
Abstract
The endocannabinoid system (ECS) plays a diverse role in human physiology ranging from the regulation of mood and appetite to immune modulation and the response to pain. Drug development that targets the cannabinoid receptors (CB1 and CB2) has been explored; however, success in the clinic has been limited by the psychoactive side effects associated with modulation of the neuronally expressed CB1 that are enriched in the CNS. CB2, however, are expressed in peripheral tissues, primarily in immune cells, and thus development of CB2-selective drugs holds the potential to modulate pain among other indications without eliciting anxiety and other undesirable side effects associated with CB1 activation. As part of a collaborative effort among industry and academic laboratories, we performed a high-throughput screen designed to discover selective agonists or positive allosteric modulators (PAMs) of CB2. Although no CB2 PAMs were identified, 167 CB2 agonists were discovered here, and further characterization of four select compounds revealed two with high selectivity for CB2 versus CB1. These results broaden drug discovery efforts aimed at the ECS and may lead to the development of novel therapies for immune modulation and pain management with improved side effect profiles.
Introduction
The endocannabinoid system (ECS) plays a central role in the regulation of a number of physiological processes in the human body, including mood, appetite, pain, memory formation, and the immune response.1,2 Therefore, modulation of the ECS has been increasingly explored for therapeutic intervention in human health and disease.3,4 Included among the biomolecular constituents of the ECS are the cannabinoid receptors CB1 and CB2, which belong to the guanine nucleotide binding protein (G protein)–coupled receptor (GPCR) superfamily of proteins, and couple to Gi/o, which inhibits the intracellular accumulation of the second messenger, cyclic adenosine monophosphate (cAMP). 5 Due to the diverse roles of the ECS and the success of targeting GPCRs in drug development, 6 the cannabinoid receptors are encouraging yet underexplored targets in the discovery of novel pharmacotherapies.
Currently marketed drugs that target the cannabinoid receptors include Cesamet (nabilone), Marinol [dronabinol/Δ9-tetrahydrocannabinol (Δ9-THC)], and Sativex (Δ9-THC and cannabidiol). Cesamet and Marinol are prescribed primarily to treat nausea and vomiting in patients undergoing chemotherapy, whereas Sativex is prescribed primarily to patients with multiple sclerosis to treat neuropathic pain. 4 The most well-known drug-containing substance with modulatory effects on the ECS, however, is marijuana, which contains a number of pharmacologically active compounds, including Δ9-THC, and elicits feelings of relaxation and analgesia.7,8 Although Δ9-THC and other cannabinoids are promising in their therapeutic effects, unfortunately, a number of negative side effects are also associated with their use, including depression, tolerance, and dependence.2,9 Although Δ9-THC and the aforementioned drugs are known to bind to CB1 and CB2 receptors, 10 the psychoactive side effects have largely been associated with modulation of the CB1 receptor expressed in neurons throughout the CNS.11,12 As a consequence, drugs that solely target the CB2 receptor, achieved by developing CB2-selective scaffolds, may abrogate these negative side effects12,13 and are attractive for future ECS modulation.
The CB2 receptors, unlike the CB1 subtype, are largely expressed peripherally in cells of the immune system,1,5 although they are also distributed in the CNS, primarily in microglia.5,14 Due to their unique expression pattern, drug discovery efforts targeting the CB2 receptor have largely been focused on the treatment of pain and inflammation, although utility for other indications is also possible. 15 For example, CB2 expression is increased by inflammatory stimuli, 16 and neuropathological findings in human brains suggest that upregulation of CB2 is a common response to chronic injury, and may play an important role in disease-associated neuroinflammation 17 and the inhibition of glial cell tumors. 18 For instance, CB2 receptors are upregulated in microglial cells located in the periphery of senile plaques of people with Alzheimer’s disease and in the striatum of a rat model of Huntington’s disease. In microglia, this upregulation is thought to control the production of neurotoxic factors, including nitric oxide, proinflammatory cytokines, and reactive oxygen species. Furthermore, cannabinoid receptor knockout mice reveal that analgesia is likely mediated by CB2 and not by CB1.19,20 Thus, identification of compounds that activate the CB2 receptor to the exclusion of the CB1 receptor may promote analgesia and modulate the body’s response to inflammation, without the psychoactive side effects associated with CB1 activation.
Because CB1 and CB2 receptor subtypes share sequence homology (~44% overall similarity, ~68% within the transmembrane regions),21,22 identification of ligands with a high degree of selectivity for CB2 versus CB1 has proven challenging. 22 One emerging approach for improving selectivity is the discovery and development of allosteric modulators. 23 Allosteric modulators are compounds that bind to an allosteric site that is topographically distinct from the binding site of the endogenous ligands (the orthosteric site). Allosteric binding sites are often less evolutionarily conserved than orthosteric sites, providing greater opportunity for small molecules to exhibit selectivity between receptor subtypes, as well as providing other potential therapeutic advantages over traditional orthosteric agonists. 24 Although positive allosteric modulators (PAMs) of the CB1 receptor have previously been identified,25–29 there are no known CB2 allosteric modulators. Structural modeling of the CB2 receptor, however, suggests the existence of an allosteric binding pocket adjacent to the orthosteric site, 30 supporting the idea that it may be possible to discover CB2 PAMs.
