
Abstract
UBC13 is a noncanonical ubiquitin conjugating enzyme (E2) that has been implicated in a variety of cellular signaling processes due to its ability to catalyze formation of lysine 63–linked polyubiquitin chains on various substrates. In particular, UBC13 is required for signaling by a variety of receptors important in immune regulation, making it a candidate target for inflammatory diseases. UBC13 is also critical for double-strand DNA repair and thus a potential radiosensitizer and chemosensitizer target for oncology. The authors developed a high-throughput screening (HTS) assay for UBC13 based on the method of time-resolved fluorescence resonance energy transfer (TR-FRET). The TR-FRET assay combines fluorochrome (Fl)–conjugated ubiquitin (fluorescence acceptor) with terbium (Tb)–conjugated ubiquitin (fluorescence donor), such that the assembly of mixed chains of Fl- and Tb-ubiquitin creates a robust TR-FRET signal. The authors defined conditions for optimized performance of the TR-FRET assay in both 384- and 1536-well formats. Chemical library screens (total 456 865 compounds) were conducted in high-throughput mode using various compound collections, affording superb Z′ scores (typically >0.7) and thus validating the performance of the assays. Altogether, the HTS assays described here are suitable for large-scale, automated screening of chemical libraries in search of compounds with inhibitory activity against UBC13.
Keywords
Introduction
The chemistry of ubiquitination in eukaryotic cells typically relies on the dynamic interaction of ubiquitin (Ub) with three different classes of enzymes, termed ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3).1–3 The specificity of substrate selection for ubiquitination is dictated by E3 ligases, which have been implicated in nearly every facet of cell biology. Many E3 ligases have been causally implicated in disease mechanisms. 4
Protein ubiquitination is probably best known for its role in controlling protein degradation. 5 In this context, formation of poly-Ub chains on target proteins, where lysine48 (K48) serves as the linking residue among Ub molecules, creates a structure recognized by components of the 26S proteasome, thus targeting proteins that bear this posttranslational modification for destruction.6,7 Chaperones that recognize K48-linked Ub chains have also been described that target proteins to lysosomes for degradation via the phenomenon of chaperone-mediated autophagy. 8 However, ubiquitin-conjugating enzymes (UBCs) have been identified that catalyze other types of Ub linkages, which are not apparently signals for protein destruction but rather play a variety of regulatory roles in cellular signaling, genome maintenance, and protein trafficking, among other functions.9–12 Some UBCs uniquely catalyze the attachment not of Ub but of Ub-related proteins (e.g., SUMO, NEDD8, APG12) to target proteins.13,14 As such, these noncanonical UBCs control discrete subsets of cellular processes beyond protein degradation, some of which are important in disease.
UBC13 catalyzes the formation of poly-Ub chains linked via K63 rather than K48. 11 This E2 requires cofactor proteins, either UEV1A or MMS2, for its catalytic activity.9,11 Among the known E3 ligases that collaborate with UBC are TRAFs, a family of RING domain–containing adapter proteins that associate with various members of the tumor necrosis factor (TNF) receptor family or intermediate adapter proteins that associate with TNF family receptors, Toll-like receptors (TLRs), and NOD-like receptors (NLRs), as well as downstream components of the T cell and B cell antigen receptor signal transduction machinery.9,15–20 UBC13 mediates K63-linked ubiquitination of various protein kinases that associate with TRAFs, a posttranslational modification associated with their activation. Recently, gene ablation studies in mice have validated UBC13 as a candidate target for autoimmune and inflammatory diseases.21,22 In this regard, our laboratory has produced hemizygous ubc13 mice (ubc13+/–), observing that reduced levels of UBC13 protein are associated with significantly impaired signaling by TNF and lipopolysaccharide (LPS), resulting in reduced TRAF ubiquitination in vivo, accompanied by reduced activation of NF-κB and stress kinases. 23 Reduced levels of UBC13 activity in these mice correlate, for example, with resistance to septic shock, implying that chemical inhibitors of this unique E2 might find utility for certain diseases.
UBC13 also plays an important role in double-strand DNA break repair in eukaryotes from yeast to mammals. 24 Ablation of the gene encoding the ortholog of UBC13 in yeast, for example, results in marked sensitivity to ionizing radiation. Thus, UBC13 may also represent a radiosensitizer or chemosensitizer target for oncology. An antimicrobial natural product, leucettamol A was reported to inhibit heterodimer formation of UBC13-UEV1A by enzyme-linked immunosorbent assay (ELISA) at 50 µg/mL. 25 Yet another study reported identification of a compound from a combinatorial peptoid library as an UBC13-UEV1A interaction antagonist. 26 However, neither of these reported UBC13 inhibitors has drug-like properties that would provide a starting point for a hit-to-lead optimization campaign.
Methods to quantitatively assay the activity of the UBCs in a high-throughput mode would therefore enable drug discovery and chemical biology explorations of these enzymes. We employed lanthanide donor terbium-chelate conjugated to ubiquitin (Tb-Ub) and its fluorescence resonance energy transfer (FRET) pair acceptor fluorescein-conjugated ubiquitin (Fl-Ub) to create robust homogeneous high-throughput screening (HTS) assays for UBC13 based on the time-resolved FRET (TR-FRET) method. 27 We report here optimized high-throughput TR-FRET-based screening assays for UBC13 and validate them by conducting automated screens of various chemical libraries.
Results
TR-FRET method for monitoring formation of poly-Ub chains produced by UBC13
Ubiquitination reactions catalyzed by UBC13 in collaboration with its cofactor UEV1A were monitored by a novel TR-FRET assay that combined commercially available Fl- and Tb-conjugated ubiquitin molecules. The excitation of donor terbium chelate (360 nm) followed by its emission (480 nm) results in energy transfer to proximal acceptor fluorescein via FRET, 28 where the energy transfer occurs when Tb-Ub and Fl-Ub are in close proximity (10–100 Å) following their co-assembly into poly-Ub chains ( Fig. 1 ). The resulting interaction is quantified in terms of ratiometric measurement of fluorescein (520 nm) to terbium (480 nm) emission, constituting the TR-FRET signal.
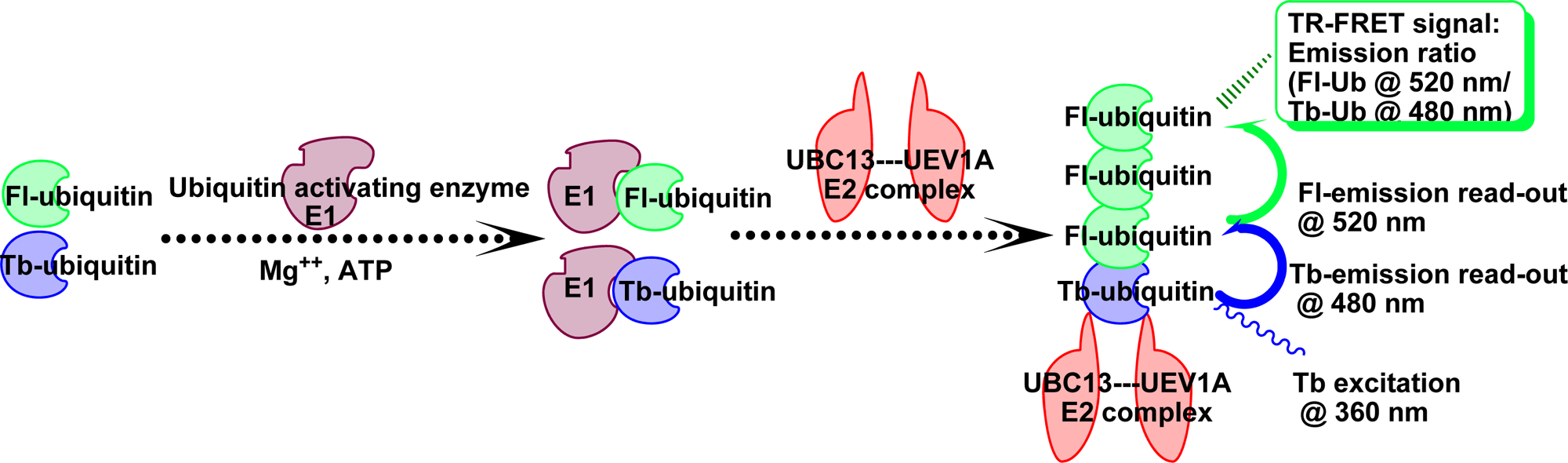
Schematic representation of time-resolved fluorescence resonance energy transfer (TR-FRET)–based assay for UBC13-UEV1A-mediated ubiquitination. Diagram shows use of terbium-ubiquitin and fluorescein-ubiquitin to generate a FRET reaction. In the presence of Mg2+ and adenosine triphosphate (ATP), labeled ubiquitin attaches to ubiquitin activating enzyme (E1) followed by transfer to ubiquitin conjugating enzyme complex (E2, UBC13-UEV1A). This event triggers ubiquitin chain buildup, which is monitored by TR-FRET, which occurs when terbium-ubiquitin and fluorescein-ubiquitin are in close proximity to each other. Terbium is excited at ~360 nm light emitting at a wavelength (~480 nm) suitable for excitation of fluorescein, which in turn emits at ~520 nm. The TR-FRET signal is measured as an emission ratio (520 nm/480 nm).
