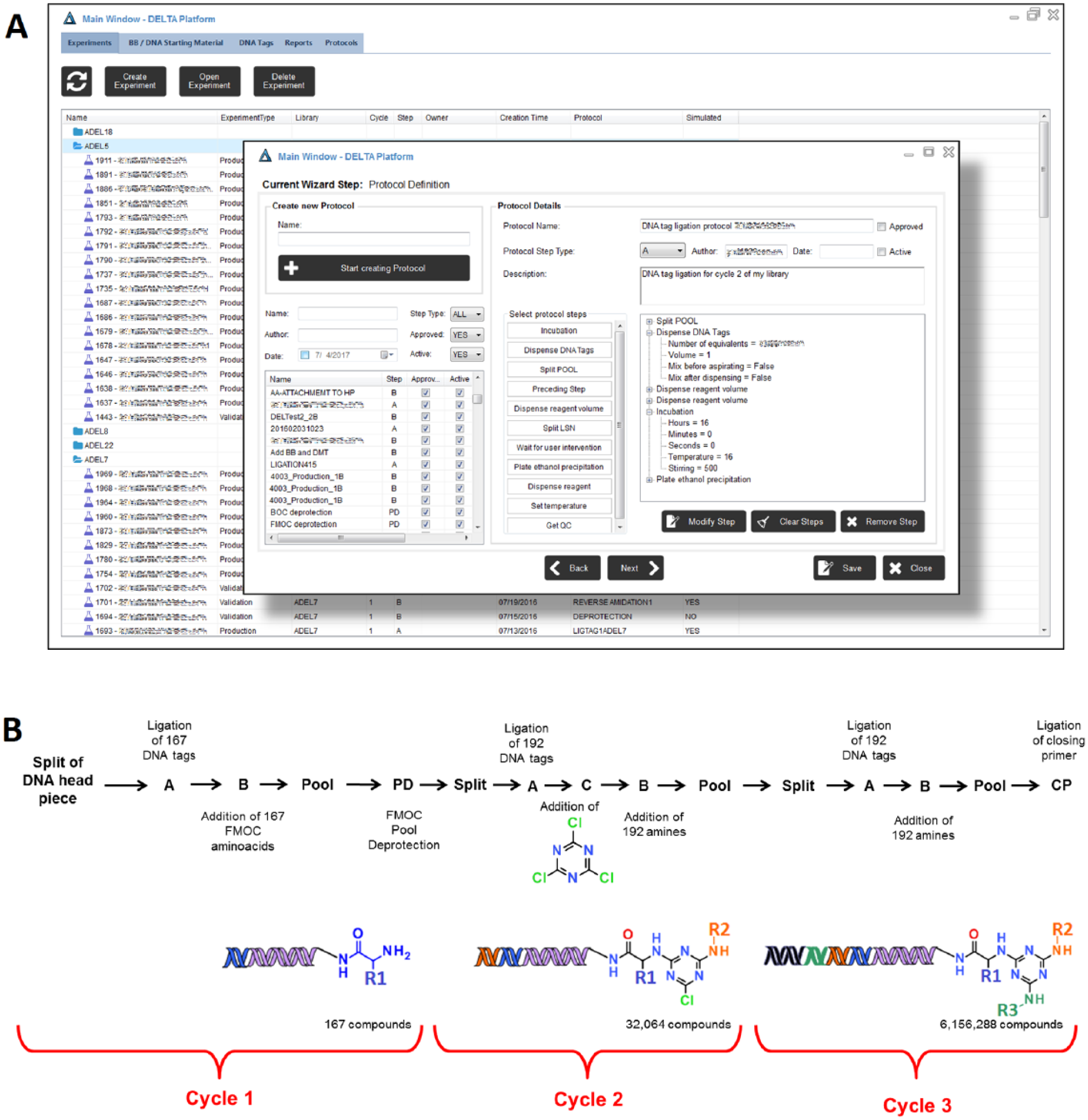
(
DEL-6 production started with a DNA headpiece functionalized with a free amine group. The first experiment comprised the ligation of 167 DNA tags, and the second one consisted of the amidation of the same number of Fmoc-protected amino acids. To deprotect the amines for the next reactions, DELTA allows the chemist to either create a Type D experiment to remove the Fmoc groups in all the wells or introduce a Type PD experiment to pool the amino acids after the amidation and deprotect all the products in a single pot. The latter approach was chosen for DEL-6. The sequence of linked experiments for the first cycle of DEL-6 was A > B > Pool > PD (see library synthesis flow scheme in Fig. 2B ).