Here, we performed a high-throughput screening (HTS) of a 60,000-compound library in mammalian cells to identify agonists or PAMs of the CB2 receptor. Identification of potent, CB2-selective compounds will not only contribute to the pharmacological toolkit available to probe the physiological role of the cannabinoid receptors, but also provide potential starting points for the development of novel drugs to treat pain and inflammation without the psychoactive side effects associated with CB1 drugs.
Materials and Methods
High-Throughput Screening
The HTS was performed using a collaboration deck that consisted of approximately 60,000 commercially available compounds arranged in 1536-well Echo Qualified source plates (Labcyte, Sunnyvale, CA). Each compound was dissolved in DMSO at a concentration of 3 mM. Compounds were then transferred to white, tissue culture–treated, 1536-well assay plates (3727BC; Corning, Corning, NY) by acoustic dispense using an Echo-550 (Labcyte) to yield a final concentration of 15 µM in the screening assay. Two separate screens were performed, and each of the 60,000 compounds were tested a single time in each of the two screens. To discover any PAMs present in the compound set, screening was performed in the presence of the CB2 orthosteric agonist CP 55,940 (Sigma-Aldrich, St. Louis, MO). CP 55,940 was dissolved in DMSO and transferred to the assay plates by acoustic dispense. In Screen 1, we used an approximate EC10 concentration of CP 55,940 that was determined by testing the compound in a 10-point concentration–response curve using an assay measuring the inhibition of cAMP accumulation, described herein (30 pM; Suppl. Fig. 1 ). In Screen 2, we used an EC25 concentration of CP 55,940 (50 pM; Suppl. Fig. 1 ). Both the screening compounds and CP 55,940 were added prior to HTS assay initiation.
Cryopreserved Chinese hamster ovary (CHO-K1) cells expressing recombinant human CB2 receptors (CHO-K1-CB2) were then added to the assay plates to yield 500 cells per well. Prior to cryopreservation, cells were grown to confluence in F-12 media supplemented with 10% fetal bovine serum (FBS), 100 IU/mL penicillin, 100 µg/mL streptomycin, and 250 µg/mL G418, in T-175 tissue culture flasks, and harvested with TrypLE Express. For use in the screens, cells were thawed, washed in F-12 media with 1% FBS, and then resuspended in assay buffer before being added to the assay plates. The assay buffer was Hanks Balanced Salt Solution (HBSS) with calcium and magnesium supplemented with 25 mM HEPES, 100 IU/mL penicillin, 100 µg/mL streptomycin, and 0.05% bovine serum albumin (BSA; Sigma-Aldrich). All media and supplements were purchased from Life Technologies (Carlsbad, CA) unless otherwise indicated.
Finally, forskolin (Sigma-Aldrich) was added to the assay plates (5 µM final concentration) to stimulate adenylyl cyclase activity. Forskolin was dissolved in DMSO and diluted in assay buffer. The final concentration of DMSO in the assay was 0.67%. Assay plates were then incubated for 45 min at room temperature, followed by lysis and cAMP detection. Unlike many other cAMP accumulation assays, the inhibition of the forskolin-stimulated cAMP window in CHO-K1-CB2 cells was more robust in the absence of the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX), so IBMX was not added to the assay conditions.
As a positive control, each assay plate contained two columns that were treated with a maximally effective concentration of CP 55,940 (33 nM; Suppl. Fig. 1 ) to define maximal (100%) inhibition of forskolin-stimulated cAMP accumulation. As a negative control, each assay plate also contained two columns that were not treated with CP 55,940 (DMSO alone) to define control levels (0% inhibition) of forskolin-stimulated cAMP accumulation.
Hit Assessment
Concentration–Response Curves
Ten-point concentration–response curves were performed for hits in both the presence (PAM mode) and absence (agonist mode) of CP 55,940, and in CHO-K1 cells lacking the expression of the CB2 receptor (null mode). Compounds were serially diluted in DMSO, with threefold dilutions between concentrations, ranging from 20 µM to 1 nM, and then transferred by acoustic dispense into 1536-well assay plates. For PAM mode, concentration–response curves were performed as in the primary screens. For agonist mode, the addition of the EC10 concentration of CP 55,940 was replaced with DMSO. For null mode, agonist mode was performed in CHO-K1 rather than CHO-K1-CB2 cells, and the positive control was assay buffer without forskolin because a maximally effective concentration of CP 55,940 was not expected to produce a response in the parental cells.
Each concentration–response curve was performed in triplicate. Concentration–response data were plotted as percentage inhibition of forskolin-stimulated cAMP accumulation and fit to a logistic equation using nonlinear regression analysis to provide estimates of Emax (top) and potency (EC50) in GraphPad Prism 7 [log(agonist) versus response (three parameters)]; bottom is constrained at 0, as defined by vehicle control wells (GraphPad Software, La Jolla, CA).