Recombinant His-tagged proteins (hereafter referred to as UBC13, UEV1A, and MMS2) for the assay were bacterially expressed and purified, then analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and size exclusion chromatography for UBC13, UEV1A, and the heterodimeric complex of UBC13-UEV1A (
A UBC13-dependent increase in TR-FRET signal was observed, representing a ~5-fold elevation under the reaction conditions employed here in 384-well plates (
Fig. 2A
). This TR-FRET signal was correlated with formation of poly-Ub chains in complete reactions, as demonstrated by SDS-PAGE analysis of the reaction products using fluorimager analysis to detect Fl-Ub polymers (
Fig. 2B
). Reactions lacking E1, UBC13, or UEV1A afforded little TR-FRET signal or Ub polymerization. Mass spectrometry analysis of the reaction products confirmed the presence of K63-linked ubiquitin (
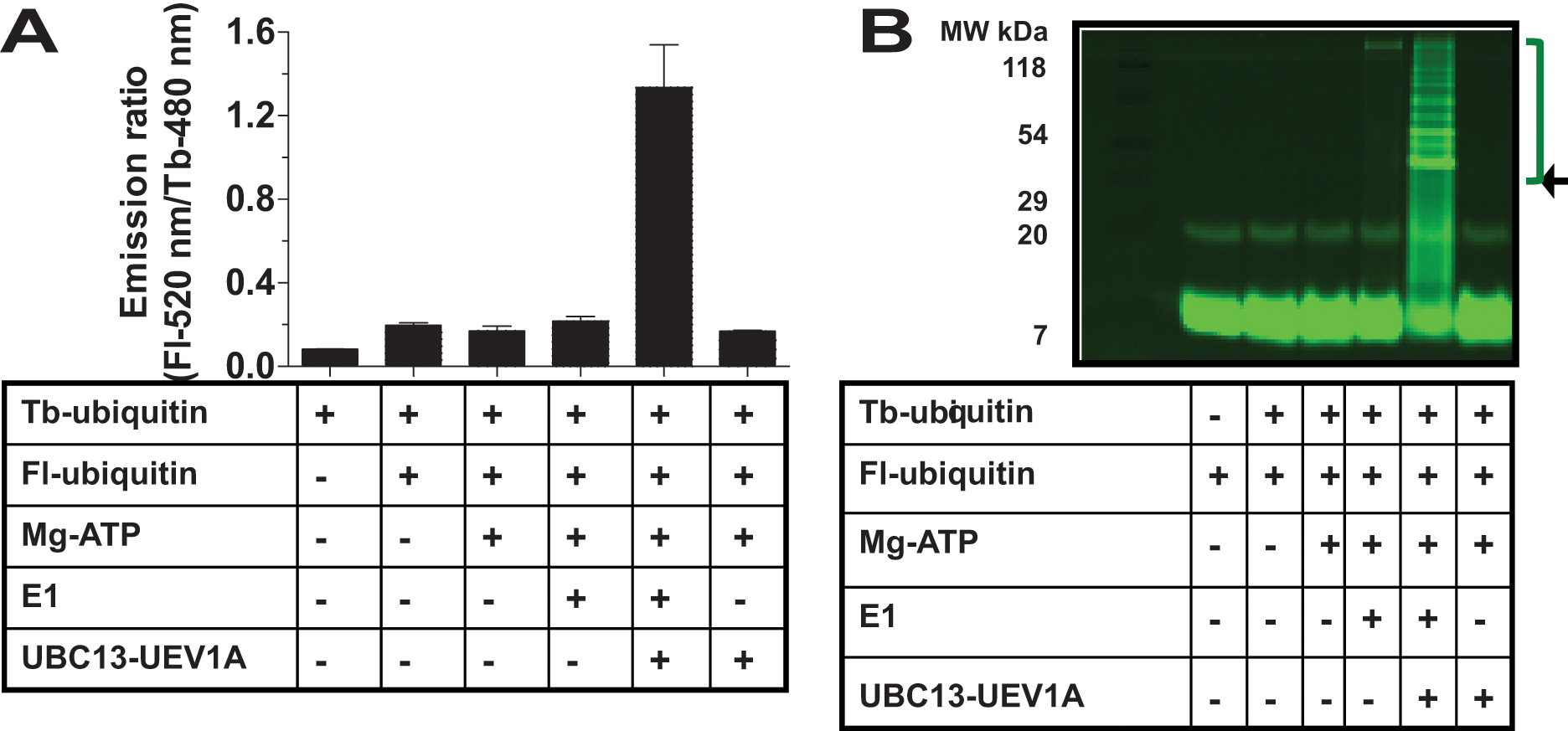
Demonstration of time-resolved fluorescence resonance energy transfer (TR-FRET) reaction for UBC13-UEV1A reactions. (
Our initial attempts to optimize TR-FRET reactions in a 384-well format addressed several variables, including concentration of substrate (molar ratio of Fl-Ub:Tb-Ub), concentration of UBC13-UEV1A heterodimeric complex, reaction time, reaction temperature, and buffer composition. A variety of assay buffers were compared, settling on 50 mM HEPES (pH 7.5) containing 0.005% Empigen BB and 0.1 mM dithiothreitol (DTT). We empirically determined the molar ratio of Fl-Ub and Tb-Ub that produced the strongest TR-FRET signal, which was approximately 15:1 in a 384-well format ( Fig. 3A ). Regardless of the Fl-Ub:Tb-Ub ratio, it should be recognized that the total ubiquitin concentration used for reactions may bias the types of inhibitors identified by the assay (competitive vs. noncompetitive), which was not addressed here.