The second cycle of DEL-6 started by splitting the pool generated in cycle 1 into 192 wells. After a ligation step, the free amines reacted through a NAS reaction with a common building block (cyanuric chloride). This was captured by DELTA as an experiment Type C. In the next step, introduced as Type B, the resulting intermediate reacted with 192 amines. The products were pooled and split again for cycle 3. After ligation, a new NAS step was performed with another 192 amines. In summary, the sequence of backward-linked experiments for this library was Split > A > B > Pool > PD > Split > A > C > B > Pool > Split A > B > Pool.
The last experiment in the production of the library was the ligation of a closing primer. The sequence of the closing primer encodes a unique library ID and, as explained below, drives deconvolution of DNA reads into final compound structures. The closing primer ligation was performed on the final pool of the library and was captured in DELTA as an experiment Type CP.
After each step of the library production, the process includes a purification (reaction quench) protocol using cold EtOH (EtOH crash). Taking advantage of the inherent characteristic of DNA of precipitating in EtOH at low temperatures (<5 °C), their addition to each well (or pool) within the process allows the separation of DNA-linked chemical entities from the excess of reagents, building blocks, buffers, or enzymes used in the different steps.
DELTA also allows for the registration of other experiment types that are frequently used in library productions, such as the combination of pools of reactions that result in intermediates featuring the same reactive group. This experiment is called pool of pools (PP) and facilitates the introduction of diversity through the inclusion of new building block sets.
By default, a pool in DELTA includes all the wells considered in an experiment. However, low synthetic yields, poor analytic results, or any other reason may suggest that some wells should be discarded. DELTA allows scientists to flag those wells and run the pooling operation selectively through a Type P experiment. Importantly, selective flagging of wells for further inclusion (or not) in the next step of the production is straightforward at the first cycle of a library, and possible in many cases at later points in the library synthesis by utilization of both UV retention times and Q-TOF of mass spectra of the complex mixtures.
Each experiment in DELTA is executed according to a protocol defined by the user. The protocol specifies the synthetic details of the reaction, including amounts of building blocks, DNA tags, additional reagents, catalysts or solvents, and times and temperatures of incubations and purification steps (ethanol precipitations). The protocols include the preparation of quality control (QC) plates with aliquots of each well for analysis by HPLC/MS. The generated data are used to monitor each reaction and ensure that the production meets the required quality standards. For every new experiment, the DEL scientist can write a custom protocol or load a preexisting one from the DELTA database ( Fig. 2 ).
Once the protocol is defined and the materials are ready, the DEL scientist prepares the deck of the liquid handler according to the layout predefined by DELTA. As plates are placed on the deck, plate barcodes are read and their contents are retrieved from the database and displayed on the user interface.
The reactor plates and the QC plates are also barcoded and introduced into the system ( Fig. 3 ). The software includes an experiment consumable and reagent validation function that visually indicates all of the items that must be configured on the liquid handler’s deck, and assists the user in registering the position of all materials before allowing the experiment to proceed. This facilitates full experimental traceability throughout the library production process.
