
Abstract
Protein mono-ADP-ribosylation is a posttranslational modification involved in the regulation of several cellular signaling pathways. Cellular ADP-ribosylation is regulated by ADP-ribose hydrolases via a hydrolysis of the protein-linked ADP-ribose. Most of the ADP-ribose hydrolases share a macrodomain fold. Macrodomains have been linked to several diseases, such as cancer, but their cellular roles are mostly unknown. Currently, there are no inhibitors available targeting the mono-ADP-ribose hydrolyzing macrodomains. We have developed a robust AlphaScreen assay for the screening of inhibitors against macrodomains having mono-ADP-ribose hydrolysis activity. We utilized this assay for validatory screening against human MacroD1 and identified five compounds inhibiting the macrodomain. Dose–response measurements and an orthogonal assay further validated four of these compounds as MacroD1 inhibitors. The developed assay is homogenous, easy to execute, and suitable for the screening of large compound libraries. The assay principle can also be adapted for other ADP-ribose hydrolyzing macrodomains, which can utilize a biotin-mono-ADP-ribosylated protein as a substrate.
Introduction
Macrodomains are ubiquitous protein modules recognizing and in some cases hydrolyzing mono-ADP-ribose (MAR) and poly-ADP-ribose (PAR) units attached to proteins.1,2 During ADP-ribosylation, an ADP-ribose is transferred from NAD+ to the target protein or to the growing ADP-ribose chain by ADP-ribosyltransferases (ARTDs or PARPs). 3 The modification regulates various signaling cascades and also serves as a docking site for other proteins, including macrodomains. ADP-ribosylation has essential roles in the regulation of cellular processes, such as DNA repair, transcription, and chromatin biology. In addition to hydrolysis of protein-linked ADP-ribose, some macrodomains also hydrolyze O-acetyl ADP-ribose, a product of sirtuin-mediated lysine deacetylation. 4 O-acetyl ADP-ribose functions as a signaling molecule and has been linked to the regulation of gene silencing and ion channel gating. 5
Macrodomains are found in more than 5000 proteins in eukaryotes, prokaryotes, and viruses (Pfam: PF01661). 6 The domain can be a part of a larger protein or a stand-alone domain (ca. 25 kDa) containing a mixed α/β fold. The overall fold and the ligand binding site are highly conserved but the binding and hydrolysis properties of MAR and PAR vary greatly between human macrodomains ( Suppl. Table S1 ).1,2 It is evident that even small structural differences have an impact on the domain function, although the activities of macrodomains are generally poorly known. Clear views of the various roles of macrodomains in cellular processes are missing.1,6,7 To date, macrodomains have been proposed to regulate protein–protein interactions and enzymatic activities in various cellular pathways.8,9 Macrodomain inhibition has been suggested to improve radiotherapy and chemotherapy in cancer treatment through inhibition of DNA repair, leading to apoptosis.1,10 However, this hypothesis has not been tested in practice due to the lack of macrodomain inhibitors.
Here we have developed an assay to screen inhibitors against MAR hydrolyzing macrodomains ( Fig. 1 ). The assay was tested with four human macrodomains (MacroD1, MacroD2, TARG1, and the third macrodomain of PARP14) and two trypanosomatid macrodomains. We optimized the assay parameters for MacroD1, screened a validatory library for inhibitors, and identified compounds inhibiting MacroD1.
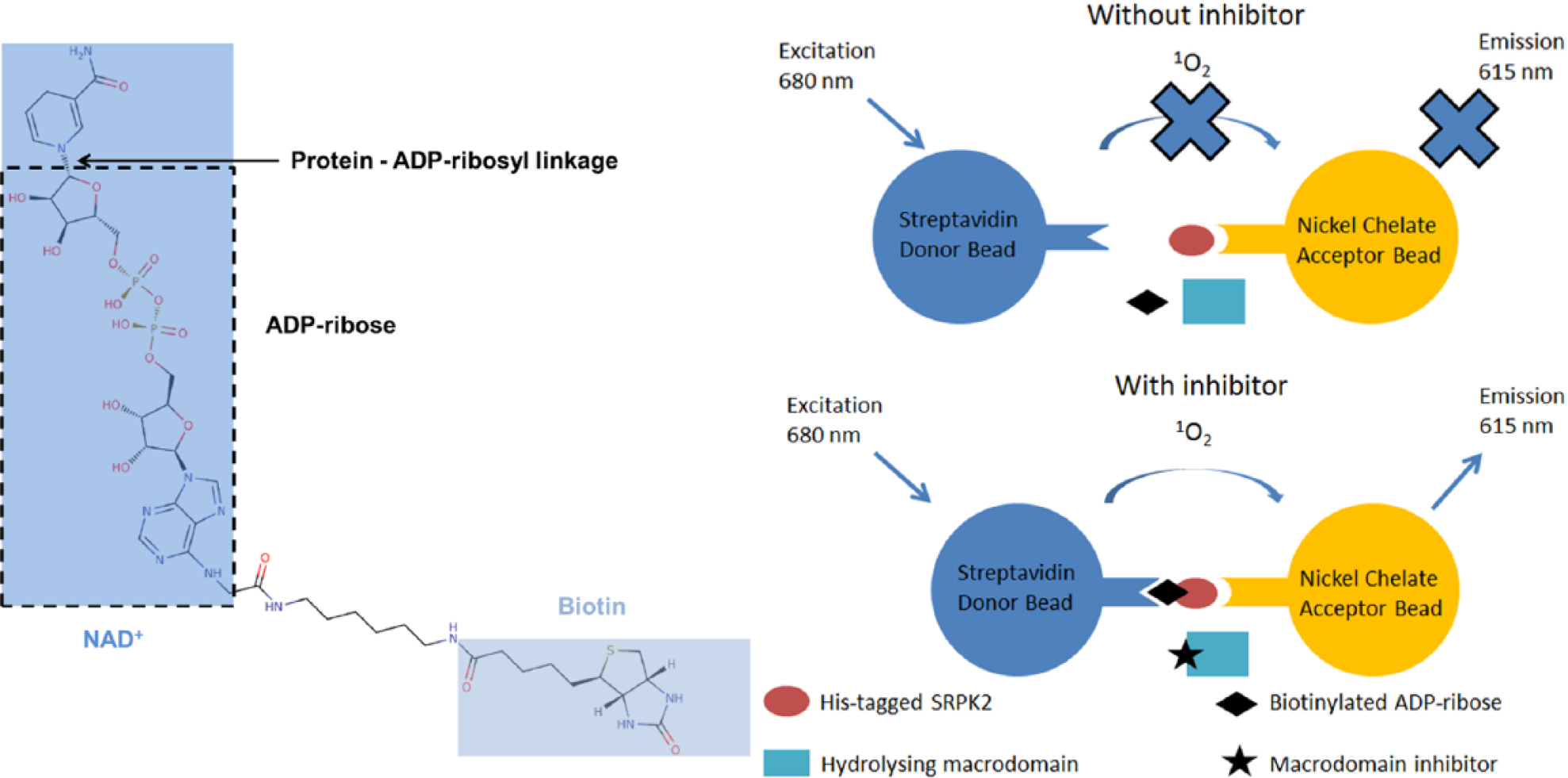
AlphaScreen assay principle. Without an inhibitor, the macrodomain hydrolyzes biotinylated ADP-ribosyl from His-tagged SRPK2. Acceptor bead-bound SRPK2 cannot interact with streptavidin-coated donor beads, and there is no singlet oxygen transfer and no emission. With a macrodomain inhibitor, biotinylated ADP-ribosyl is not hydrolyzed from SRPK2. This leads to proximity-generated singlet oxygen transfer and emission.