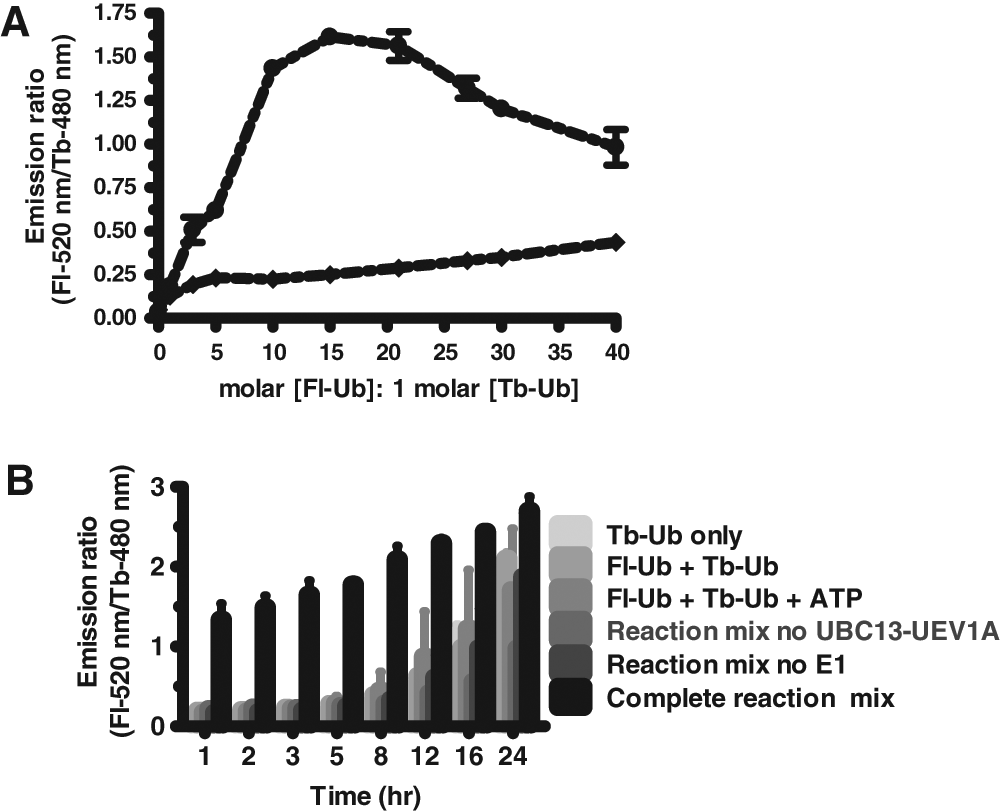
Assay optimization. (
Ubiquitination reactions were performed at 37 °C and at room temperature (RT) to assess whether it is feasible to run the assay at RT, and the stability of TR-FRET signal over time was determined at RT and at 37 °C (
Fig. 3B
and
Cofactor-dependent polyubiquitin chain formation monitored by TR-FRET
Previous data have demonstrated the physical association of UBC13 with its cofactor UEV1A or MMS2 to form heterodimeric complexes with 1:1 stoichiometry.29,30 We used the TR-FRET assay and gel electrophoresis of reaction products to explore the dependence of UBC13 on the cofactors UEV1A and MMS2. UBC13 stimulated modest TR-FRET signals when reactions were established without cofactor UEV1A or MMS2 ( Fig. 4A , Fig. 4B ). Addition of UEV1A or MMS2 at an equimolar ratio relative to UBC13 increased the TR-FRET signal by an additional six- to eightfold beyond UBC13 alone ( Fig. 4A , Fig. 4B ).
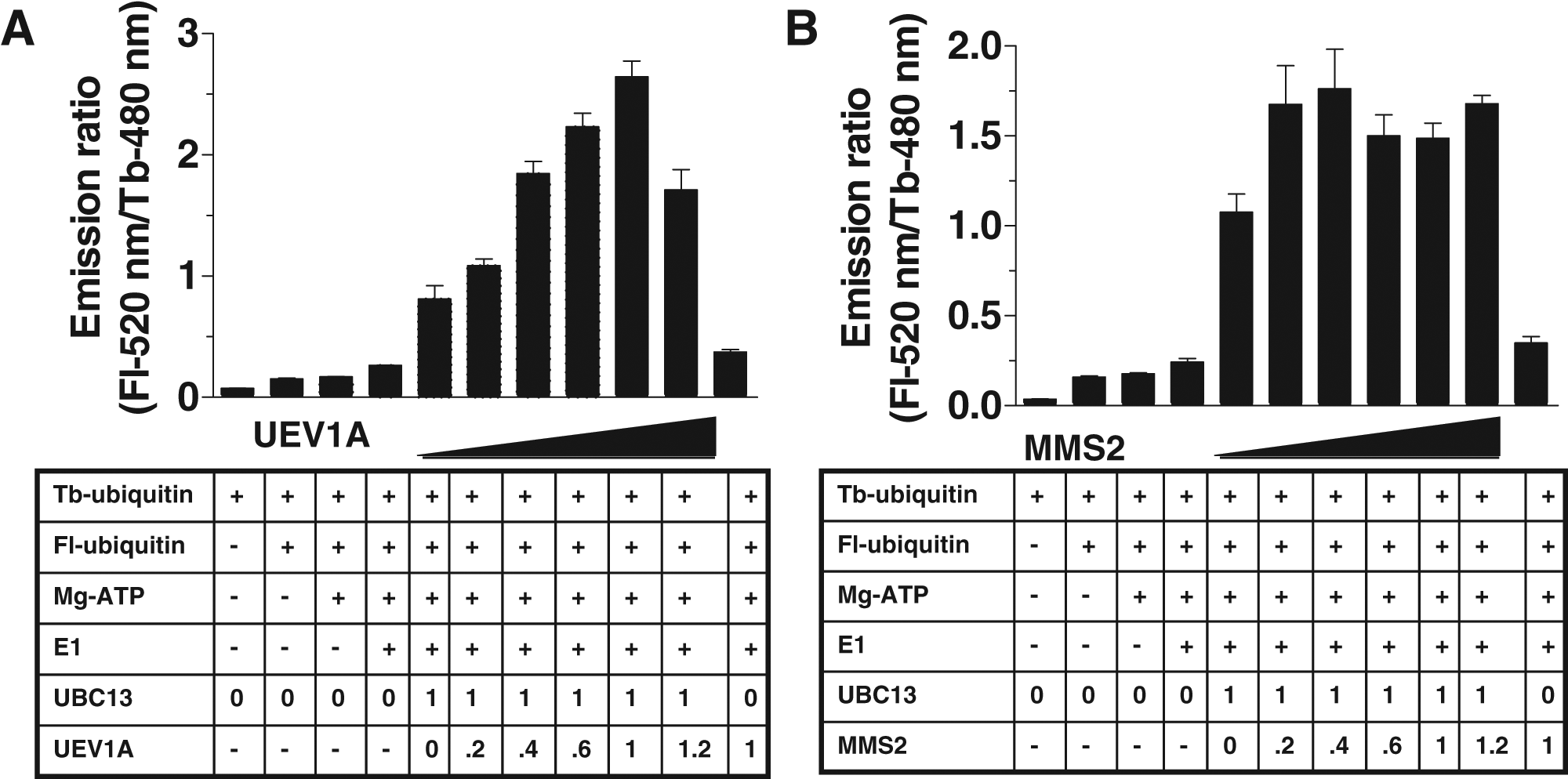
Cofactor dependence of UBC13-catalyzed poly-ubiquitination. Time-resolved fluorescence resonance energy transfer (TR-FRET) reactions were performed without or with various amounts of cofactor (
The concentration dependence of cofactor-mediated enhancement of UBC13 activity was examined. Titrating various concentrations of UEV1A into reactions with fixed concentration of UBC13 resulted in a concentration-dependent increase in TR-FRET signal up to a molar ratio of 1:1 (UEV1A:UBC13), above which the signal declined modestly ( Fig. 4A ). Although shown only for 3 h, similar results were obtained for reactions of a 1- or 5-h duration (not shown). Without UBC13, only background levels of TR-FRET signal were generated, consistent with reports that UEV1A lacks the ability to catalyze Ub polymerization by itself ( Fig. 4A ). Similar results were obtained for MMS2 ( Fig. 4B ). Sub-stoichiometric concentrations of MMS2 in complex with UBC13 yielded concentration-dependent TR-FRET signals up to 0.4:1 (MMS2:UBC13), beyond which a stable TR-FRET signal was observed. Analysis of reaction products by SDS-PAGE showed that in the absence of UEV1A or MMS2, low molecular weight conjugates containing Fl-Ub were produced in reactions containing UBC13 (not shown). Addition of various amounts of UEV1A or MMS2 resulted in the concentration-dependent appearance of higher molecular weight Fl-Ub-containing conjugates, suggesting that these cofactors are especially important for Ub chain elongation but perhaps not initiation. We conclude that the TR-FRET-based Ub polymerization method described here is suitable for monitoring the effects of cofactors on UBC13-catalyzed Ub chain assembly.