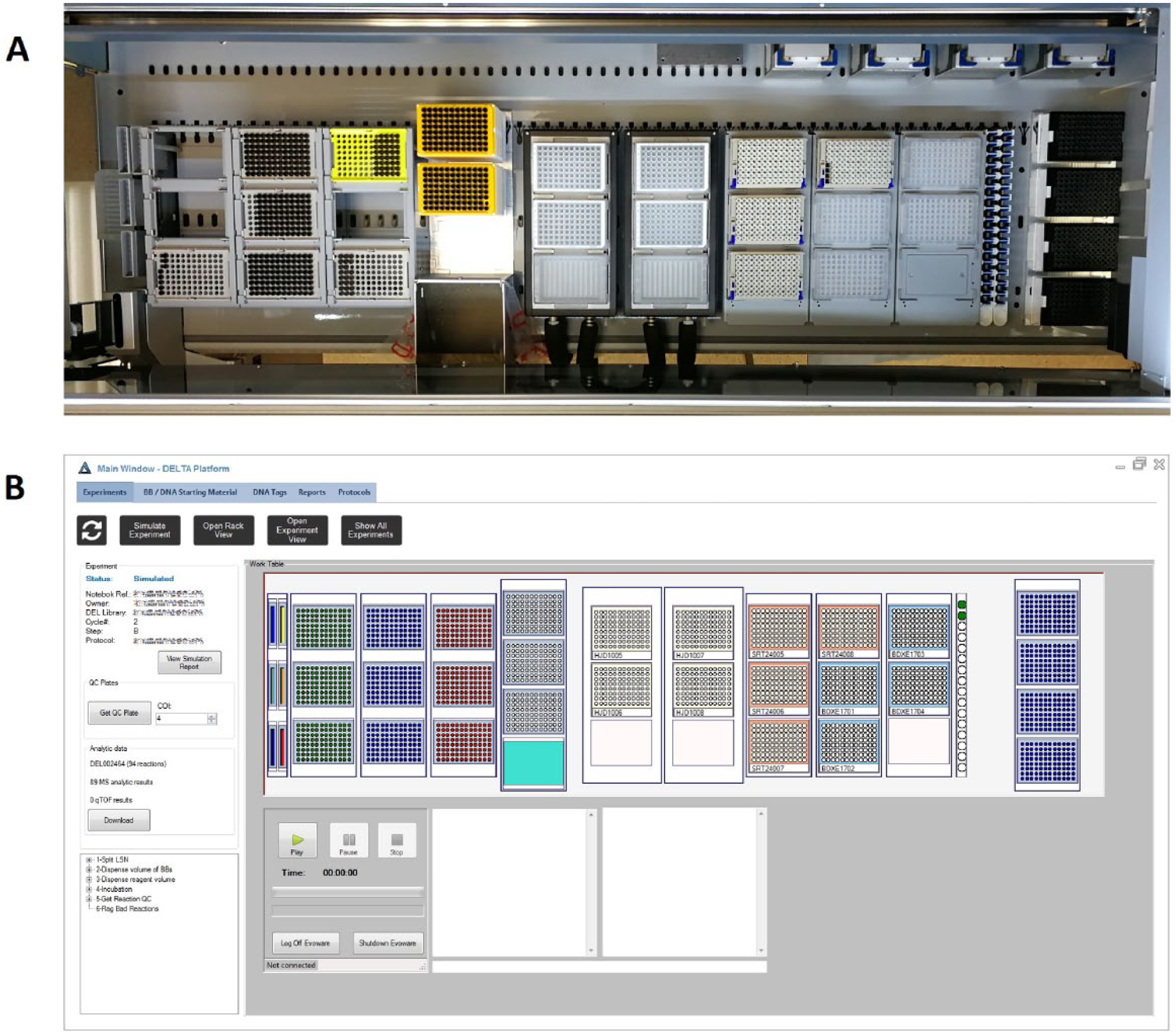
Experiment setup and execution. (
When an experiment is launched, DELTA compiles the protocol into a script and sends it to the liquid handler for execution. The progress of the run is displayed in real time, and any alert or error message the instrument may produce is echoed on DELTA’s application immediately. The use of the platform speeds up the liquid handling steps significantly. Besides, in terms of the overall library production process, automation provides relevant value regarding quality of work and ability to free up the scientist from tedious and error-prone tasks, like data tracking and manual manipulation.