Materials and Methods
Cloning and Expression Constructs
The MacroD1 (GenBank: NP_054786.2) construct (residues 58–325) cloned in pNH-TrxT vector and the SRPK2 construct (residues 81–699) cloned in pNIC28-Bsa4 were from Structural Genomics Consortiums (Oxford, UK). cDNA of TARG1 (GenBank: BC011709.2) was purchased from GenScript and cloned in pNH-TrxT vector. MacroD2 (GenBank: NM_080676.5) DNA was a kind gift from Dr. Ahola (University of Helsinki, Finland), and the DNA of the PARP14 macrodomain (residues 1207–1388; GenBank: NG_051076.1) was from Dr. Schüler (Karolinska Institut, Sweden). The construct of the PARP10 catalytic domain (residues 809–1017) with a C-terminal His-tag was used as a template for cloning. 11 The trypanosomatid macrodomains from Trypanosoma brucei (TbMDO) and Trypanosoma cruzi (TcMDO) were previously cloned to pNH-TrxT vectors. 12
MacroD2 (residues 7–243) and PARP10 (residues 819–1008) were cloned by PCR extension cloning to pNH-TrxT and pNIC28-Bsa4 vectors, respectively, and they were sequenced. All the vectors contain a TEV-protease cleavage site after the N-terminal affinity/solubility tags.
Protein Expression and Purification
SRPK2, TbMDO, and TcMDO were expressed and purified as described previously.11,12 MacroD1, MacroD2, PARP10, and PARP14 were expressed in Escherichia coli Rosetta 2 (DE3) cells using Terrific broth autoinduction media (Formedium, Norfolk, UK) supplemented with 8 g/L of glycerol, 50 µg/mL kanamycin, and 34 µg/mL chloramphenicol. TARG1 was expressed in E. coli BL21 (DE3) cells using Terrific broth autoinduction media (Formedium) supplemented with 8 g/L glycerol and 50 µg/mL kanamycin. The cells were grown until OD600 reached 1.0, and the temperature was lowered to 18 °C for protein expression. After 16 h of incubation, the cells were collected by centrifugation, suspended in lysis buffer (50 mM HEPES, pH 7.5, 0.5 M NaCl, 10% glycerol, 10 mM imidazole, and 0.5 mM TCEP), and stored at −20 °C.
The proteins were purified with immobilized metal ion affinity chromatography (IMAC) and gel filtration. Briefly, the cell suspension was supplemented by 0.1 mM Pefabloc SC (Sigma-Aldrich, St. Louis, MO), 20 µg/mL DNaseI (Roche, Basel, Switzerland), and 0.5 mg/mL lysozyme (Sigma-Aldrich). The cells were broken by sonication, and the lysate was centrifuged at 31,000g at 4 °C for 45 min to remove the insoluble material. The supernatant was filtered through a 0.45 µm filter and loaded onto a 1 mL HisTrap HP column (GE Healthcare, Pittsburgh, PA). The column was washed with lysis buffer, followed by wash buffer with 25 mM imidazole. The proteins were eluted by increasing the buffer imidazole concentration to 350 mM. The fusion tags were cleaved by incubating with TEV protease at 4 °C overnight.
The proteins were further purified with gel filtration using a HiLoad 16/600 Superdex 75 preequilibrated with 20 mM HEPES, pH 7.5, 0.3 M NaCl, and 0.5 mM TCEP. Finally, the proteins were polished by passing them through the HisTrap HP column and collecting the proteins in the flow-through. The proteins were flash frozen in small aliquots in liquid N2 and stored at −70 °C.
Production of Mono-ADP-Ribosylated SRPK2
His-tagged SRPK2 (10 µM) was ADP-ribosylated with 5 µM PARP10 using 10 µM biotinylated NAD+ as a substrate (Trevigen, Gaithersburg, MD). The reaction was incubated in 50 mM Tris-HCl, pH 7.2, at room temperature for 3.5 h and purified using Ni-NTA Superflow (Qiagen, Venlo, Netherlands) resin. The resin was incubated with the reaction solution for 20 min and washed three times using 50 mM Tris-HCl pH 7.2 and 300 mM NaCl. The bound modified SRPK2 was eluted with 50 mM Tris-HCl, pH 7.2, 300 mM NaCl, and 500 mM imidazole and dialyzed against 50 mM HEPES, pH 7, and 300 mM NaCl at 4 °C overnight. The preparation was flash frozen in small aliquots in liquid N2 and stored at −70 °C.