Implementation of HTS assays for UBC13
The TR-FRET-based ubiquitination assay for UBC13 was implemented as an HTS assay for chemical library screening in a 384-well format at RT (for convenience) to assess the assay window (signal-to-noise ratio) and reproducibility. First, TR-FRET assays were performed at RT in which multiple replicates were prepared of the complete reaction (maximum signal: 830.8 ± 28.9, mean ± SEM; n = 60) and compared with multiple replicates of reactions containing all assay components except UBC13-UEV1A (minimum signal: 377.9 ± 14.7, mean ± SEM; n = 56). Data from these experiments allowed Z′ factor determination, which was ~0.7 at 1 h at RT (
Fig. 5A
) and ~0.66 at 37 °C (
Next, the DMSO tolerance of the HTS assay was assessed, in preparation for compound library screening, owing to the standard practice of suspending compounds in DMSO for HTS screening of compound libraries. Although DMSO diminished the UBC13-dependent TR-FRET signal, the fold difference in specific signal still remained ≥5-fold when compared to control reactions at DMSO concentrations as high as 5% v:v ( Fig. 5B ).
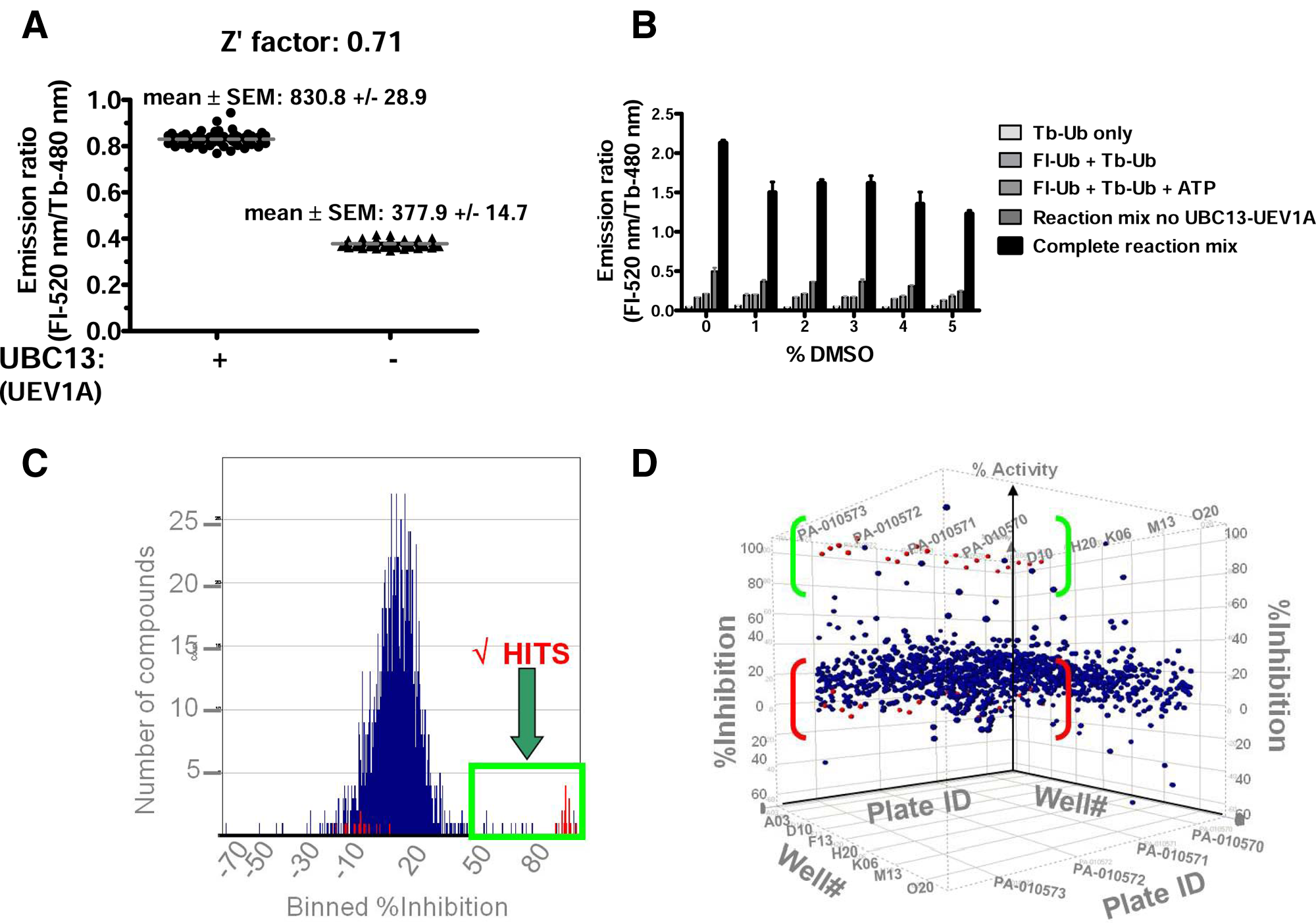
High-throughput screening (HTS) implementation of time-resolved fluorescence resonance energy transfer (TR-FRET)–based ubiquitination assay. (
A small pilot chemical library screen was then undertaken using the TR-FRET assay in the 384-well format. To this end, a library of 1280 pharmacologically active compounds (LOPAC; Sigma, St. Louis, MO) suspended in DMSO was screened (20 nL DMSO per well, equating to 0.1% v:v, and a final in-well compound concentration of 10 µM). Each plate contained a row of wells that received complete reaction mix with 0.1% DMSO (maximum) and a row of wells with reaction mix lacking UBC13-UEV1A (minimum), again with 0.1% DMSO. These positive and negative control wells were used to calculate a Z′ statistic for each plate. Presentation of the primary screening data in histogram format ( Fig. 5C ) showed a Gaussian distribution of TR-FRET signals for most of the compounds, with the peak of the distribution coinciding approximately with the control wells comprising complete reactions without compounds. The slight shift to the right (~10% inhibition) of the peak of the Gaussian distribution obtained for library compounds relative to the positive control likely results from “aging” of DMSO that occurs with long-term storage of compounds in this solvent and suggests that using fresh stock of the chemical library would be advisable for some compound collections. From this screen of 1280 compounds, 21 suppressed the UBC13-catalyzed TR-FRET reactions by >50%, yielding a hit rate of 1.6%. These compounds appear as a tail on the Gaussian distribution ( Fig. 5C ). Of these 21 compounds, 6 confirmed upon repeat testing (>50% inhibition), for an overall hit rate of 0.29%. Results from this pilot screening campaign are presented in Figure 5D , where TR-FRET signals for each well are displayed on the z-axis and the plate and well coordinates are depicted on the x- and y-axes. The positive and negative control wells are shown in red.