Inhibition of cAMP Accumulation Assay
To test for CB2 receptor activation, we measured inhibition of forskolin-stimulated cAMP accumulation. Assay plates with test compounds, forskolin, and cells were incubated at room temperature for 45 min, and reactions were terminated by the sequential addition of Eu-cAMP and ULight reagents from the LANCE Ultra cAMP Detection Kit (PerkinElmer, Cambridge, MA) in a 1:1 ratio. Following a 1 h incubation at room temperature, time-resolved fluorescence resonance energy transfer (TR-FRET) was measured on an Envision plate reader (PerkinElmer) with excitation at 337 nm and emission reads at 615 nm and 665 nm. The ratiometric data [(665 nm read / 615 nm read) × 10,000] were then converted to [cAMP] based on a standard curve for cAMP run at the same time and under identical conditions to the assay.
Radioligand Binding Assay
To determine the binding affinity of test compounds for cannabinoid receptors, human embryonic kidney (HEK293) cells were transfected with plasmid DNA encoding the human CB1 or human CB2 receptor, and 24 h posttransfection membranes were prepared as described previously. 31 Ligand binding was also determined essentially as described previously.31,32 In brief, nine different concentrations of the test compounds (from 100 pM to 100 µM) were incubated with cell membranes and the tracer [3H]CP 55,940 (150.2 Ci/mmol; PerkinElmer) at 1.5 nM to achieve a robust signal while staying lower than the KD determined for this system. 33 Nonspecific binding was determined using 1 µM unlabeled CP 55,940. Experiments were performed in duplicate three times, and only two times when no binding was detected.
GTPγS Binding Assay
Stimulation of guanosine-5′-O-(3-[ 35 S]thio)triphosphate ([ 35 S]GTPγS) binding was used as a direct measure of G protein activation produced by agonists acting through the human CB2 receptor. Membranes prepared from HEK293 cells expressing recombinant human CB2 receptor (8 µg/well) were incubated with 0.1 nM [ 35 S]GTPγS (1250 Ci/mmol; PerkinElmer), 10 µM guanosine 5′-diphosphate (GDP) (Sigma-Aldrich), and 0.1% (w/v) BSA in the presence and absence of varying concentrations of the test compounds (typically, 1 nM to 10 µM) in GTPγS binding assay buffer (50 mM Tris-HCl, pH 7.4, 3 mM MgCl2, 0.2 mM EGTA, and 100 mM NaCl). 10 µM unlabeled GTPγS (Sigma-Aldrich) was used to determine nonspecific binding. The binding reaction was stopped by filtration, and bound [ 35 S]GTPγS was determined as described previously. 32 Experiments were performed in duplicate three times.
Results
HTS Identifies CB2 Agonists
To identify agonists or PAMs of the CB2 receptor, a collaboration deck consisting of approximately 60,000 commercially available compounds was screened by the Lead Discovery group at Bristol-Myers Squibb (BMS) in collaboration with Yale University through its Educational Alliance program. We screened compounds in CHO-K1 cells expressing the human CB2 receptor for inhibition of forskolin-stimulated increases in cellular cAMP levels in the presence of a low (EC10) concentration (30 pM; Suppl. Fig. 1 ) of the orthosteric agonist CP 55,940. Screening in the presence of an orthosteric agonist enables detection of both agonists and PAMs in a single screen. Conditions were chosen based on assay optimization experiments, which included testing fresh versus cryopreserved cells, various cell-seeding densities, and varying concentrations of forskolin (1–20 µM). Once optimized, the screening run was completed in a single day, in workstation mode (40 × 1536-well plates). Despite assay optimization efforts, assay robustness for this screen was less than ideal, with a Z’ median of 0.45 (±0.09) and a signal-to-background ratio (S:B) of 3.2, as defined by a maximally active concentration of CP 55,940 (33 nM; Fig. 1 ).
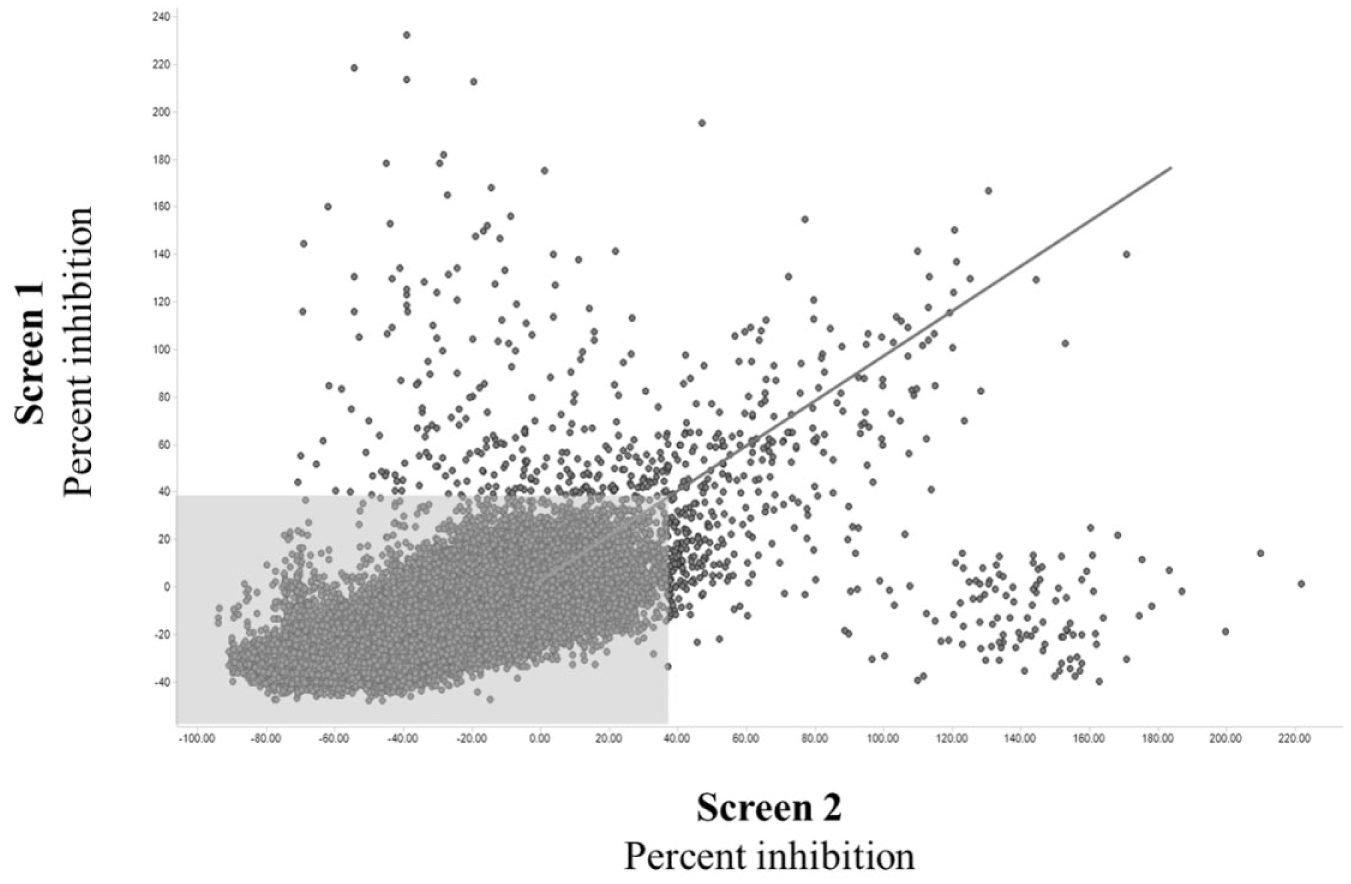
