
Abstract
The discovery of ligands via affinity-mediated selection of DNA-encoded chemical libraries is driven by the quality and concentration of the protein target. G-protein-coupled receptors (GPCRs) and other membrane-bound targets can be difficult to isolate in their functional state and at high concentrations, and therefore have been challenging for affinity-mediated selection. Here, we report a successful selection campaign against protease-activated receptor 2 (PAR2). Using a thermo-stabilized mutant of PAR2, we conducted affinity selection using our >100-billion-compound DNA-encoded library. We observed a number of putative ligands enriched upon selection, and subsequent cellular profiling revealed these ligands to comprise both agonists and antagonists. The agonist series shared structural similarity with known agonists. The antagonists were shown to bind in a novel allosteric binding site on the PAR2 protein. This report serves to demonstrate that cell-free affinity selection against GPCRs can be achieved with mutant stabilized protein targets.
Keywords
Introduction
The protease-activated receptors (PARs) are a class of G-protein-coupled receptors (GPCRs) that are activated by extracellular proteases.1–3 Proteolytic cleavage near the receptor N-terminus reveals a stimulatory sequence that can bind the receptor in an intramolecular fashion and induce signaling. Four members of the PAR family have been described (PAR1–4), and some of these have attracted interest as therapeutic targets. PAR1, for instance, is antagonized by the anticlotting drug Vorapaxar. 4
PAR2 is expressed in a variety of vascular and smooth muscle tissues, including in the gastrointestinal tract, the lung, and the skin. Proteolytic activation of PAR2 unmasks the peptide sequence SLIGKV, which then binds and induces inflammatory signaling cascades. Antagonists to PAR2 signaling could have potential application in inflammation, pain, and other diseases. Despite the therapeutic potential of such agents, relatively few PAR2 antagonists have been described.5,6 Several authors of this report previously reported the cocrystal structures of several PAR2 antagonists bound to the receptor. 7 The discovery of one of these antagonists via affinity-mediated selection of DNA-encoded chemical libraries (DECLs)8–10 is described here.
GPCRs are membrane-spanning proteins that have traditionally been difficult to isolate in a soluble, nonaggregated, and correctly folded form. Because the affinity selection of DECLs typically requires the use of highly pure and soluble protein targets, GPCRs pose obvious challenges. Several recent reports have described approaches to overcoming these challenges ( Fig. 1 ). In a model study, 11 the β2 adrenergic receptor was expressed in insect cells and extracted and purified in detergent-containing buffers. The receptor was the wild-type sequence with the exception of an N-terminal FLAG tag and point mutation of a glycosylation site. This detergent-solubilized receptor was loaded onto anti-FLAG beads and used for affinity-mediated selection with DECLs; a novel antagonist with an IC50 of 1.9 µM was discovered. Cooperative binding with an orthosteric ligand revealed this compound to be an allosteric antagonist.