The relative potency of the confirmed hits was evaluated by dose–response studies, determining the concentration required for inhibition of TR-FRET reactions by 50% (IC50). The results for three representative compounds along with their structures are shown in
Automated large-scale library screening
Automated large chemical library screens were conducted in a 384-well format. A 125 000 chemical library screen assembled at Sanford-Burnham (referred to as the SBMRI library) was undertaken in a 384-well format with a few changes made to the optimized protocol implemented for the LOPAC library screen. The concentration of E1 was raised to 50 nM from 12.5 nM used for the LOPAC library screen. This was done to prevent chances of detecting inhibitors targeting E1 when used at higher or excess concentration. In addition, E1, labeled ubiquitins, and Mg-ATP were preincubated 30 min before the 2-h incubation period of UBC13 and UEV1A for heterodimer formation. A summary of the primary HTS representing the Z′ factor for the 125K compound screen is shown in
1536-Well implementation of HTS assay for UBC13
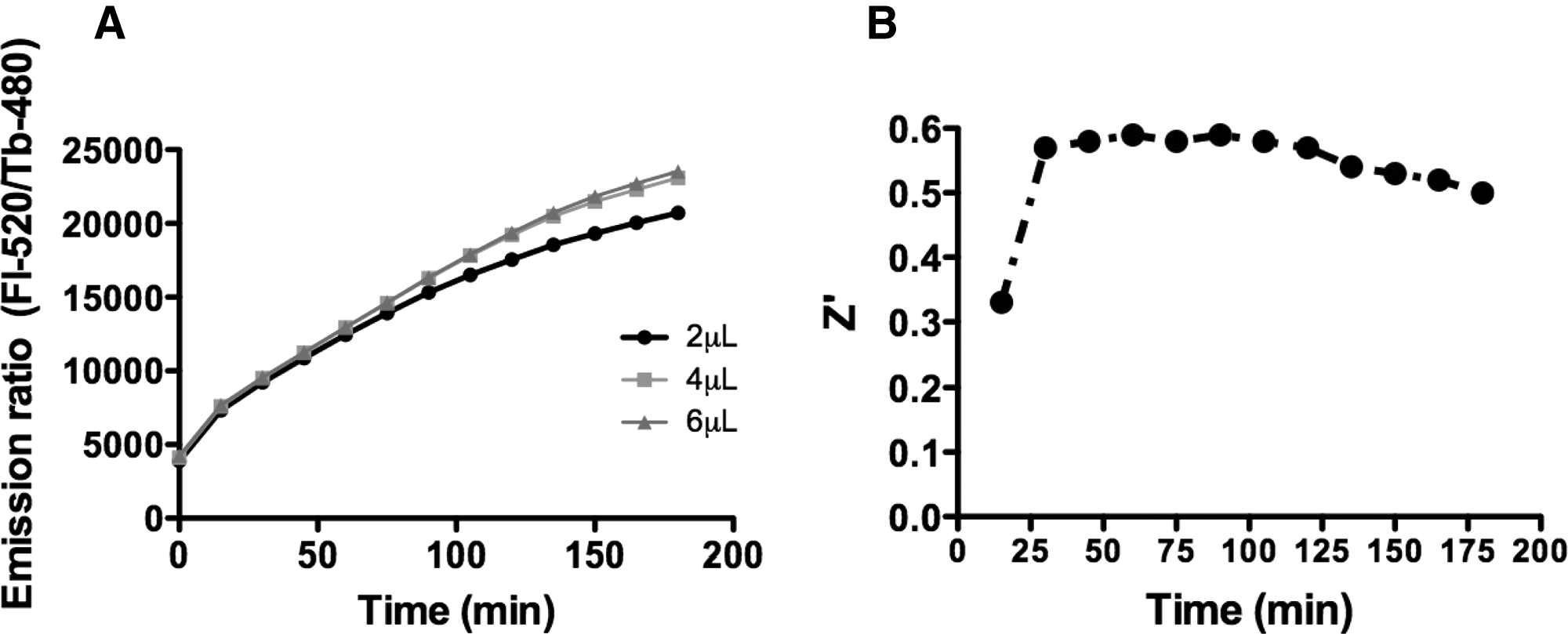
In preparation for screening large chemical libraries and to preserve assay reagents, we implemented the HTS assay in a 1536-well format. TR-FRET readouts were compared in a 384- versus 1536-well format (
Sanford-Burnham internal chemical library screen data. (
To balance assay performance against reagent consumption, the 1536-well HTS assay was conducted with reaction conditions that included (final concentrations) E1 = 100 nM, E2 = 125 nM (heterodimeric complex of UBC13 and UEV1A), and ratio of Fl/Tb-ubiquitin = 75/6 nM. Assay signal generation was nearly linear for at least 3 h. Assay performance as reflected by Z′ factor over time was also assessed in 4-µL volume reactions in a 1536-well format ( Fig. 6B ). Representative concentration–response profiles for one of the chemical inhibitors identified are shown in Figure 7 .
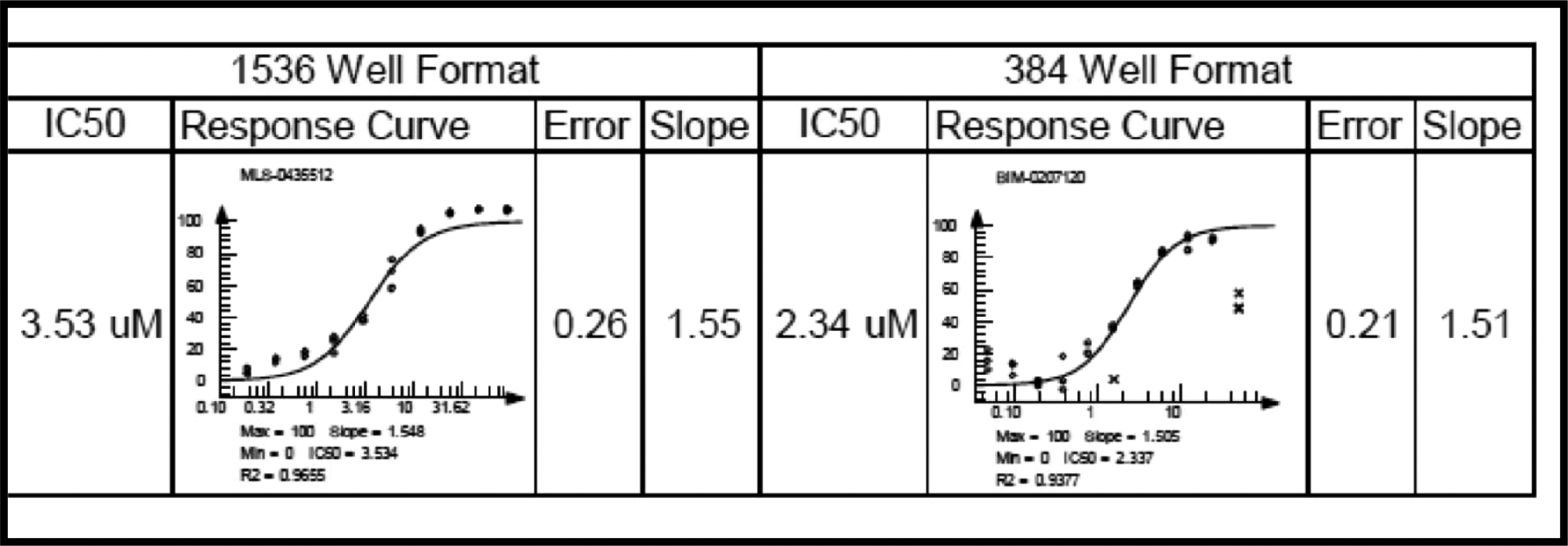
Comparison profile of the screening hit in 384- versus 1536-well formats. Percent inhibition profiles obtained for one of the inhibitors derived from a chemical library screen are represented. Data obtained from time-resolved fluorescence resonance energy transfer (TR-FRET)–based high-throughput screening (HTS) assay performed in a 384-well format (
Screening the National Institutes of Health library
The National Institutes of Health (NIH) collection of 253 plates (1536-well format), approximately 330 000 compounds, was screened at 20 µM using the 1536-well format assay protocol. The average Z′ for the assay was 0.78, suggesting a robust assay optimized for HTS ( Fig. 8A ). The signal to background was 3.8, signal to noise was 85.6, and the signal window was 14.2. A histogram representation of the NIH library screen for a subset of compounds is shown in Figure 8B , C . Results show that the assay and screen are behaving in a manner that is typical for a high-throughput screen. For inhibitors of the reaction, a hit cutoff of ≥45% activity compared to the controls was chosen, and an F ratio between 0.5 and 1.5 was applied to filter compounds that exhibit optical interference. The F ratio is defined as normalized fluorescence (F) in a plate well with a compound compared to control wells. It is calculated as a ratio of total fluorescence for fluorescence polarization (FP) assays and, in our screen, using a fluorescence of the reference channel in a TR-FRET assay system. The measure of the F ratio helped remove compounds that optically interfere with the assay result, yielding a hit rate of 0.47%. Thus, in contrast to the SBMRI library screen where the F ratio was not applied, the hit rate was lower. The NIH library-derived hits were reordered from the NIH Small Molecule Repository and retested to confirm their activity. Approximately 87% confirmed upon repeat testing, for an overall confirmed hit rate of 0.37%. Chemical hits were then tested in an alternate counterscreen TR-FRET assay employing Bfl-1. The final hit rate after reconfirming and counterscreening with the Bfl-1 assay was 0.089%.