Two high-throughput screens identify 678 potential cannabinoid receptor-2 (CB2) agonists or potentiators. Two screens of approximately 60,000 compounds identified 678 compounds that decreased forskolin-evoked cyclic adenosine monophosphate (cAMP) accumulation in CB2-expressing cells. Screen 1 was performed in the presence of an EC10 concentration (30 pM) of CP 55,940 and resulted in a signal-to-background (S:B) ratio of 3.2 and Z’ median of 0.45 (SD = ±0.09). Screen 2 was performed using an EC25 concentration (50 pM) of CP 55,940 and resulted in a S:B of 2.5 and Z’ median of 0.35 (SD = ±0.12). Hits were defined as compounds that produced ≥37% inhibition of cAMP accumulation relative to the maximum response produced by CP 55,940 (33 nM), in either of the two screens. Compounds that fall outside of the shaded gray box were considered to be hits in one or both screens. Compounds within the gray box are nonhits and were excluded from further analysis.
Because weak (low-cooperativity) PAMs exhibit greater activity as the concentration of an orthosteric agonist is increased,24,34 to enhance our chances of identifying PAMs of the CB2 receptor, we repeated the HTS using a higher EC25 (50 pM; Suppl. Fig. 1 ) concentration of CP 55,940. As the concentration of the orthosteric agonist is increased, however, the “noise” (or scatter) associated with the assay is also typically increased. 24 Thus, as expected, the higher concentration of CP 55,940 further reduced assay robustness, resulting in a Z’ median of 0.35 (±0.12) and S:B of 2.5 ( Fig. 1 ). In addition, these screening conditions revealed a number of spurious false positives (cluster of compounds in the top left corner and lower right corner of Fig. 1 ) that did not validate after further testing. Nonetheless, combining the results of these two screens provided two data points for each of the 60,000 test compounds, thereby increasing the chances of identifying any PAMs or agonists that may have been present in the compound set. “Hits” were defined as the top 1% of compounds, which corresponded to compounds that produced ≥37% inhibition of cAMP accumulation relative to the maximal inhibition produced by CP 55,940 in either screen. This cutoff yielded 678 hits.
Hit Characterization in cAMP Accumulation Assay
The activities of hits were then validated in 10-point concentration–response curves (ranging from 1 nM to 20 µM) in both the presence (PAM mode) and absence (agonist mode) of CP 55,940 to distinguish potential PAM hits from direct agonists. Compounds active in PAM mode but inactive or significantly less potent in agonist mode would be considered putative PAMs, whereas agonists would be active in both modes. Hits were also tested in parental CHO-K1 cells lacking the expression of the CB2 receptor (null mode) to confirm that observed effects on cAMP accumulation were CB2 mediated. Hits active in null mode are likely producing their effects through “off-target,” non-CB2-mediated mechanisms and were therefore excluded from further testing. Because only a small fraction of compounds elicited a response in null mode, for these compounds we compared potency and efficacy values among assays to determine whether the response was CB2 mediated or nonselective. Of the 678 initial hits, 167 validated in the 10-point concentration–response curves and were CB2 mediated (devoid of activity in null mode; Suppl. Fig. 2 ). All validated hits showed similar activity in both PAM and agonist modes (potency range: ~1 nM to >30 µM), suggesting that all of the compounds identified in our screen were direct CB2 agonists, with no PAMs being identified. Of the 167 active compounds identified, 47 displayed a potency greater than 100 nM (EC50 < 100 nM) in the cAMP accumulation assay. From these 47 hits, 4 compounds were initially selected for follow-up characterization based on a number of considerations, including observed potency in initial cAMP experiments as well as logistical factors such as compound availability. Concentration–response curves for select validated CB2 agonists ( Table 1 ), and their structures, are depicted in Figure 2 , and their EC50 and Emax values provided in Table 2 . Future efforts will include the characterization of the remaining hits, including structure–activity relationship (SAR) studies.