If the protocol includes the preparation of QC plates, DELTA produces input files for the HPLC/MS instruments. These input files are comma-separated value (CSV) files that contain the expected masses of starting materials, expected final products and by-products for every reaction, and a DELTA database unique Result ID providing data traceability. These CSV files are imported by the instrument controller software to run the analysis.
Retention time and peak area data, as well as full report data files produced by the HPLC/MS instruments, are automatically swept by DELTA from OPENLAB servers through their Result ID key, imported back into the database, and made available to the users by means of a custom-made TIBCO Spotfire file. In the resulting file, key parameters are available (e.g., UV purity of product and ratio of desired product to starting material) and DEL scientists can visualize analytical data in a variety of ways that allow a rapid analysis of the reactions. According to our internal records, the time involved in QC analysis with this Spotfire file is reduced by 90% compared with manual inspection of instrument reports. Detailed analysis of individual wells is still possible through a link to the full LCMS data file.
DEL scientists rely heavily on these semiautomated analytical features during the library validation phase to survey building block reactivity and archive yields of each reaction under specified reaction conditions. Briefly, an experiment is created where a DNA headpiece is split into as many wells as candidate building blocks are available. According to the workflow explained above, these building blocks are added to the wells under the specific reaction conditions and QC plates are analyzed by HPLC/MS. Only those building blocks with high synthetic conversion are flagged for inclusion in the library production.
Affinity Selection and DNA Sequencing
Affinity selections are conducted using the same TECAN liquid handlers utilized for library production. Different protocols and conditions for the assay are set up and compiled into a library of EVOware scripts. These scripts offer the DEL scientist a wide range of possibilities in terms of immobilization procedures, incubation times, rate of liquid flow, temperatures and shaking speed, washing protocols, and so forth. The experimental protocols used in affinity selections, along with relevant metadata of the assays (e.g., therapeutic target and protein construct), are also captured in the DELTA database.
Affinity selections typically involve three rounds of incubations with the therapeutic target. DEL binders retained in the last round are sequenced by an external supplier that provides the list of detected DNA sequences, along with the number of times each of them was found in the samples, corrected for PCR duplicates identified using a degenerated region in the closing primer. 5
DEL Deconvolution
The DNA sequences detected after affinity selection need to be translated into the corresponding chemical structures of the compounds. This process, known as deconvolution, relies on the library production records to decode the DNA sequence and reproduce the steps of the synthesis of the final compounds.
Specifically, each valid DNA sequence read obtained in the affinity selection is syntactically processed and the DNA tag and closing primer IDs are extracted. The closing primer ID is used to query the database and obtain the last experiment of the production process. This experiment is the last node in the linked list of steps that led to the synthesis of the library. Following the links, the deconvolution algorithm can traverse backwards the node sequence and reconstruct the full synthetic route of each compound, starting from the ligation of the closing primer, all the way down to the initial split of the DNA headpiece. In this sequence, the start and end points of each cycle are recognized by the split-and-pool operations. Inside each cycle, the ligation steps (Type A experiments) and the addition of building blocks (Type B experiments) are matched and the DNA tags with their corresponding building blocks are unequivocally paired based on the wells where they were dispensed. This deconvolution process is fully automated and requires no user intervention.
Both the final compound structures and the sequencing counts are inserted in the DELTA database and assigned to the affinity selection metadata.
Affinity Selection Data Analysis and Pattern Detection
Analysis of affinity selection results is carried out in a TIBCO Spotfire file highly customized for DEL needs. The file is directly connected to the DELTA database to retrieve the results of any stored selection on demand. Users can browse the list of selections, load sequencing data, run a standardized analysis pipeline, or build their own.