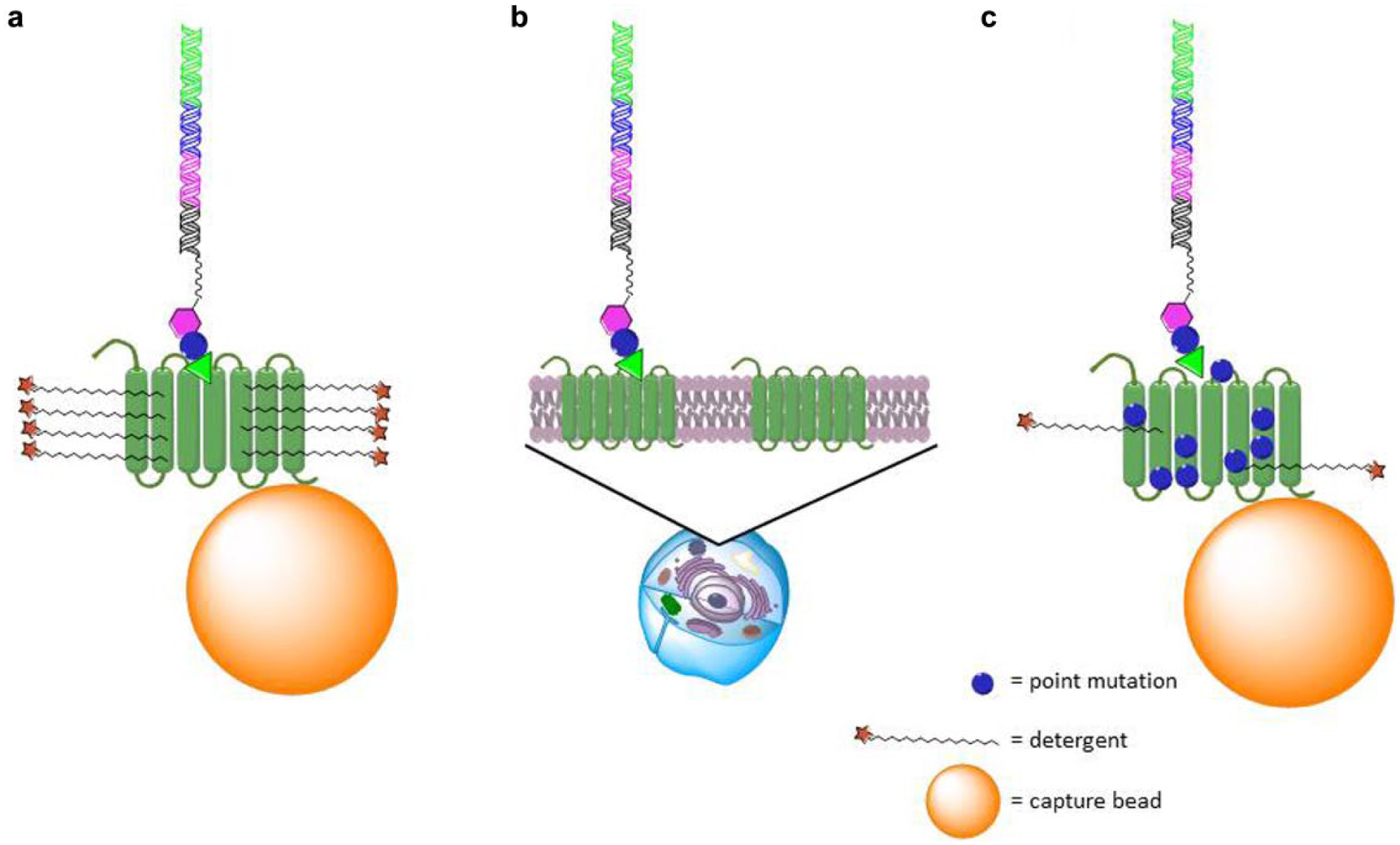
Whole cells have also been reported to be an effective means to present high-quality, high-concentration proteins to DECLs. In a seminal report, 12 workers at GSK described their efforts to discover antagonists of NK3, a GPCR of the tachykinin family. Rather than purify the receptor, they expressed it at high levels in HEK293 cells using a BacMam viral vector. Cells transduced with virus without the NK3 gene were used as negative controls. Affinity-mediated selection was conducted directly on the cells, and several families of antagonists were discovered, with potencies down to single-digit nanomolar. It should be noted that the expression levels of NK3 were high (500,000 copies/cell). High expression levels in this system are presumably needed to mimic the high concentrations of purified soluble proteins required for conventional affinity-mediated selection experiments.
For our efforts with PAR2, we turned to a third approach: mutational stabilization.7,13 Thermostabilized GPCRs (StaRs) have found utility in structural studies of GPCRs14–16 and in GPCR lead and fragment-based discovery.17,18 StaRs are generated by iterative point mutations and exhibit increased stability relative to wild-type receptors while retaining their ability to bind to known ligands. StaRs can be stabilized in particular conformations and activation states depending on whether agonist or antagonist binding is maintained through the mutational stabilization process. The utility of StaRs is evidenced by the growing number of GPCR x-ray structures obtained using StaR constructs.