CB2 Agonists Selected from the Screen for Further Characterization.

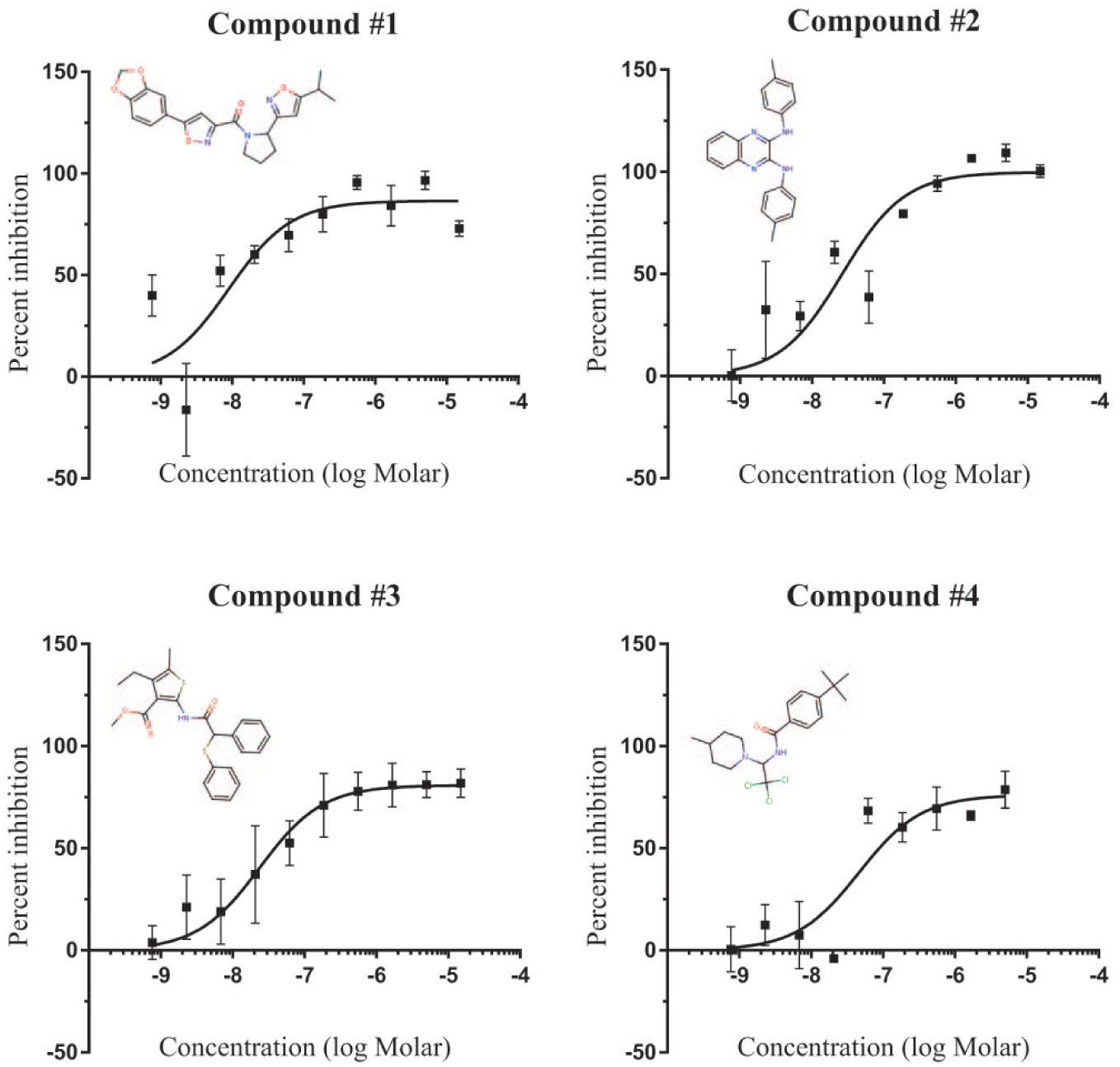
Concentration–response curves for selected CB 2 agonist hits. Each hit was tested in the inhibition of cAMP (cyclic adenosine monophosphate) accumulation assay at 10 different concentrations, ranging from 1 nM to 20 µM, in both the presence [positive allosteric modulator (PAM) mode] and absence (agonist mode) of the orthosteric ligand CP 55,940. Hits were also tested in cells that did not express the CB2 receptor (null mode) to rule out off-target effects. Although no PAMs were identified, 167 agonists were discovered, 47 of which exhibit EC50 values lower than 100 nM. Four CB2-selective agonists from the agonist mode, and their structures, are shown (mean ± SD; n = 3).