As part of the standardized analysis pipeline, this Spotfire file represents the chemical space of each library at each affinity selection as a 3D plot where each axis is an array of building blocks of one of the cycles ( Fig. 4 ). For those libraries with more than three cycles, two dimensions may be collapsed into one by considering all pairwise combinations of their building blocks. Compounds found in the affinity selection are represented by dots or spheres in the chemical space, whose size is proportional to the number of counts. The DEL scientist can hover over each of these spheres to see the chemical structure of the compound, its constituent building block IDs, the corresponding DNA tags, and the number of counts. These data are also found in an accompanying data table that can be conveniently filtered by any field. The user can introduce a threshold value to separate noise from real signals. This threshold can be determined based on the theoretical shape of the distribution of counts for each sample, positive and negative encoded tool compounds, and other experimental considerations.
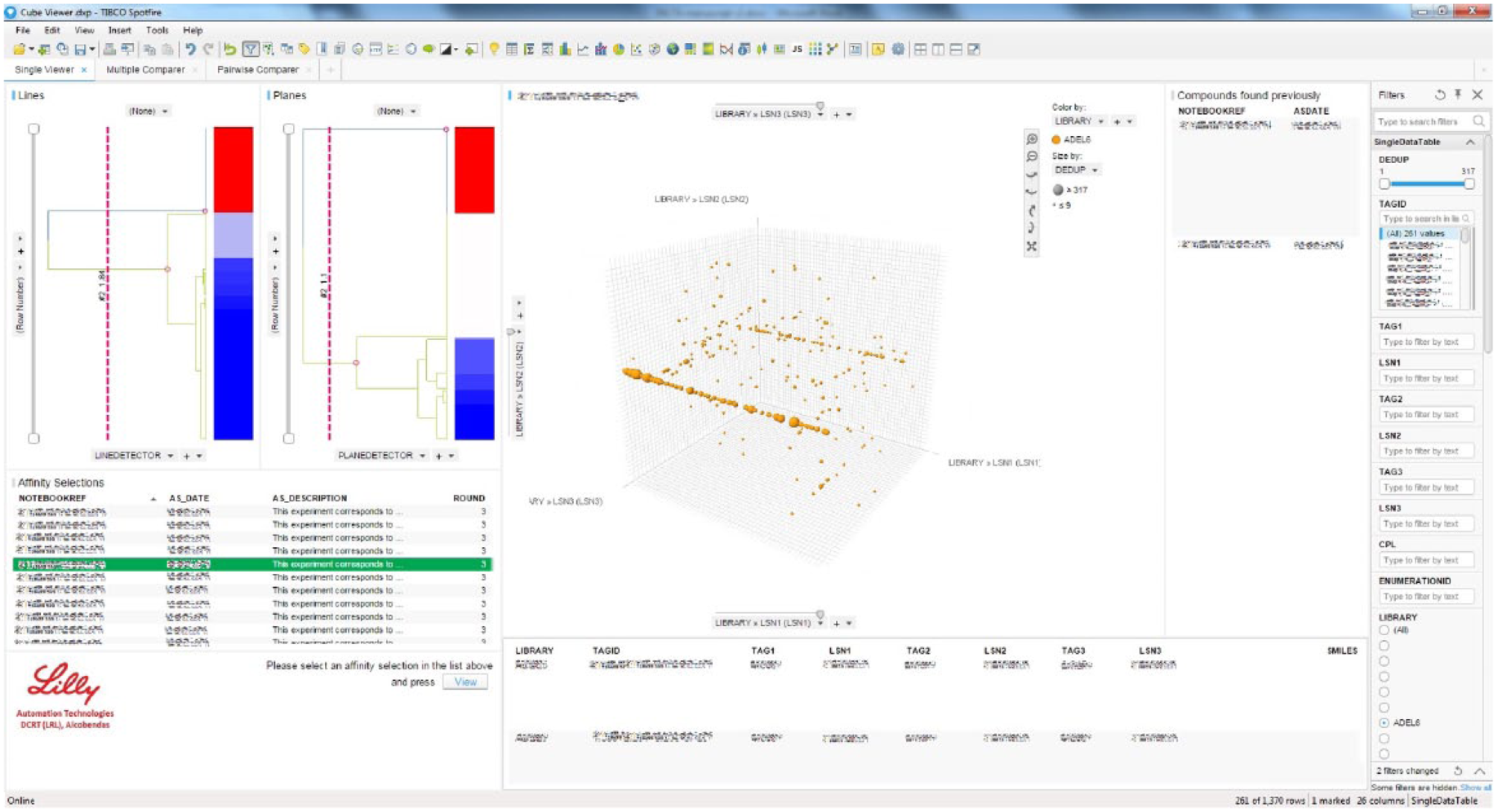
Analysis of affinity selection results. A customized TIBCO Spotfire file allows DEL scientists to retrieve, explore, and analyze affinity selection results stored in the DELTA database. On the left-hand side, the user can browse and search the list of affinity selections. The main pane displays the results of each library against the assayed therapeutic target. The user can see the sequencing counts, deconvoluted chemical structures, building block IDs, encoding DNA tags, and so forth. On the right-hand side, both the data spreadsheet and the plot can be filtered and searched by any relevant criteria. Lines and planes can be automatically detected by means of hierarchical clustering, shown on the upper left-hand side.
For any detected compound, the Spotfire file can run a database query to retrieve its sequencing counts in any previous affinity selection. This is particularly useful to obtain an estimate of the selectivity of the compound against different targets or compare results across different selection conditions against the same library and target.
This representation usually reveals sets of compounds organized along planes or lines. Planes evidence the existence of a single building block that binds the target regardless of which other building blocks or structures take part in the compound. Similarly, lines indicate that a substructure composed of the combination of two particular building blocks is capable of binding the protein. Both lines and planes are automatically detected in this Spotfire-based application by means of agglomerative hierarchical clustering. The algorithm groups together lines and planes with similar sums of counts and separates those consisting of many compounds (and/or higher counts) from others with very few or no representatives. The user can filter the chemical space visualization by cluster and see only the lines and planes that are relevant for the analysis.