In our PAR2 campaign, we wished to determine not only which library compounds bound to PAR2, but also their binding sites and competitive behavior. In order to derive this information, an affinity-mediated selection campaign was designed in which binders were identified by incubation with the target in the absence of any ligands, and parallel selections were also undertaken in which binding sites were fully occupied using known ligands as competitors. Competition is often used in DECL affinity selection to classify library members on the basis of their competitiveness, and to form hypotheses around binding site and modulatory function. Examples include the use of dasatinib to probe the ATP site of BTK, 19 SB-235375 to identify NK3 antagonists, 12 and ZSTK474 to identify ATP-competitive ligands for PI3Kα. 20 Use of competitors in biophysical screening is not limited to DECL selection; affinity selection–mass spectrometry (AS-MS) screens routinely utilize competitors to characterize binding sites of hits, 21 including when screening GPCRs such as the M2 acetylcholine receptor. 22
Materials and Methods
Small-Molecule Pull-Down by Immobilized PAR2
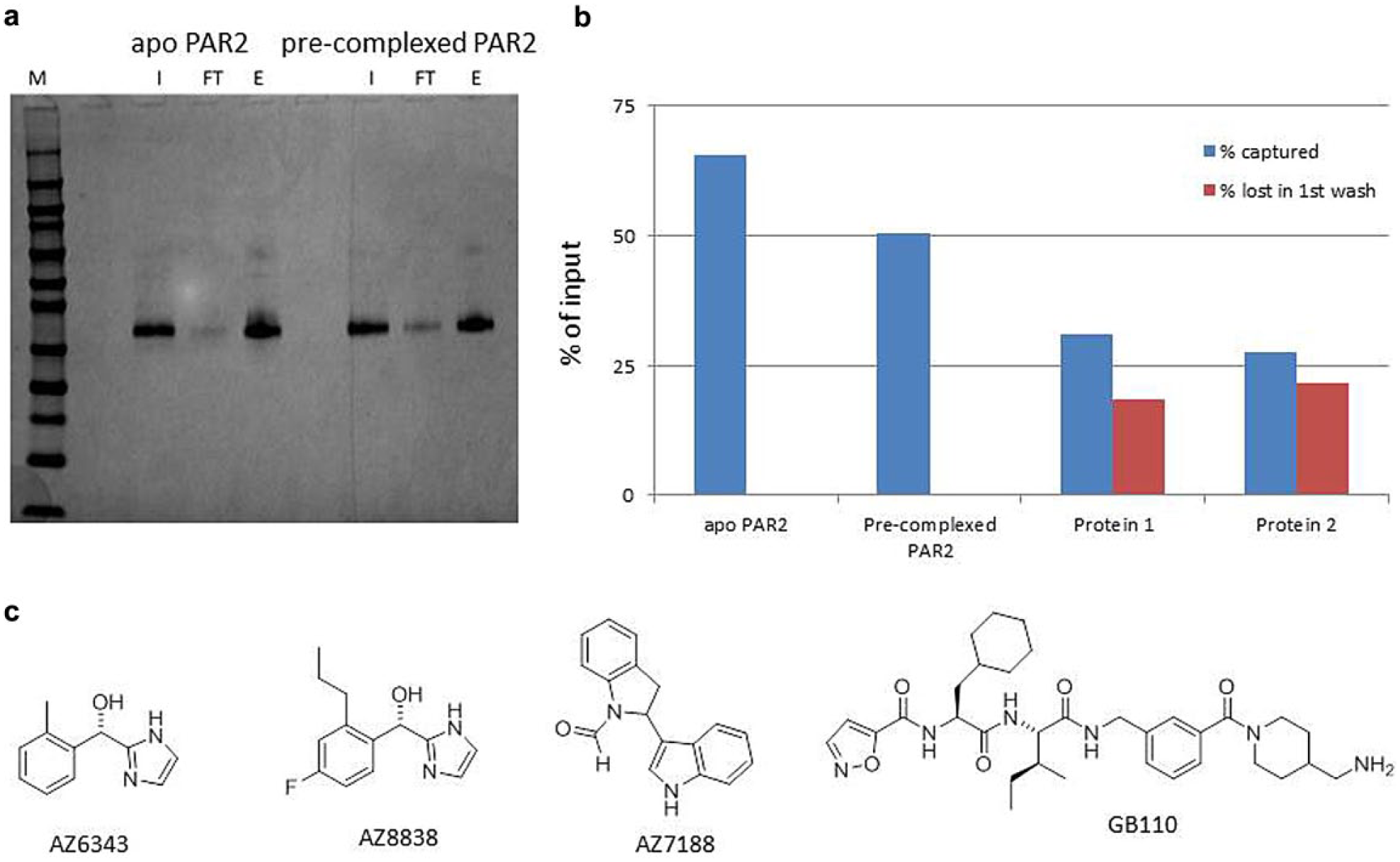
PAR2 (final concentration 5 µM) and a small molecule (AZ8838 or AZ7188, final concentration 5 µM) were incubated in selection buffer (50 mM HEPES, 150 mM NaCl, 0.004% lauryl maltose neopentyl glycol, 0.002% cholesteryl hemisuccinate, 0.02% DMSO, 1 mg/mL sheared salmon sperm DNA, pH 7.4) in a total volume of 60 µL for 1 h at room temperature. Protein immobilization was then conducted using a 5 µL bed of nickel affinity matrix (His-Select High-Flow Nickel Affinity Gel; Phynexus, San Jose, CA) and 20 passages followed by three washes with 200 µL of incubation buffer aliquots. Elution was performed by heating the matrix to 95 °C in the presence of NuPAGE LDS sample buffer (Thermo Fisher, Waltham, MA). Protein capture was assessed using denaturing polyacrylamide gel electrophoresis (PAGE) for the input, flow-through, and elution. Relative concentrations of small molecule were assessed for the input, flow-through, and washes using liquid chromatography–mass spectrometry (LCMS). No-protein control and off-target capture assays were also run for comparative purposes.