The log EC50, log EC50 95% Confidence Intervals, and ECmax Values for the Four CB2-Selective Agonists (Compounds 1–4) in Figure 2 .
Radioligand Binding Reveals CB2-Selective Agonists
The selected CB2 agonists were further characterized using a radioligand binding assay to determine their affinities for CB1 and CB2 receptors based on each test compound’s ability to compete with [3H]CP 55,940 for receptor binding. Using membranes prepared from HEK293 cells expressing the CB2 receptor, Compound 1 displaced the binding of [3H]CP 55,940 with a Ki value of 2.3 µM; pKi: −5.6 [95% confidence interval (CI): lower, −6.2; upper, −5.1] ( Table 3 ). In membranes prepared expressing the CB1 receptor, however, Compound 1 produced no measurable inhibition of [3H]CP 55,940 binding. Compound 4 also inhibited [3H]CP 55,940 binding to CB2 receptors (Ki: 0.2 µM; log Ki: −6.6; 95% CI: −6.9, −6.3) with no detectable inhibition of [3H]CP 55,940 binding to CB1 receptors. These data suggest that both Compounds 1 and 4 selectively bind CB2 receptors while showing no appreciable binding to CB1 receptors at concentrations up to 10 µM. Compound 2, in contrast, inhibited binding of [3H]CP 55,940 to CB2 (Ki: 1.1 µM; log Ki: −6.0; 95% CI: −6.2, −5.7) but also displayed measurable affinity for CB1 receptors (Ki: 1.8 µM; log Ki: −5.7; 95% CI: −6.7, −4.8). Compound 3 also inhibited [3H]CP 55,940 binding to both CB2 receptors (Ki: 0.8 µM; log Ki: −6.1; 95% CI: −6.5, −5.7) and CB1 receptors (Ki: 1.6 µM; log Ki: −5.8; 95% CI: −6.8, −4.8). Thus, Compounds 2 and 3 are only weakly selective for CB2 versus CB1.
The Ki Values, log Ki Values, and Corresponding 95% Confidence Intervals of Selected Compounds for CB1 and CB2.
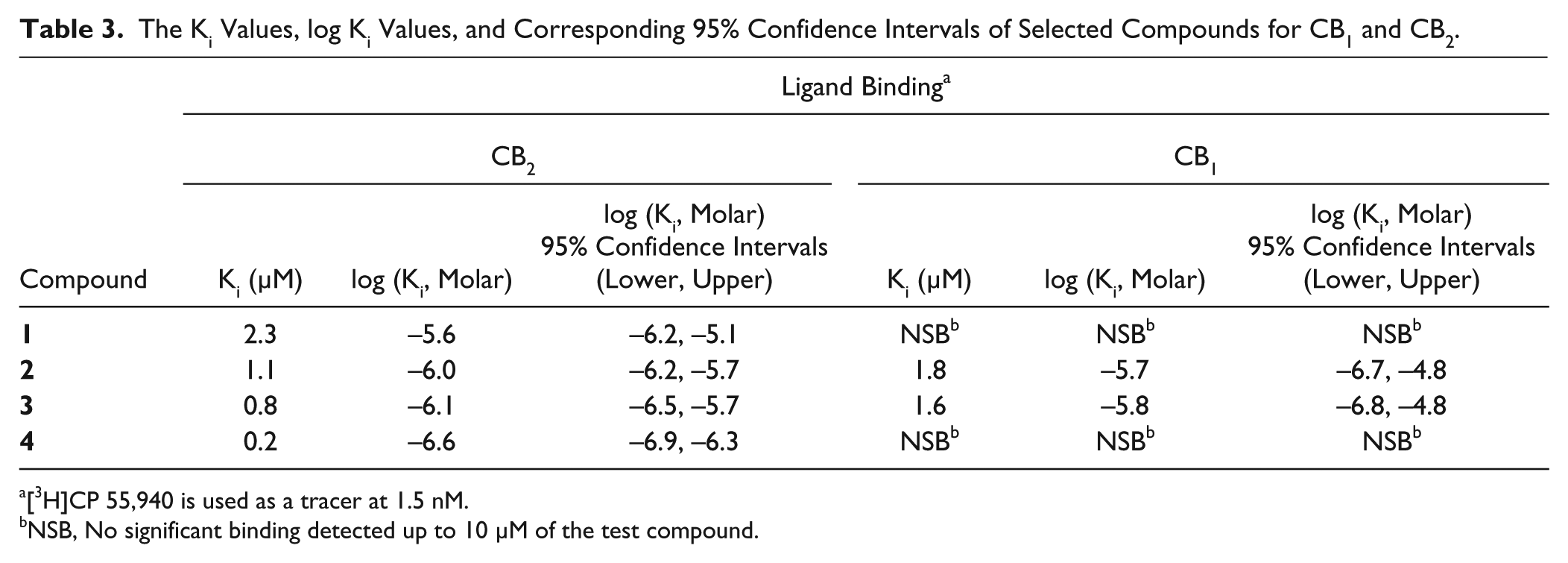
[3H]CP 55,940 is used as a tracer at 1.5 nM.