Finally, DEL scientists can build their own data analysis pipelines with the built-in functionality of Spotfire. They can insert new calculated columns with different data normalizations, create new visualizations for results, or run customized comparisons with the embedded TIBCO enterprise runtime for R.
DELTA has been validated through the production of the Lilly DEL collection and affinity selection screening against a variety of therapeutic targets. Its database-centric approach, combined with its web service–based architecture and integration with laboratory equipment, ensures process reliability, efficiency, and traceability.
Registration of the whole synthetic process as a sequence of backward-linked experiments, together with a high degree of flexibility for capturing a wide range of experiment types, allows full and automatic reconstruction of the synthetic route of every compound regardless of the complexity of the library design.
Compared with the common practice of storing process data in spreadsheets, DELTA represents a superior standard of data confidentiality, integrity, and availability. 21 Access to sensitive data, such as building block IDs, library designs, laboratory process records, or affinity selection results, is restricted according to the company’s policies, such as password-based authentication, roster review controls, and so forth. The use of a high-end relational database and validated implementations of data management procedures guarantees atomicity, consistency, isolation, and durability. 22
DELTA extends these features to laboratory automation by integrating with liquid handlers and HPLC/MS equipment. Execution scripts and instrument input files automatically generated by the platform reduce human intervention, prevent data handling errors, and contribute to the consistency and reliability of results.
Finally, DELTA aids in the design and execution of experiments while collecting high-quality experimental data and metadata in a central repository. Mining this information in context with state-of-the-art analytical and reporting tools allows users to extract meaningful conclusions from experimental results to improve the productivity of the DEL platform.
Footnotes
Acknowledgements
The authors thank Jesús Ezquerra, Miguel Ángel Toledo, and Alfonso Rivera for their review of the manuscript and all the stimulating discussions.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.