Affinity-Mediated Selection of DECLs
This campaign comprised seven parallel selection conditions in which 10 nmol of a mix of 20 different DECLs was diluted into selection buffer (50 mM HEPES, 150 mM NaCl, 0.004% lauryl maltose neopentyl glycol, 0.002% cholesteryl hemisuccinate, 0.02% DMSO, 1 mg/mL sheared salmon sperm DNA, pH 7.4) and incubated with no protein or 5 µM PAR2 supplied either precomplexed or in the apo form. For the selections conducted with PAR2 supplied in the apo form, library mix and competitors, where included, were incubated together in solution in a 60 µL volume for 1 h at room temperature prior to capture on a 5 µL bed of nickel affinity matrix (His-Select High-Flow Nickel Affinity Gel) by 20 passages, followed by eight washes with 200 µL of incubation buffer aliquots. For the selections initiating with precomplexed PAR2, the protein and competitors, where appropriate, were preincubated without the library and captured on the tip, as described above, followed by three washes of the capture matrix. The library mix was then added to the captured proteinm and then followed by eight additional washes. For selections that included competitors, these were present in all incubations and washes. In all cases, retained library members were eluted by incubation with 60 µL of selection buffer at 72 °C for 5 min, followed by a further incubation with a second 5 µL resin bed of His-Select High-Flow Nickel Affinity Gel to remove any eluted protein. After the first round of selection, the selection protocol was repeated using the library eluted from round 1 for each of the seven conditions. Encoding oligonucleotides present in the output of the second selection round were amplified using Platinum PCR Supermix (Invitrogen, Waltham, MA) with denaturation at 94 °C, annealing at 55 °C, and extension at 72 °C for 24 cycles using 5′ and 3′ primer oligonucleotides (each at 0.5 µM), which each incorporate sequences complementary to the tailpiece or headpiece, along with Illumina READ1 or READ2 sequences, required to support clustering and subsequent single-read 100 base-pair sequencing on an Illumina HiSeq 2500. Sequencing was also performed for PCR-amplified samples of the naïve library for normalization purposes.