NSB, No significant binding detected up to 10 µM of the test compound.
Characterization of Selected Hits Using a [ 35 S]GTPγS Binding Assay
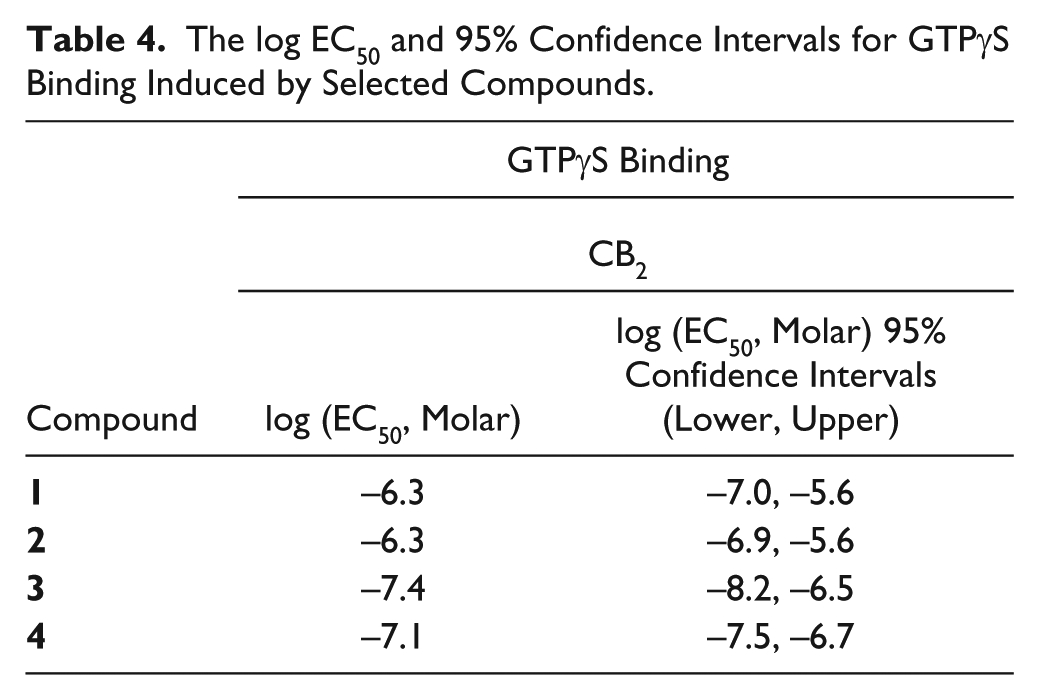
The CB2 receptor couples to G proteins, predominantly the adenylyl cyclase inhibitory G proteins Gαi/o. Therefore, agonist-stimulated binding of [ 35 S]GTPγS was used to characterize agonist potency in a direct measure of receptor/G protein activation. 35 Cell membranes prepared from HEK293 cells expressing the CB2 receptor were incubated with [ 35 S]GTPγS and varying concentrations of each compound. The EC50 values for Compounds 1–4 were 0.5 µM, log EC50: −6.3 (95% CI: −7.0, −5.6); 0.6 µM, log EC50: −6.3 (95% CI: −6.9, −5.6); 0.03 µM, log EC50: −7.4 (95% CI: −8.2, −6.5); and 0.08 µM, log EC50: −7.1 (95% CI: −7.5, −6.7), respectively ( Table 4 ).
The log EC50 and 95% Confidence Intervals for GTPγS Binding Induced by Selected Compounds.
Discussion
In this study, we aimed to identify compounds with selectivity for CB2 versus CB1 receptors. The identification of allosteric modulators is one strategy for attaining selectivity, because allosteric ligands can sometimes exhibit superior selectivity between related receptor subtypes compared to orthosteric ligands, which bind to a site that is often more evolutionarily conserved. 24 We therefore performed a HTS of approximately 60,000 compounds in the presence of the orthosteric agonist CP 55,940, to allow the identification of agonists and PAMs of CB2. However, the two screens of the 60,000 compounds that were performed, differing only in the concentration of CP 55,940 (EC10 and EC25), did not reveal PAMs. Given that PAMs have previously been identified for the CB1 receptor, these results were somewhat surprising. There are several possible explanations for the lack of PAM hits – one is that, despite structural modeling that suggests otherwise, 30 “druggable” allosteric sites simply may not exist on CB2.