Assay Development
Activity Assay
We have previously discovered that SRPK2, mono-ADP-ribosylated by PARP10, can serve as a substrate in the macrodomain-catalyzed hydrolysis of protein-linked ADP-ribosyl. 11 This principle was transformed to Alpha-Screen format using His-tagged SRPK2 ADP-ribosylated by PARP10 with biotinylated NAD+. The assay measures the proximity-generated luminescence using the Alpha-Screen technology. SRPK2 modified with biotinylated NAD+ binds to nickel chelate acceptor beads via poly-His-tag and to the streptavidin donor beads via biotin. A signal decrease is achieved through hydrolyzing the biotinylated ADP-ribose from SRPK2 with a MAR hydrolyzing macrodomain.
The reactions were carried out in 384-well plates (Alphaplate, PerkinElmer, Waltham, MA) in a total volume of 25 µL. The reactions consisted of SRPK2, macrodomain (assay was optimized for MacroD1), and nickel chelate acceptor and streptavidin donor beads (PerkinElmer) in assay buffer (25 mM HEPES, pH 7.5, 100 mM NaCl, and 1 mg/mL bovine serum albumin [BSA]). The plates were incubated at room temperature and protected from light after bead addition, and luminescence was read with a Tecan Infinite M1000 Pro plate reader using the AlphaScreen detection module. Macrodomain activity was read as a decrease in total luminescence with respect to the control wells (buffer instead of macrodomain).
Assay sensitivity was first tested to establish the optimal SRPK2 concentration and a maximal signal (hook point). 13 Modified SRPK2 (15 µL) and nickel chelate acceptor beads (5 µL) were added to the well, and after 30 min of incubation, streptavidin donor beads (5 µL) were added, followed by 60 min of incubation. The final concentrations were 0–500 nM modified SRPK2 and 15 µg/mL acceptor and donor beads ( Fig. 2A ). To establish whether MacroD1 is able to hydrolyze the biotinylated substrate, a concentration series of MacroD1 was titrated against SRPK2 and ADP-ribosyl hydrolysis activity was measured by setting the conversion of the control wells (wells with SRPK2 only) to 0%. Modified SRPK2 (7.5 µL) and MacroD1 (7.5 µL) were added to the wells and incubated at room temperature for 180 min. Next, nickel chelate acceptor beads (5 µL) were added to the well, and after 30 min of incubation streptavidin donor beads (5 µL) were added, followed by 60 min of incubation. Concentrations of modified SRPK2 and MacroD1 were 50 and 0–600 nM, respectively (before bead addition) ( Fig. 2B ).
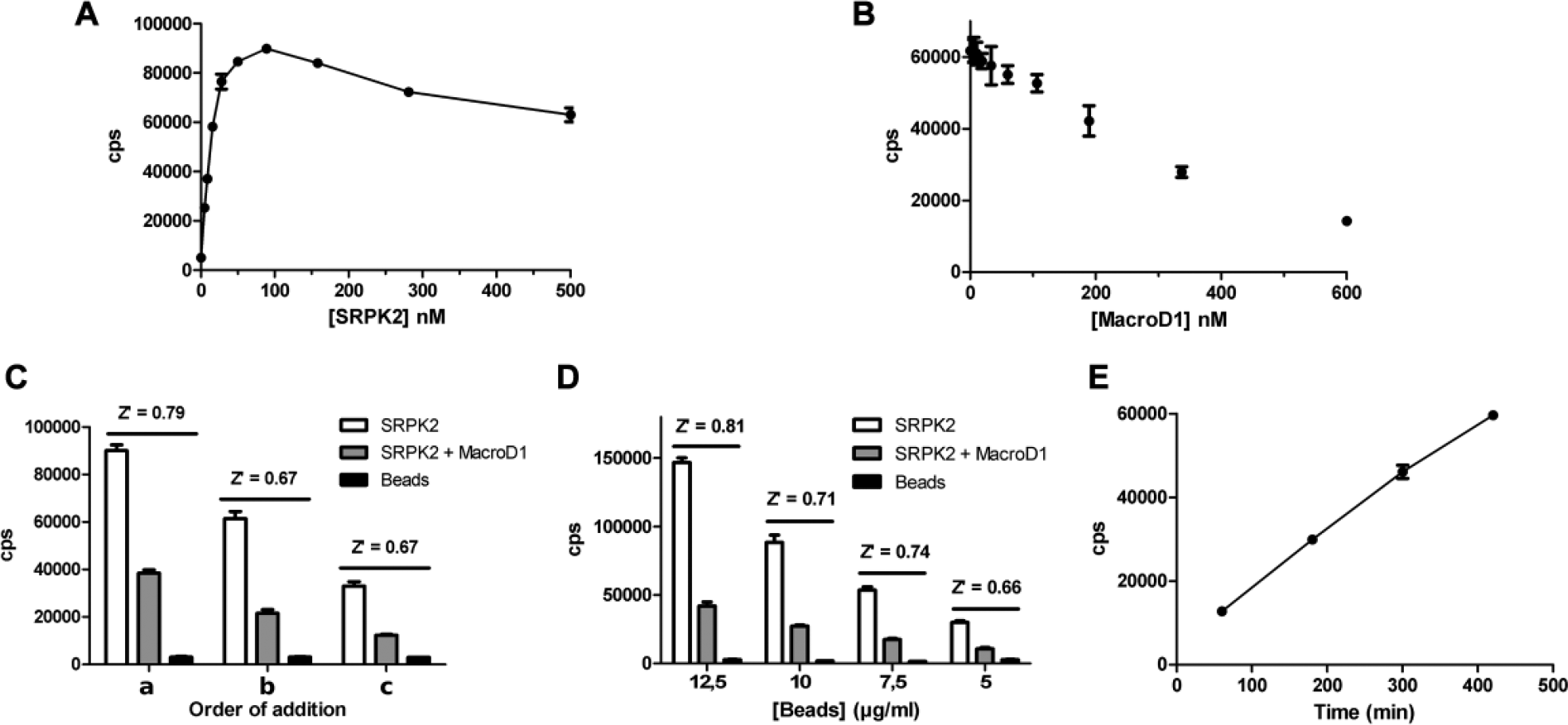
Assay sensitivity, ADP-ribosyl hydrolysis, and AlphaScreen bead optimization. (
Assay Optimization
Once we had demonstrated that the assay principle works, we began to optimize various assay parameters. We first optimized the addition order of the AlphaScreen beads using three sequences ( Fig. 2C ): (a) Modified SRPK2 was incubated with MacroD1 for 180 min, followed by the addition of both beads and 90 min of incubation; (b) modified SRPK2 was incubated with MacroD1 for 180 min, followed by the addition of acceptor beads and 45 min of incubation, and then the addition of donor beads and 45 min of incubation; and (c) modified SRPK2 was incubated with MacroD1 for 180 min, followed by the addition of donor beads and 45 min of incubation, and then the addition of acceptor beads and 45 min of incubation.