Cell Culture
1321N1 cells stably expressing the human PAR2 receptor were cultured in Dulbecco’s modified Eagle’s medium (DMEM) + GlutaMAX supplemented with 10% heat-inactivated fetal bovine serum (FBS), 1% pen/strep, 1% G418, and 1% nonessential amino acids. HEK293 cells were cultured in DMEM supplemented with 10% heat-inactivated FBS. To detach cells, PBS containing 1 mM EDTA was used because trypsin-based solutions would cause receptor activation.
Functional Inositol Phosphate Assays
Stimulation of Gαq by PAR2 induced phosphalipase C (PLC) activation and triggered an inositol phosphate (IP) signaling cascade. Inositol-1-phosphate (IP1) is a downstream metabolite of inositol-3-phosphate (IP3), which accumulates in cells following Gαq activation and is stable in the presence of LiCl. Cell-based IP1 assays (Cisbio BioAssys, Codolet, France) were performed in 384-well plates. 1321N1 cells stably expressing the human PAR2 receptor were thawed in media, centrifuged at 1000 rpm for 4 min, and resuspended in preassay buffer (HBSS buffer containing 20 mM HEPES, pH 7.4). In agonist mode, test compound or DMSO controls were prepared in stimulation buffer (HBSS buffer containing 20 mM HEPES, 100 mM LiCl, pH 7.4), serially diluted, and added to wells as required. Subsequently, cells were seeded at a density of 15,000 cells/well into plates and incubated for 30 min at 37 °C, 5% CO2. Accumulation of IP1 was measured by addition of the IP1-d2 conjugate, followed by the Ab-cryptate according to the manufacturer’s instructions, and incubated at room temperature for 1 h with gentle agitation. Assay plates were read using a PHERAstar homogeneous time-resolved fluorescence (HTRF) cAMP protocol under a time-resolved fluorescence program at 620 and 665 nM. This is a competition-based assay, so that the HTRF ratio is directly inversely proportional to the IP1 concentration. In antagonist mode, test compound or DMSO controls were prepared in preassay buffer, serially diluted as required, and added to the plate. Cells were seeded at a density of 15,000 cells/well into plates and incubated at 37 °C, 5% CO2. The agonist peptide, SLIGRL, was prepared as a 2 × EC80 concentration (30 µM) in stimulation buffer. After 30 min, the agonist peptide was added to wells to a final assay concentration of 15 µM and cells were incubated for a further 60 min at 37 °C, 5% CO2.
Results and Discussion
The PAR2 StaR used in affinity-mediated selection experiments has been described previously. 7 Binding experiments with tool compounds indicated that the PAR2 StaR was in an antagonist-stabilized conformation; the receptor was not able to bind the known agonist GB110. 12 Two preparations of the PAR2 StaR were used in selection: an apo form (a-PAR2), which lacked a bound ligand, and a precomplexed form (pc-PAR2), which was supplied as a complex with the antagonist AZ6343. Previous work had shown that expression and purification in the presence of AZ6343 substantially increased the thermal stability of the purified PAR2 (data not shown). For the selection experiments, pc-PAR2 was captured on the immobilization matrix and then washed to remove AZ6343. During our protein quality control (QC) process, we used MS to determine that both a-PAR2 and pc-PAR2 could bind each of the competitors AZ8838 and AZ7188 following immobilization ( Fig. 2 ).