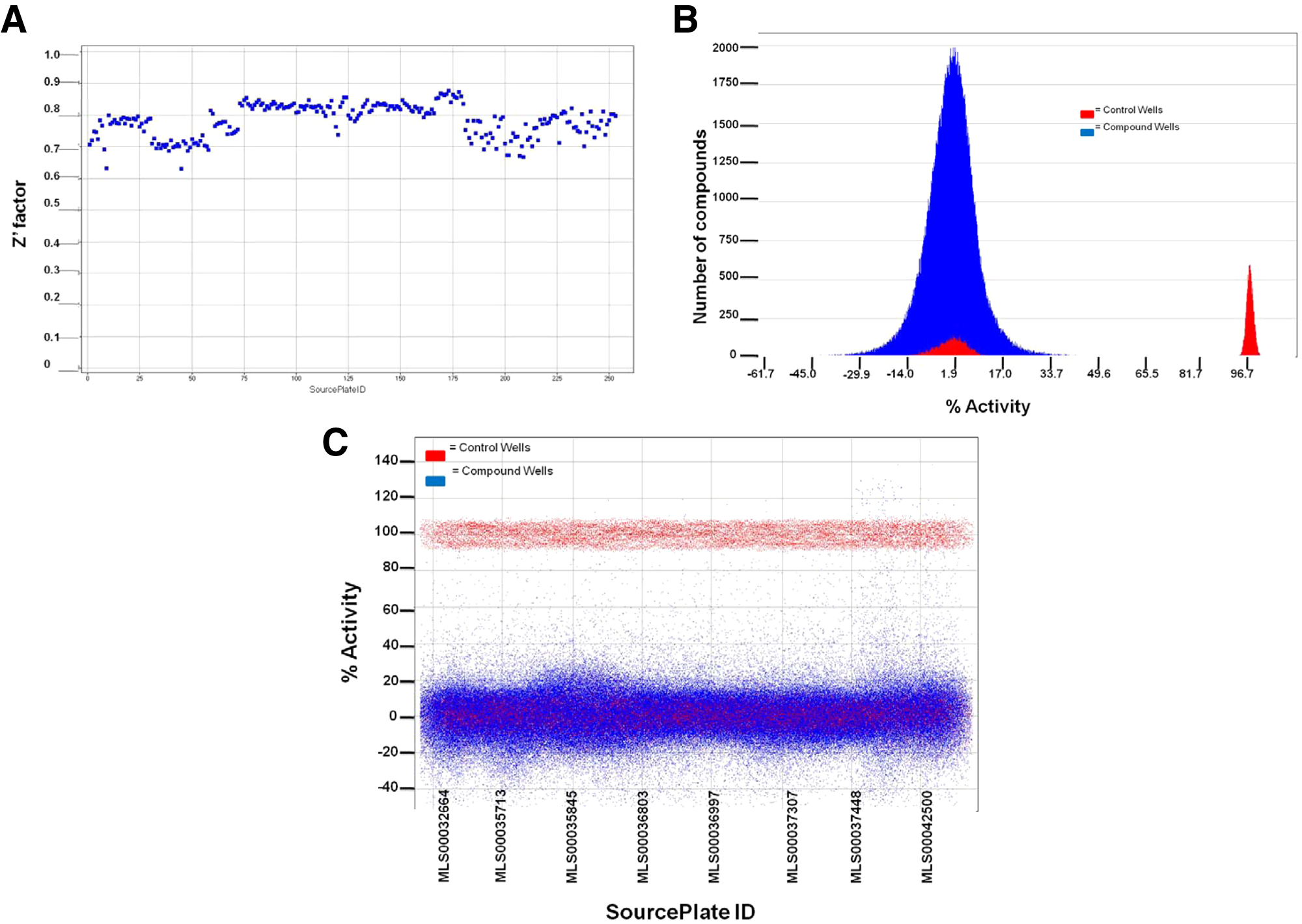
National Institutes of Health (NIH) chemical library high-throughput screening (HTS) data. (
Discussion
Components of the cellular ubiquitination machinery are critical regulators of many biological processes of medical importance, thus making them of interest as targets for drug discovery. Previous efforts to develop HTS assays that address the ubiquitin system have resulted in several successful assay formats for monitoring in vitro ubiquitination reactions. 31 Typically, these alternative methods have measured the addition of ubiquitin to a substrate protein, either detecting the presence of the ubiquitin conjugate or monitoring the formation of a polyubiquitin chain in the presence of an E3 ligase. 31 For example, TRAF6 ubiquitination was studied using a mixture of ubiquitin conjugated with europium chelate or biotin in a reconstituted ubiquitination reaction mixture containing E1, E2, and ATP. FRET was observed upon addition of streptavidin linked to the fluorescence acceptor, allophycocyanin (APC). 32 Another example is a dissociation-enhanced lanthanide fluoroimmunoassay, a heterogeneous methodology developed for studying ubiquitination of an HECT E3 ligase, Rsc. 33 This procedure involved transfer of biotin-Ub from an E2 (UBC4) to GST-Rsc followed by immobilization of biotin-Ub-Rsc-GST to streptavidin-coated plates. Anti-GST europium antibody was then added, followed by washing and measurement of fluorescence signal. Here we describe an in vitro method for measuring the activity of E2-mediated polyubiquitination that is independent of the presence of an E3, using complementary FRET donor/acceptor pairs of ubiquitin, which are incorporated into polyubiquitin chains. The TR-FRET method described is homogeneous, without requirement for washing steps, and uses all components in solution phase, thus avoiding the disadvantages inherent in mixed-phase (solid-phase/solution-phase) reactions commonly used to detect ubiquitin conjugates in other forms of in vitro assays. Our TR-FRET-based assay method was implemented for HTS in a 384- and 1536-well format, demonstrating its robust performance, with acceptable Z′ scores. The reaction method was demonstrated for UBC13, an ubiquitin-conjugating enzyme that we empirically determined is capable of catalyzing polyubiquitin chain formation in the absence of an E3 ligase, provided cofactors UEV1A or MMS2 are present. The TR-FRET assay method described therefore should be extendable to essentially any E2, if the activity of that UBC is sufficient to generate an adequate signal above background—a criterion that may require supplying reactions with an E3 ligase that stimulates the UBC’s activity.