We then optimized the AlphaScreen bead concentration (12.5, 10, 7.5, and 5 µg/mL) ( Fig. 2D ) and bead incubation time ( Fig. 2E ). Once we had established the optimal parameters for the beads, we optimized the time and concentration dependency of the MacroD1 catalytic reaction. Finally, we measured the DMSO tolerance of the assay with 0%–10% DMSO concentrations.
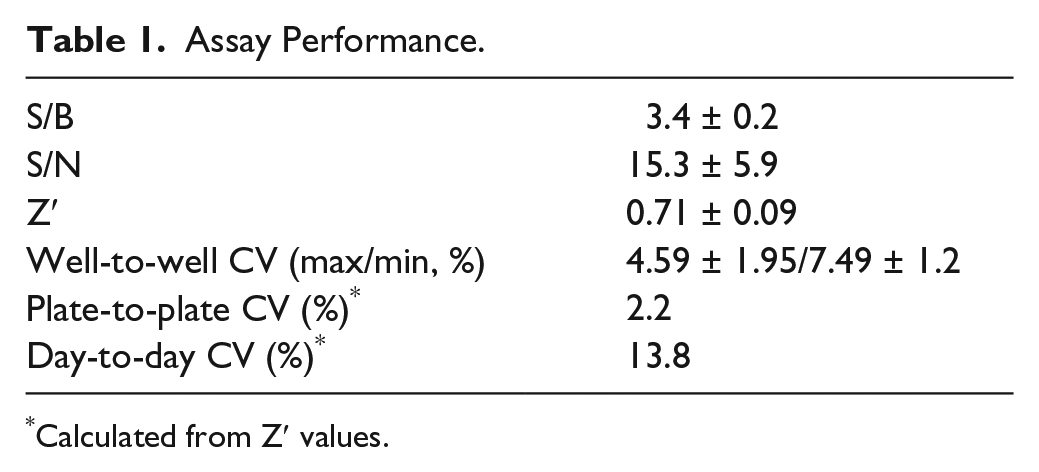
The assay was validated by measuring the repeatability of the maximal and minimal signals between different wells, plates, and days ( Table 1 ). Altogether, five plates containing maximal and minimal signals were measured during 3 days, one plate on days 1 and 2, and three plates on day 3. Forty maximal and minimal signal points were included in each of the plates and well-to-well, plate-to-plate, and day-to-day variations were calculated as coefficients of variation (CVs). The quality of the assay was measured with common statistical parameters: signal-to-noise ratio (S/N), signal-to-background ratio (S/B), and screening window coefficient (Z′).14,15
Assay Performance.
Calculated from Z′ values.
Library Screening
MacroD1 was screened for inhibition against the MicroSource Spectrum collection. Two thousand compounds were screened at a single concentration (100 µM). The compounds (0.15 µL) were transferred to the assay plates with an Echo acoustic dispenser (Labcyte, Sunnyvale, CA). Then 7.5 µL of MacroD1 and SRPK2 (final concentrations of 800 and 50 nM, respectively) was added to the assay plates. The plates were incubated for 1 h 20 min at room temperature, followed by the addition of the acceptor and donor bead mixture (10 µL, final concentration 5 µg/mL) and an additional incubation of 3 h. Each screening plate contained blank wells (AlphaScreen beads only), negative controls (0% inhibition) with no inhibitor, and positive controls (100% inhibition) with no MacroD1.
Potency Measurements
Dose–response curves for hit compounds were measured in quadruplicates from 320 µM to 3.2 nM using half-logarithmic dilutions. The compounds were transferred to the assay plates with Echo, followed by the addition of 7.5 µL of both MacroD1 (final concentration 800 nM) and SRPK2 (final concentration 50 nM). The plates were incubated for 40 min at room temperature, followed by the addition of the acceptor and donor bead mixture (10 µL, final concentration 5 µg/mL) and an additional incubation of 3 h. The dose–response curves were fitted using a four-parameter nonlinear regression analysis (sigmoidal dose–response fitting with variable slope) with GraphPad Prism version 5.03 for Windows (GraphPad Software, La Jolla, CA).