(
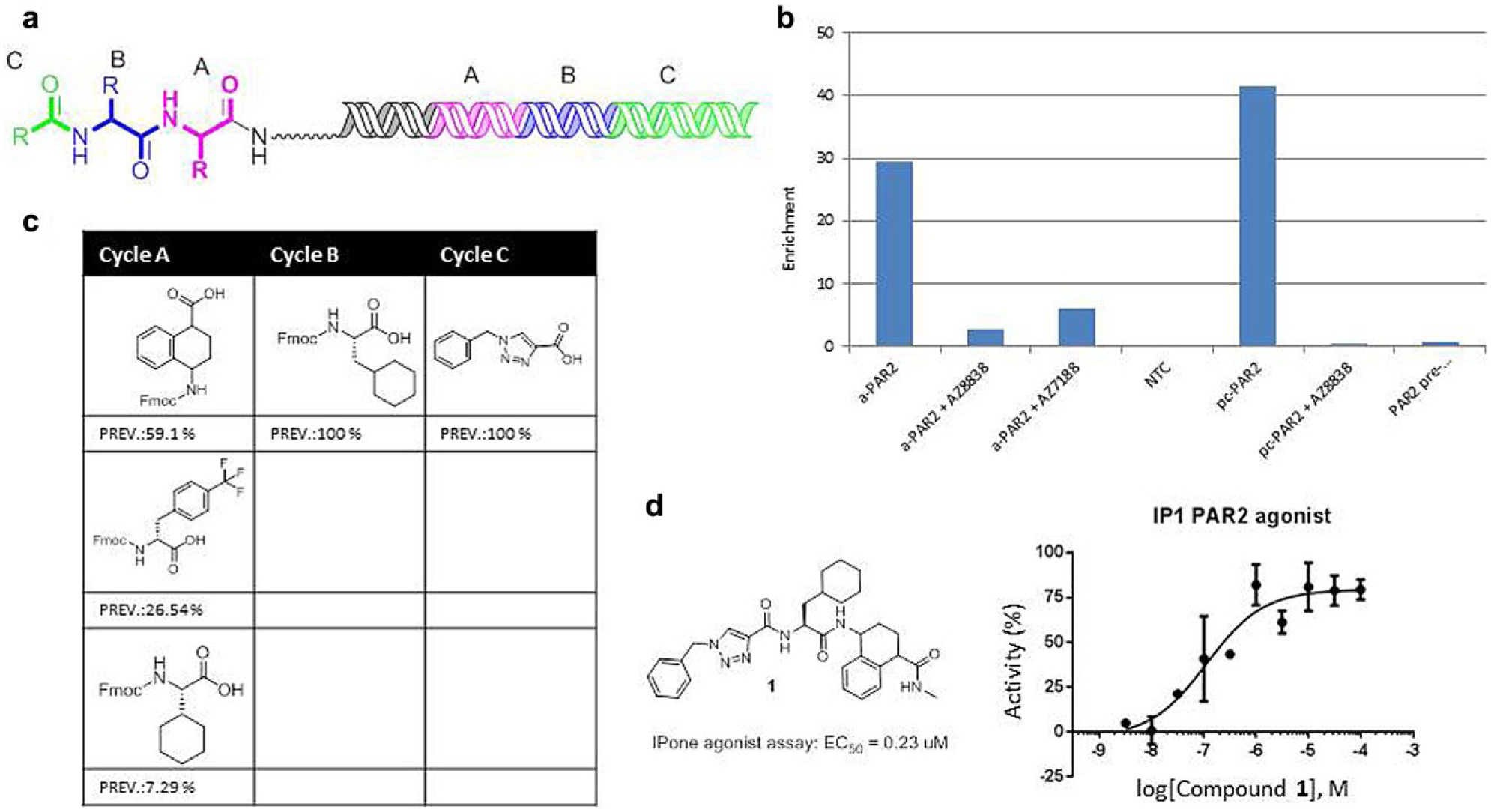
An affinity-mediated selection campaign was designed to identify library compounds able to bind to PAR2 and to provide competition information with respect to the PAR2 antagonists AZ8838 and AZ7188. This campaign was comprised of seven parallel selection conditions in which a mix of 20 different DECLs were diluted into selection buffer and incubated with no protein (as a negative control) or 5 µM of either a-PAR2 or pc-PAR2. A further variable employed was the presence or absence of AZ8838 or AZ7188. For the selections conducted with a-PAR2, library mix and competitors (when included) were incubated with protein in solution prior to capture. For the selections initiating with pc-PAR2, the protein was captured on the immobilization matrix, followed by washing. The library mix was then added to the immobilized protein. For selections that included competitors, these were present in incubations and washes. In all cases, retained library members were eluted by heat-induced target denaturation at 72 °C. After the first round of selection, the selection protocol was repeated using the library eluted from round 1 for each of the seven conditions. Encoding oligonucleotides present in the output of the second selection round were then amplified, clustered, and sequenced using Illumina/Solexa technology, and each sequence was then translated to identify individual library members. The visualization and analysis of DECL selection outputs have been described previously,19,23 and were applied to the PAR2 output. Identified