Although the TR-FRET assay for UBC13 described here did not require an accompanying E3 ligase (such as a TRAF protein) for achieving performance characteristics suitable for HTS, it is possible that addition of an appropriate E3 ligase would stimulate the catalytic activity of UBC13 and further improve the signal-to-noise ratio. Production of well-folded recombinant full-length TRAF proteins in high yield, however, has not been trivial, although fragments of TRAFs either lacking the E2-binding RING domain or containing only the RING domain have been produced.34–36 Regardless, the E3-independent activity of UBC13 is consistent with prior models, suggesting that some E2s can catalyze multiubiquitin chain preassembly prior to E3-mediated transfer of that multiubiquitin chain to a substrate.37–40
Because of the multicomponent nature of our TR-FRET reactions, it will be necessary to deconvolute the target of compound hits obtained from HTS. In this regard, the compounds could target the E1 in these reactions, UBC13, or the cofactor (UEV1A or MMS2). Methods for excluding compounds that attack the E1 could include counterscreens using the same E1 in TR-FRET reactions with a different unrelated E2. For distinguishing compounds that bind the E1, UBC13, or cofactors UEV1A and MMS2, we routinely employ micro–isothermal titration calorimetry (µITC) or use either 1D–nuclear magnetic resonance (NMR; measuring the compound’s 1H spectrum in the presence vs. absence of protein) or 2D-NMR (using 15N uniformly labeled UBC13 and measuring chemical shifts caused by addition of compounds). Also of note, because of the nucleophilic attack on ubiquitin substrate by the active site cysteine in both E1 and E2, compounds with redox-active properties and electrophilic compounds such as Michael acceptors are prone to score as hits. The redox modulators can be eliminated by comparing the activity of compounds using the TR-FRET assay in the presence and absence of a reducing agent such as DTT. Alternatively, counterscreening with an unrelated cysteine-dependent enzyme would permit this distinction. 41
NMR and x-ray crystallography studies have revealed the structures of UBC13, its catalytic site, and its interface with obligatory cofactor proteins.30,42–45 These structural studies of UBC13-UEV1A complexes show a deep, narrow, and long hydrophobic binding pocket at the interface of UBC13-UEV1A. 30 UBC13 therefore has two potential sites of attack by small molecules—the active site and the cofactor-binding site. Thus, compounds identified by the TR-FRET assay described here could bind UBC13 either at the catalytic site or at the UEV1A interaction site. Achieving selective inhibitors of UBC13 by targeting the active site of the enzyme may be challenging because of the overall conserved features of the active sites of many UBC family enzymes. Nevertheless, exploiting compound interactions with regions adjacent to the active site as an approach for achieving selectivity has been successfully applied to many other classes of enzyme families. The unique binding pocket involved in obligatory association with cofactors UEV1A or MMS2 has the advantage of selectivity for UBC13. However, it suffers the challenge of all protein–protein interaction targets, in as much as the intermolecular interactions are spread over a relatively large surface area. Nevertheless, these structural studies of UBC13 and the UBC13-UEV1A complex lay the foundation for the potential rational design of small-molecule inhibitors, the activity of which could be then tested using a novel TR-FRET assay. Hence, the TR-FRET methodology described here could be applied for HTS for identification of chemical inhibitors from libraries or used to profile compounds derived by in silico docking or structure-based rational design.
Methods
Plasmids and reagents
The cDNAs encoding human UBC13, UEV1A, and MMS2 were amplified and subcloned into pET vectors for expression of proteins. Human ubiquitin activating enzyme (E1) was obtained from Boston Biochemicals (Cambridge, MA). Terbium-ubiquitin and fluorescein-ubiquitin were purchased from Invitrogen (Carlsbad, CA). Stock solutions of ubiquitination reagents were prepared in 50 mM HEPES (pH 7.5).
Protein expression and purification of ubiquitin-conjugating enzymes
Plasmids containing UBC13, UEV1A, and MMS2 were expressed in BL21/DE3 Escherichia coli cells. Cells were grown at 37 °C to exponential phase (optical density [OD] ~0.6) and induced with 0.4 mM IPTG (BioVectra, Charlottetown, Canada). Briefly, cells were resuspended in lysis buffer consisting of 1 mM Tris (pH 8.0), 1 mM DTT, 0.2 mg/mL lysozyme, and protease inhibitors (Roche, Basel, Switzerland). Lysates were incubated with Ni+-NTA agarose (Qiagen, Valencia, CA) previously prepared with washing buffer consisting of 1 mM Tris (pH 8.0) and 1 mM DTT. His-tagged proteins were eluted with imidazole (Sigma) under linear gradient conditions followed by fast protein liquid chromatography (FPLC) purification (Pharmacia BioTech, Uppsala, Sweden) on a MonoQ column using high-salt gradient conditions (1 M NaCl). Proteins of >90% purity were obtained. Further purification of proteins was obtained by size exclusion chromatography on a Superdex 200 (GE Healthcare, Piscataway, NJ) column. Representative Coomasie-stained 15% SDS-PAGE gel images of purified His-UBC13 (~17 kDa), His-MMS2 (~19 kDa), and His-UEV1A (~26 kDa) are shown in
TR-FRET-based ubiquitination assay methodology
Ubiquitin chain assembly mediated by UBC13-UEV1A complex was monitored based on the principle of TR-FRET. An equimolar ratio of UBC13 and UEV1A (or UBC13 and MMS2) was prepared in 50 mM HEPES (pH 7.5) buffer and incubated for about 2 h on ice to afford the UBC13-UEV1A or UBC13-MMS2 heterodimeric complex, which was confirmed by size exclusion chromatography using the Superdex 200. Ubiquitination reactions were performed in black 384-well round-bottom plates (Corning 3676; Corning, Inc., Corning, NY). The reaction buffer consisted of 50 mM HEPES (pH 7.5)/0.005% Empigen BB detergent/0.1 mM DTT/1% DMSO. Stock solutions of ubiquitin, E1, UBC13-UEV1A or UBC13-MMS2 complex, and Mg-ATP were prepared in 50 mM HEPES (pH 7.5). Complete ubiquitination reaction mixture consisting of ubiquitin-activating enzyme (E1, 12.5 nM), UBC13-UEV1A (E2, 250 nM each), terbium-labeled ubiquitin (10 nM), fluorescein-labeled ubiquitin (150 nM), and Mg-ATP regenerating system (consisting of 1 mM ATP and 1.25 mM MgCl2) were sequentially added along with buffer components to a 384-well plate. Control reaction mixtures included either terbium-Ub + fluorescein-Ub or terbium-Ub + fluorescein-Ub + Mg-ATP or a reaction mixture lacking UBC13-UEV1A complex in it. The reaction plate was incubated at 37 °C or RT with TR-FRET readings at regular time intervals (1, 3, and 5 h, etc.) on an Analyst instrument (Molecular Devices, Sunnyvale, CA). TR-FRET measurements were taken by setting the 360/480-nm filter for recording terbium emission, the 360/520-nm filter for fluorescein emission, and using a 50:50 dichroic beamsplitter. A graphical analysis was generated by plotting the ratio of emission intensities of the acceptor and donor fluorophores (emission ratio 520 nm/480 nm) for each set of reaction mixtures. Data are represented as mean ± SEM. The assay was optimized for UBC13-UEV1A concentration, Fl-Ub:Tb-Ub ratio, substrate concentration, reaction buffer, reaction time, and temperature. With regard to chemical inhibitors, data were represented in a graphical format with concentration of the inhibitors on the x-axis and emission ratio on the y-axis from which IC50s were determined for chemical inhibitors.
Gel-based fluorimager analysis of ubiquitination reactions
Aliquots of samples from TR-FRET analysis were analyzed for ubiquitination on 15% Tris-glycine gels. Gels were scanned with an FLA 5100 scanner (Fujifilm, Tokyo, Japan) and analyzed with MultiGauge imager 3.0 software (Fujifilm). For fluorescence assessment, gels were scanned using a laser excitation beam at 473 nm and a long pass blue (LPB, Y510) filter. Ubiquitinated protein was identified either as a smear or as series of discrete bands of increasing molecular weight in SDS-PAGE gels.