Western Blot
The time dependency of the removal of ADP-ribose from SRPK2 catalyzed by MacroD1 was tested by incubating the modified SRPK2 (0.5 μM) with MacroD1 (4 μM) in 25 mM HEPES, pH 7.5, 100 mM NaCl, and 1 mg/mL BSA for 15 s to 20 min. Validation of the hit molecules as potential MacroD1 inhibitors was performed using Western blot as an orthogonal assay. The modified SRPK2 (1 µM) and MacroD1 (4 µM) with a hit compound (100 µM) were incubated in 25 mM HEPES, pH 7.5, and 100 mM NaCl for 2 h 30 min. All the reactions were stopped by adding 2× Laemmli buffer (Bio-Rad) and incubating at 95 °C for 5 min. The samples were analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose membrane (Whatman, Little Chalfont, UK). After transfer, the membrane was stained with 0.1% (w/v) Ponceau S in 5% (v/v) acetic acid to verify equal sample loading. The membrane was blocked using 1% casein in 1× TB (Bio-Rad, Hercules, CA). The samples were visualized with 1:15,000 streptavidin-conjugated horseradish peroxidase (PerkinElmer) using a chemiluminescent substrate (WesternBright ECL, Advansta Corporation, Menlo Park, CA).
Results
Activity Assay for ADP-Ribosyl Hydrolyzing Macrodomains
No screening assays have been described for mono-ADP-ribosyl hydrolyzing macrodomains to date. Recently, however, a high-throughput screening (HTS) assay for the PAR degrading macrodomain, PARG, was developed and utilized for inhibitor screening. 16 Here we aimed to establish an AlphaScreen-based screening assay applicable to mono-ADP-ribosyl hydrolyzing macrodomains that measures the hydrolysis of protein-linked ADP-ribosyl by macrodomains. To this end, we used poly-His-tagged SRPK2, which was ADP-ribosylated by PARP10 with biotin-NAD+. This produces SRPK2 that has both an N-terminal poly-His fusion tag and biotin-ADP-ribose as a covalent modification. The modified SRPK2 interacts with nickel-chelate acceptor beads via the poly-His-tag and with streptavidin donor beads via the biotin ( Fig. 1 ). The interaction can be relieved by hydrolyzing the ADP-ribose from SRPK2 by ADP-ribosyl hydrolases ( Fig. 1 ).
The assay sensitivity was first tested to establish the optimal SRPK2 concentration and maximal signal (hook point) ( Fig. 2A ). 13 We found that maximal signal was reached between 50 to 100 nM SRPK2 concentration. We chose the 50 nM SRPK2 concentration to be used in the subsequent assays. Next, we titrated 50 nM SRPK2 with MacroD1 to find the optimal concentration for the assay ( Fig. 2B ). The aim was to achieve a robust screening assay with the ability to clearly differentiate between actives and nonactives in the screening stage, so a protein concentration resulting in a 60% signal decrease (60% theoretical ADP-ribosyl hydrolysis) was aimed for. Based on the experiment, we decided to use 400 nM MacroD1 (180 min incubation time) in the following experiments.
We had added the AlphaScreen beads in the initial experiments based on the recommended protocol (PerkinElmer): after the enzymatic reaction, the acceptor beads were added, followed by 30 min of incubation and addition of the donor beads, followed by 60 min of incubation. The effect of the addition order of the beads was tested to improve assay sensitivity and for ease of use ( Fig. 2C ). We kept the total incubation time in the experiments to 90 min. The maximal signal was achieved when both beads were added together, and this sequence also resulted in the highest Z′. Z′ was the same when either acceptor or donor beads were added first, but a higher signal was achieved when acceptor beads were added first. Based on these results, we decided to add both beads simultaneously, and we used this sequence in subsequent experiments.
To reduce the running costs of the assay, we optimized the AlphaScreen bead concentration required for an acceptable signal window for screening. The reactions were run with four bead concentrations ( Fig. 2D ). As expected, the AlphaScreen signal, together with Z′, decreased with bead concentration. However, even the lowest concentration (5 µg/mL) resulted in an acceptable signal level and Z′, and it was selected as the bead concentration for the follow-up experiments.
The length of bead incubation time can have a significant effect on the assay sensitivity, and therefore we optimized it using the 5 µg/mL bead concentration ( Fig. 2E ). The AlphaScreen signal increased almost linearly with incubation time to the highest time point (7 h) tested. To compromise between signal strength and assay time, we chose a 3 h bead incubation for the assay.
We further optimized the MacroD1 concentration and incubation time ( Fig. 3A ). A clear MacroD1 concentration dependency in the ADP-ribosyl hydrolysis is evident. However, the hydrolysis shows poor time dependency, as most of the hydrolysis already takes place at the first time point and incubation time does not significantly increase the hydrolysis. This was further studied with Western blotting using higher protein concentrations, which showed significant hydrolysis after 10 min of incubation ( Fig. 3B ). It is possible that biotin-mono-ADP-ribosylation may not be an ideal substrate for MacroD1, and therefore a high enzyme concentration is required for the assay. In addition, SRPK2 may have several modification sites, some of which could be poor substrates for MacroD1 explaining the high concentration of MacroD1 needed for the reaction. In order to ensure efficient 60% hydrolysis, we chose 800 nM MacroD1 and an 80 min incubation time for the assay. Since DMSO is the commonly used solvent for compound libraries, we tested the DMSO tolerance of the assay ( Fig. 3C ). The assay was found to be insensitive to DMSO up to the 10% concentration tested. No statistically significant deviation between the 0% DMSO control reaction and the DMSO reactions was found. Finally, we optimized the excitation and integration times of the plate reader and selected 600 ms excitation and 300 ms integration times for further experiments ( Fig. 3D ).