families were then profiled for their behavior across the various selection conditions. We find that selection profiles can be conveniently visualized as bar plots, 19 in which each bar represents a selection condition and the height of each bar represents a family’s enrichment under that condition.
Analysis of the output sequence set revealed two families of structurally related compounds whose structures and selection behavior led to their prioritization for further study. The first (family 1) bore a striking structural similarity to GB110 and the native peptide sequence SLIGKV (
Fig. 3
). Family 1 showed enrichment across both forms of the receptor, and exhibited competitive behavior with both competitors (
Fig. 3b
). Family 1 was discovered within a 225-million-member capped dipeptide library. The most prevalent building blocks at each cycle of family 1 are shown in
Figure 3c
. Cycles B and C showed completely specific selection, with
(
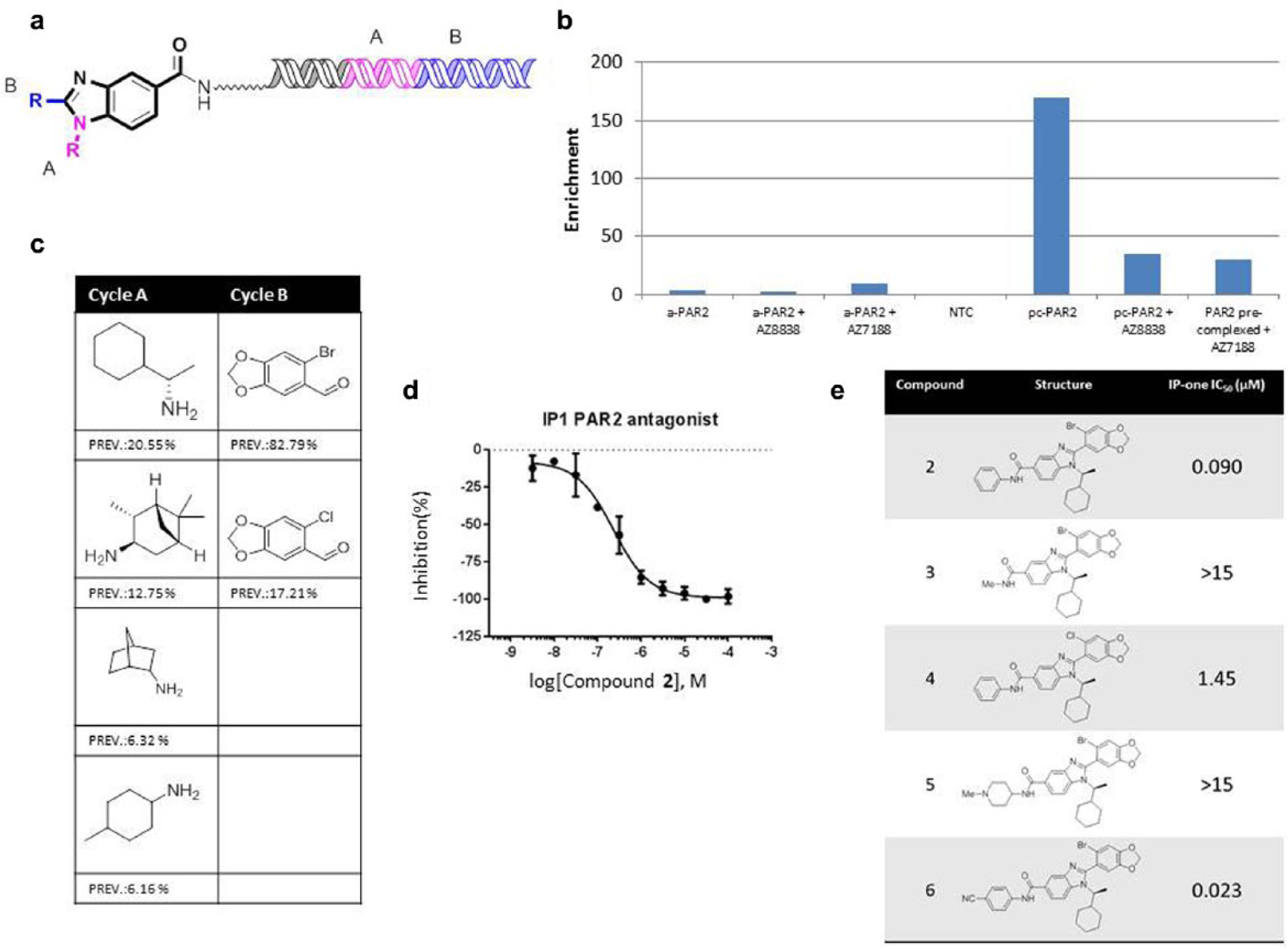
A second cluster of compounds, family 2, was also prioritized (
Fig. 4
). In contrast to family 1, the second family showed much stronger enrichment with pc-PAR2 than with a-PAR2. Family 2 was also competitive with both added ligands. Structurally, family 2 bore no resemblance to known PAR2 modulators. It was discovered within a 7.1-million-member two-cycle library based on a benzimidazole core. Cycle A showed a marked preference for α-branched cycloalkyl amines (
Fig. 4c