There are, however, many other potential explanations for the lack of PAM hits from this screening effort. As discussed above, the assay robustness for this HTS campaign was less than ideal, and therefore it is possible that weak PAM hits may have been obscured by the relatively high noise level in the assay. It is also possible that our results were limited by the screening conditions we used and that screening using a different assay to detect G protein–dependent signaling, 36 or an alternative signaling pathway,37,38 would reveal PAMs. For instance, a label-free technology, such as a cell-impedance-based assay, has been used to evaluate CB2 agonists, including the selective JWH133. 36 This avoided use of markers or dyes for detection, which were used here for detection of cAMP changes. Furthermore, the cannabinoid receptors display a range of signaling bias, or the ligand-dependent differential activation of one signaling cascade over another. Cannabinoid ligands have been documented to harbor β-arrestin bias as well as G protein bias, including bias among the various G proteins.39–41 Allosteric modulators of the cannabinoid receptors, too, have been shown to elicit biased responses.42,43 For example, the allosteric modulator ORG27569 inhibits G protein coupling, while activating β-arrestin-2–mediated receptor internalization and β-arrestin-1–mediated ERK1/2 signaling.42,43 Furthermore, CB1 PAMs at this point have largely been shown to potentiate β-arrestin signaling rather than G protein signaling. 25 Thus, although our screen focused on the identification of allosteric modulators that potentiate G protein signaling, screening compounds with an assay to detect changes in β-arrestin activity, like the PathHunter β-galactosidase fragment complementation assay (DiscoverX Corporation, Fremont, CA), which has been validated with the CB1 receptor for HTS campaigns, 37 may be more likely to lead to the discovery of CB2 PAMs. Because CB2 typically undergoes desensitization and internalization after activation, an assay that is independent of the coupling partner’s composition may be warranted. 37
Another factor to consider is that, due to technical considerations, the current screens were run using the synthetic agonist CP 55,940 as the orthosteric agonist, rather than a more physiologically relevant endocannabinoid. PAMs for CB1 have been identified using CP 55,940 as the orthosteric agonist probe. Nonetheless, because of the phenomenon of “probe dependence” that allosteric modulators can exhibit, 24 we could have success identifying PAMs for CB2 screening with a different orthosteric agonist (e.g., 2-AG or anandamide).
A more trivial explanation is that PAMs of CB2-mediated cAMP inhibition may exist but were not present in the compound collection that was screened. The current screens used a relatively small deck of 60,000 commercially available compounds that, although diverse, may simply not have contained any compounds that are PAMs for CB2. Therefore, we cannot conclude that PAMs of CB2 do not exist simply because none were identified from the current HTS. Nonetheless, the fact that hundreds of CB2 agonists were identified from this screen suggests that PAMs of CB2 may be rarer than orthosteric agonists.
The screens did reveal a number of CB2 agonists, 47 of which had a potency of greater than 100 nM (EC50 < 100 nM), a relatively large number of high-potency hits from a screen of this size. Of these novel agonists, four selected for further characterization (Compounds 1–4) also displayed efficient G protein activation based on GTPγS binding data, validating the cAMP accumulation results. The potencies obtained in the cAMP accumulation assay were, however, somewhat different from those obtained in the GTPγS binding assay. Compounds 1 and 2, for instance, had log EC50 values of −8.0 and −7.6 (EC50: 9 nM and 25 nM), respectively, in the cAMP accumulation assay, but log EC50 values of −6.3 and −6.3 (EC50: 0.5 µM and 0.5 µM), respectively, in the GTPγS binding assay. These differences likely reflect the number of spare receptors that exist for the two different assays used. The cAMP inhibition assay is a more downstream readout that is well known to exhibit signal amplification. GTPγS binding, however, directly measures the initial event (G protein activation) produced in response to receptor activation and is known to exhibit little signal amplification (i.e., a lack of spare receptors relative to the cAMP accumulation assay). It is, however, also possible that these differences may originate from differences in stable versus transient receptor expression systems. Regardless, these data from orthogonal assays confirm that the discovered CB2-selective compounds are in fact functionally relevant and stimulate G protein activation as well as inhibition of cAMP accumulation through the inhibition of adenylyl cyclase.
Two of the compounds analyzed in further detail (Compounds 1 and 4) were racemic mixes. In the case of these compounds, it is likely that one form is inactive, and the other stimulates the binding and G protein coupling activity; consequently, these parameters are likely better for the pure enantiomeric form that is active. To test this possibility, efforts are underway to synthesize, purify, and separately test the two enantiomeric forms. This next step and pharmacological characterization through SAR of the lead series are important next steps.
In this study, we performed HTS designed to identify both agonists and PAMs of the CB2 receptor. This HTS used a collaboration deck that was built at BMS and contains 60,000 commercially available compounds. This screen was run at BMS by a Yale graduate student as part of a summer internship program, and follow-up work was performed by researchers at the University of Connecticut. This HTS and follow-up therefore represent an efficient and successful collaboration between industry and academia, a paradigm with growing importance in drug discovery, especially at very early (exploratory) stages.
No CB2 PAMs were identified in this study, but it does not rule out the possibility that CB2 PAMs may exist. Because a large number of CB2 agonists were identified, however, it suggests that CB2 PAMs are rare. On further characterization of only 4 of 167 agonists identified in this study, we were able to identify compounds with CB2 selectivity. Because compounds that target the cannabinoid receptors may have analgesic properties, and because of the distinct expression patterns of the CB2 receptors relative to the CB1 receptors, the identification of CB2-selective compounds carries the potential to be developed into effective pain medications with improved side effect profiles. This study has therefore not only broadened drug discovery efforts aimed at the ECS but also provided critical starting points toward the development of novel CB2 therapeutics that could lead to improved therapies for pain management and inflammation.
Footnotes
Acknowledgements
We would like to thank the collaborative efforts of Bristol-Myers Squibb, Yale University, and the University of Connecticut, and the coordinated efforts of all those involved, that enabled the execution and publication of this study.
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by the National Institutes of Health grants T32GM007223 (LMO) and DA020763 (DAK).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.