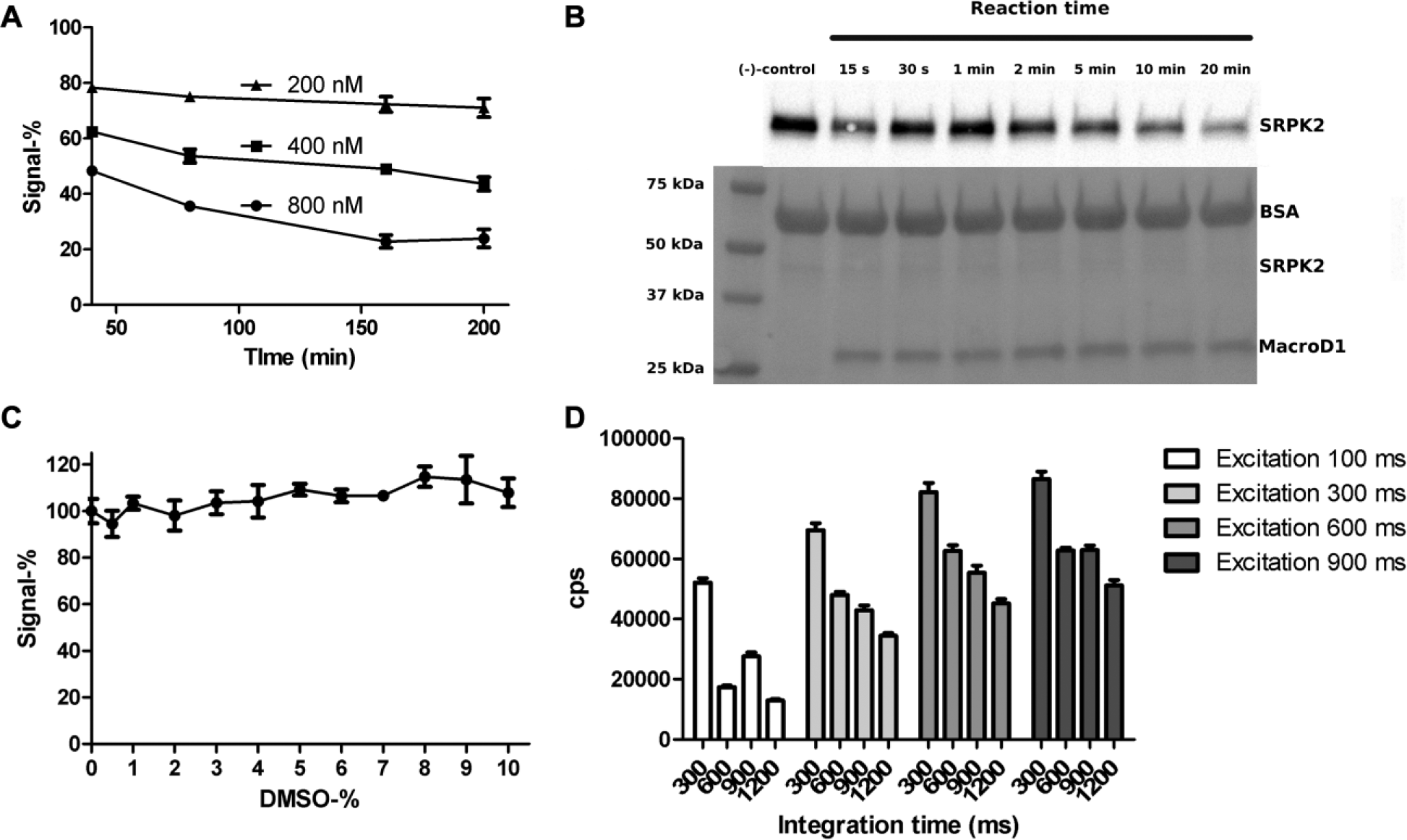
Time and concentration dependency of MacroD1, DMSO tolerance, and plate reader parameters. (
To validate the quality of the assay for screening, we tested the changes in plate-to-plate and day-to-day minimal and maximal signals with five different plates. The average Z′ value for all the plates was 0.7, indicating a robust screening assay ( Table 1 ).
To test the wider usability of the assay and especially SRPK2 as a general substrate for MAR hydrolysis, we measured the activity of five other macrodomains with the assay ( Suppl. Fig. S1 ). Human MacroD2 had higher activity than MacroD1 and showed better time dependency in the assay ( Suppl. Fig. S1A ). The trypanosomatid macrodomains TbMDO and TcMDO displayed much higher activity than the human macrodomains, and even the lowest concentration used (200 nM) hydrolyzed almost all the substrate ( Suppl. Fig. S1B,C ). However, TARG1 and the third macrodomain of PARP14 did not show any activity in the assay, not even with the highest concentration (800 nM). This was surprising, as TARG1 is classified as hydrolyzing enzyme of mono-ADP-ribosylation, 17 whereas the third macrodomain of PARP14 is a reader of mono-ADP-ribosylation and was used as a control. 18 The binding of these macrodomains to the SRPK2 was further studied with surface plasmon resonance. Based on the studies, TARG interacted unspecifically with the SRPK2, and this was not dependent on the modification ( Suppl. Fig. S2 ). The third macrodomain of PARP14 did not show binding to SRPK2 at all.