). Cycle B was exclusively 2-halo-4,5-methylenedioxybenzaldehyde, with the bromo analog showing higher prevalence than the chloro one. In the library, the benzimidazole core was attached by either aliphatic or aromatic linkers. Family 2 showed a strong preference for the aromatic linker (data not shown), and so we designed compound
(
Compound
More thorough biophysical characterization of this class of compounds is ongoing, and will be reported in due course. It is interesting to note that in contrast to the family 1 agonists, the family 2 antagonists enriched much more strongly against pc-PAR2 than against a-PAR2. The stronger enrichment with pc-PAR2 would seem to indicate that it retained an antagonist-binding conformation to a greater extent than a-PAR2. Whether this observation is due to the stabilizing effect of the precomplexed ligand or some stabilizing effect of immobilization of the receptor (prior to library incubation) cannot be determined from the available data.
In conclusion, this study illustrates the power of StaRs for affinity-mediated selection of DECLs. Selection using a PAR2 StaR yielded both the first example of a DECL-derived GPCR agonist and a novel series of allosteric antagonists. We suggest that these results indicate that future use of StaRs with DECL technology will be a fruitful approach to the discovery of novel modulators of GPCRs.
Footnotes
Acknowledgements
The authors thank Dr. Daniel Resnicow for his help with preparing this manuscript.
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors acknowledge AstraZeneca, Heptares, and X-Chem Pharmaceuticals for financial support.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.