Monitoring ubiquitin chains catalyzed by UBC13
The catalytic role of UBC13 on the synthesis of the ubiquitin chain built up on the UBC13-UEV1A or UBC13-MMS2 complex was studied by TR-FRET analysis and confirmed by gel-based fluorimager analysis. Ubiquitination reactions were performed as described above except that the heterodimeric complex of UBC13-UEV1A or UBC13-MMS2 was prepared in varying molar ratios of UBC13 (250 nM) and UEV1A (0–500 nM) or MMS2 (0–500 nM). The molar ratios of UBC13:UEV1A and UBC13:MMS2 in our experiments were 1:0, 1:0.2, 1:0.4, 1:0.6, 1:0.8, 1:1, and 1:2. Optimal molar stoichiometry of UBC13-UEV1A or UBC13-MMS2 was determined based on TR-FRET signal generated and ubiquitin chain formation observed in fluorimager analyses.
Linkage specificity of ubiquitination reactions mediated by the UBC13-UEV1A complex
Ubiquitin chains formed in the presence of UBC13-UEV1A were studied for type of chain linkage using the NanoLC-LTQ mass spectrometry–based proteomics strategy, which involved the following steps:
In-gel digestion
Ubiquitinated E1 and UBC13-UEV1A proteins were analyzed after SDS-PAGE and Coomassie blue staining. Bands were cut and gel slices incubated with 0.25 ng/µL trypsin at 37 °C overnight. Samples were cleaved and concentrated by C18 ziptip and resuspended in Nano liquid chromatography/tandem mass spectrometry (LC/MS/MS) loading buffer A.
NanoLC-LTQ MS analysis
The automated NanoLC-LTQ MS setup consisted of an Eksigent Nano 2D LC system, a switch valve, a C18 trap column (Agilent, Santa Clara, CA), and a capillary reverse-phase column (10 cm in length, 75 mm id) packed with 5-mm C18 AQUASIL resin with an integral spray tip (Picofrit, 15-mm tip; New Objective, Woburn, MA). A reverse-phase LC directly coupled to an LTQ mass spectrometer (Thermo Electron, Waltham, MA) was performed using linear gradient elution from buffer A (H2O plus 0.1% formic acid) to 15% buffer A plus 85% buffer B (ACN plus 0.1% formic acid) in 45 min. The LC/MS run was operated in the data-dependent mode. Data on the four strongest ions above an intensity of 50 × 10e4 were collected with dynamic exclusion enabled and the collision energy set at 35%.
Protein database searching and data analysis
The MS/MS spectra were analyzed by Sorcerer 2 system with SEQUEST (v.27, rev. 11; Sage-N Research, Milpitas, CA) as the search program for protein identification. SEQUEST was set up to search the target-decoy ipi.HUMAN.v3.22 database containing protein sequences using semi-trypsin as the digestion enzyme with the allowance of up to two missed cleavages. The mass tolerances of a fragment ion and a parent ion were set as 0.5 and 1.0 Da, respectively. A molecular mass of 57 Da was added to all cysteines to account for carboxyamidomethylation in case of alkylation of cysteines. Differential search included 16 Da for methionine oxidation and lysine ubiquitination with a GG tag (114 Da) to reflect the mass difference between ubiquitinated and un-ubiquitinated peptides. The search results were viewed, sorted, filtered, and statistically analyzed by using comprehensive proteomics data analysis software, Peptide/Protein prophet (Institute for Systems Biology [ISB], Seattle WA). The relative abundance of each identified protein in each sample was calculated by in-house developed QTools automated differential peptide/protein spectral counting software.
HTS implementation of UBC13 assay: LOPAC library screen
Optimization of the TR-FRET assay for HTS (as reflected by Z′ score)
The screening window coefficient, termed the Z′ factor, was determined for assessing the reliability and reproducibility of the UBC13/UEV1A assay in a 384-well format (or 1536-well format as required). The Z′ score was calculated for the complete reaction mixture, and this factor was compared to a reaction lacking the UBC13-UEV1A complex in the mixture. 46 Each well contained 20 µL of solution consisting of complete reaction mixture with or without UBC13-UEV1A. Initially, a pilot screen was conducted employing a library of pharmacologically active compounds (LOPAC). Compounds were dispensed at a final concentration of 10 µM into 384-well assay plates. Afterwards, the ubiquitination reaction mixture consisting of Tb-Ub, Fl-Ub, E1, UBC13-UEV1A complex, and ATP was added. The final concentrations of the assay components were as follows: 12.5 nM E1, 250 nM Ubc13, 250 nM Uev1a, 150 nM Fl-Ub, 10 nM Tb-Ub, 1 mM ATP, and 1.25 mM MgCl2 in 50 mM HEPES buffer (pH 7.5). Positive and negative controls in the experiments included wells with complete reaction mixture and reaction mixture with no UBC13-UEV1A. Plates were read after incubating for 1 and 3 h using Analyst (Molecular Devices). LOPAC primary screen data were formatted as a histogram. The cutoff for hits was set at 50% of maximum average signal (IC50 values). Data were represented in a graphical format with log inhibitor (µM) on the x-axis versus TR-FRET ratiometric readings on the y-axis. Analysis was performed using GraphPad Prism version 5.0 (GraphPad Software, La Jolla, CA) by nonlinear regression analysis using least squares fit method from which IC50 values were determined.
Automated large-scale library screening
Automated large chemical library screens were conducted in both 384- and 1536-well formats. In the addition to the LOPAC library, the Sanford-Burnham chemical library (consisting of a ~125 000–chemical compound library) was also screened in a 384-well format following the optimized TR-FRET HTS protocol. The HTS assay, optimized to operate in a 1536-well format, was also implemented to screen the NIH chemical library (consisting of a ~330 000–chemical compound library). The final concentrations of the assay components employed for the 384-well HTS assay were as follows: 12.5 nM E1, 250 nM Ubc13, 250 nM Uev1a, 150 nM Fl-Ub, 10 nM Tb-Ub, 1 mM ATP, and 1.25 mM MgCl2, in 50 mM HEPES buffer (pH 7.5). The final concentrations of the assay components for the 1536-well HTS assay were as follows: 100 nM E1, 125 nM Ubc13, 125 nM Uev1a, 75 nM Fl-Ub, 6 nM Tb-Ub, 1 mM ATP, and 1.25 mM MgCl2, in 50 mM HEPES buffer (pH 7.5).
Footnotes
Acknowledgements
We thank Drs. Thomas Chung and Chris Hassig for project management, Dr. Khatereh Motamedchaboki for mass spectrometry, and Tessa Siegfried and Melanie Hanaii for manuscript preparation. This work was supported by postdoctoral fellowship and research fellow awards to C.M. from the National Multiple Sclerosis Society (FG-1760-A-1) and Multiple Myeloma Research Foundation, respectively, and by NIH grants R03 MH085677 and U54 HG005033.
C.M. designed, performed, and interpreted experiments and drafted the manuscript; K.W. performed initial protein purification studies; M.P.C. worked on cloning some constructs; S.V. performed data analysis for the LOPAC library; P.G. participated in protein purification and discussions pertaining to structural studies; I.P. designed and optimized the assay for the Sanford-Burnham internal chemical library screen; T.N. performed the Sanford-Burnham internal chemical library screen; E.A.S. collaborated on experiments and offered suggestions regarding the assay; P.D. performed LOPAC library robotic titrations in 384-well plates; S.-I.M. contributed and offered suggestions regarding cloning the constructs and designing in vitro ubiquitination assays; and J.C.R. conceived of the assay, designed experiments, interpreted data, and edited the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.