Validatory Screening
To test the assay in real compound screening, we used a MicroSource Spectrum library of 2000 compounds, which includes drugs, natural products, and bioactive compounds. The screening was done with 100 µM compound concentration in singlets. The assay performed well in screening, with an average Z′ of 0.73 ± 0.07 over six plates. Altogether, five hits were identified (0.25% hit rate) ( Fig. 4 ) that inhibited MacroD1 activity using a hit limit set to 50% activity. Two additional compounds, namely, protoporphyrin IX and hematoporphyrin, were below the 50% activity limit, having activities of –190% and –51%, respectively. These activity values were clear outliers, and both of these compounds interfere with the AlphaScreen signal by acting as singlet oxygen quenchers. 19
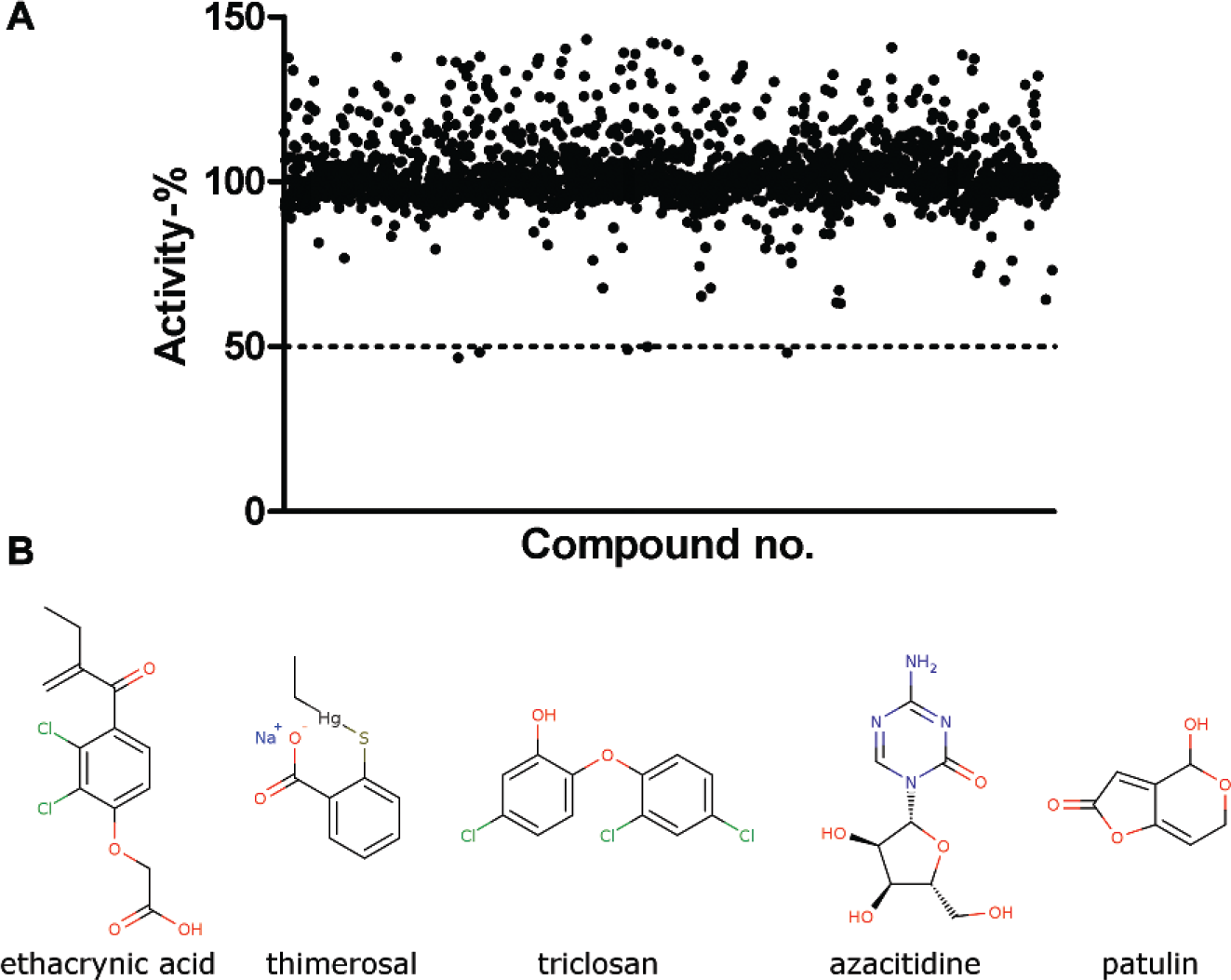
Screening of MacroD1 with MicroSource Spectrum library. (
Dose–response curves were measured for the hit compounds ( Fig. 5A–E ), which revealed one of the initial hits (azacitidine) as a false positive. The IC50 values were 100 µM for triclosan, 27 µM for ethacrynic acid, 5.2 µM for thimerosal, and 2.5 µM for patulin.
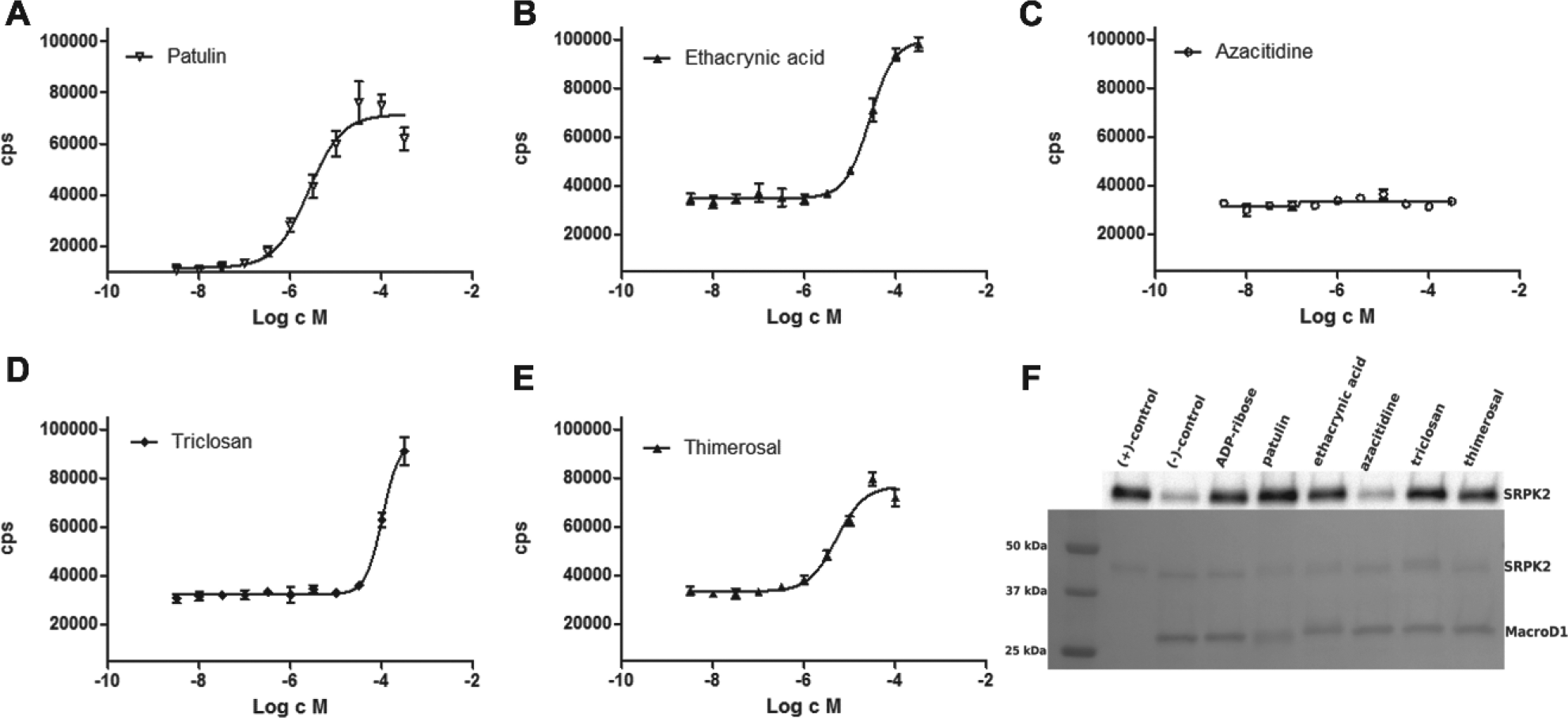
Dose–response measurements and validation of the hit compounds. Dose–response measurements (
We also tested the hits using an orthogonal Western blot assay ( Fig. 5F ). ADP-ribose was used as a control, and it inhibited ADP-ribose hydrolysis, as expected. Azacitidine did not inhibit ADP-ribose hydrolysis, and the result is consistent with dose–response measurements ( Fig. 5C ). More potent hit compounds, ethacrynic acid, triclosan, thimerosal, and patulin, inhibited the macrodomain-catalyzed reaction in the orthogonal assay: patulin had the highest potency, whereas ethacrynic acid, triclosan, and thimerosal showed slightly weaker inhibition. Results were overall in agreement with the AlphaScreen assay.
Discussion
ADP-ribosylation is used in cells for various functions, such as DNA-repair processes and cell signaling. A substantial amount of inhibitors have been developed for poly-ADP-ribosylating ARTDs via HTS methods, fragment-based screening, and virtual screening. Despite functioning in the same pathways, proteins reversing the modification have not been targeted by small-molecule inhibitors. Recently, an HTS method was developed against PAR hydrolyzing PARG, 16 but no screening assays for mono-ADP-ribosyl hydrolyzing macrodomains have been reported to date.
In this article, we described an AlphaScreen-based screening assay for inhibitors against mono-ADP-ribosyl hydrolyzing macrodomains. The assay was tested with four macrodomains, namely human MacroD1 and MacroD2, as well as two trypanosomatid macrodomains. Generally, the assay should be adaptable to other mono-ADP-ribosyl hydrolyzing macrodomains provided that they can utilize biotin-mono-ADP-ribosylated SRPK2 or some other protein as a substrate. However, this was not the case for PARP14 macrodomain and TARG, which both were inactive in the assay. Despite the potential limitations due to substrate protein and unnatural biotin-ADP-ribosylation, the assay is well suited for inhibitor screening, as it tolerates at least 10% DMSO, allowing the screening of compounds using high concentrations. The high DMSO tolerance also allows the screening of fragment libraries, which usually require high compound (and DMSO) concentrations due to low binding affinity. The assay was developed using a 384-well plate format but should be further adaptable to a 1536-well plate format by utilizing an acoustic liquid dispenser.
The assay was validated for human MacroD1, and validatory screening was conducted with a library of 2000 compounds. The assay performance was good, as indicated by Z′ (0.71) and S/N (15.3) values. The validatory screening yielded five hits, and four were further validated as hits using dose–response measurements and an orthogonal assay. Currently, there are no inhibitors reported for mono-ADP-ribosyl hydrolyzing macrodomains. Therefore, the micromolar inhibitors identified in the validatory screening, patulin and ethacrynic acid, belonging to different chemical classes, could serve as starting points in future inhibitor development efforts. Both of these compounds have a range of activities and are also used as medicines. Patulin has been recently identified as an antimicrobial agent against Salmonella, 20 and ethacrynic acid has been shown to improve the antitumor effects of epidermal growth factor receptor tyrosine kinase inhibitors in breast cancer. 21 Taking into account the modest potency and reported activities of the compounds, we foresee that the hit molecules could be useful starting compounds for the development of macrodomain inhibitors when medicinal chemistry and structural biology methods are combined, especially as there are already several crystal structures of macrodomains, such as MacroD1 4 and MacroD2, 8 available in the Protein Data Bank.
The assay described for hydrolyzing macrodomains is simple and utilizes materials available commercially, and therefore it will facilitate discovery of chemical probes for mono-ADP-ribosyl hydrolyzing macrodomains.
Footnotes
Acknowledgements
We thank Structural Genomics Consortium and Dr. Tero Ahola for providing some expression constructs, and we thank Dr. Jani Saarela and FIMM for help in preparing the assay plates. The use of the facilities and expertise of the Biocenter Oulu sequencing and crystallography core facilities are gratefully acknowledged.
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by Biocenter Oulu, Orion Research Foundation, and Academy of Finland (grant nos. 287063 and 294085 for L.L. and grant no. 266922 for T.H.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.