
Abstract
Trypanosoma cruzi is the causative agent of Chagas disease. The lack of an efficient and safe treatment supports the research into novel metabolic targets, with the malic enzyme (ME) representing one such potential candidate. T. cruzi expresses a cytosolic (TcMEc) and a mitochondrial (TcMEm) ME isoform, with these activities functioning to generate NADPH, a key source of reducing equivalents that drives a range of anabolic and protective processes. To identify specific inhibitors that target TcMEs, two independent high-throughput screening strategies using a diversity library containing 30,000 compounds were employed. IC50 values of 262 molecules were determined for both TcMEs, as well as for three human ME isoforms, with the inhibitors clustered into six groups according to their chemical similarity. The most potent hits belonged to a sulfonamide group that specifically target TcMEc. Moreover, several selected inhibitors of both TcMEs showed a trypanocidal effect against the replicative forms of T. cruzi. The chemical diversity observed among those compounds that inhibit TcMEs activity emphasizes the druggability of these enzymes, with a sulfonamide-based subset of compounds readily able to block TcMEc function at a low nanomolar range.
Introduction
American trypanosomiasis, also known as Chagas disease, is caused by the protozoan Trypanosoma cruzi. According to the WHO, up to 7 million people across Latin America are infected by this pathogen, with the disease now emerging as a problem at nonendemic sites.1,2 The available treatments are based on the use of two nitroheterocycle prodrugs, nifurtimox and benznidazole. Although less toxic than nifurtimox, benznidazole displays side effects leading to discontinuation of treatment for about 30% of patients.3,4 These prodrugs are curative against the initial acute stage of the disease, with strains refractory to therapy being frequently encountered.5,6 The use of benznidazole in chronic phase is still under investigation.7,8 Against this backdrop, there is a need for the development of safer, efficacious drug treatments that target all stages of the disease.
Reduced nicotinamide adenine dinucleotide phosphate (NADPH) has a crucial role in parasite survival. This cofactor is used as source of reducing equivalents to maintain trypanothione, the main free thiol found within this organism, in its reduced dihydro state. In this form, trypanothione can drive a series of redox cascades that facilitate the detoxification of reactive oxygen species (ROS) produced from endogenous metabolic reactions or from exogenous immune insults generated by mammalian host cells. 9 Additionally, NADPH participates in a number of biosynthetic pathways, such as in fatty acid and nucleic acid synthesis. 10 As this cofactor plays an essential role in maintaining the reduced environment found within a cell and in the production of the basic building blocks required for cell viability, the enzymes responsible for its generation are considered potential drug targets. In most organisms, the enzymes responsible for NADPH production depend on intermediates derived from highly reduced substrates or with high energy content. For instance, this is the case of the pentose phosphate pathway (PPP), 11 which includes the reactions catalyzed by glucose-6-phosphate dehydrogenase (G6PDH) and 6-phosphogluconate dehydrogenase (6PGDH). The operability of this route relies on the supply of glucose-6-phosphate (G6P) to maintain the flux of NADPH generation. On the other hand, enzymes such as malic enzymes (MEs), 12 isocitrate dehydrogenase (IDH), 13 and glutamate dehydrogenase (GLDH) 14 also represent important candidates for NADPH production, mainly when glucose is scarce and amino acids are utilized as an alternative source for energy production. 15
T. cruzi energy metabolism changes according to the carbon source available in the different microenvironments the parasite finds itself during its life cycle. 16 In the infectious, nonreplicative bloodstream trypomastigote form, the parasite is able to utilize the abundant glucose supply found in its mammalian host’s bloodstream. In contrast, the replicative intracellular amastigotes grow in almost free glucose medium, and are predicted to depend on amino acid catabolism for energy production. Considering the nutrient availability in the amastigotes stage, enzymes such as ME, IDH, and GDH are likely to gain importance in NADPH production.
ME, in the presence of a divalent cation ion (Mg2+ or Mn2+), catalyzes the oxidative decarboxylation of malate to pyruvate, concomitantly reducing NAD(P)+ to NAD(P)H. T. cruzi and Trypanosoma brucei express two ME isoforms, one located in the cytosol (TcMEc and TbMEc, respectively) and the other present in the mitochondrion (TcMEm and TbMEm, respectively).
12
Downregulation of TbMEm expression using RNAi on T. brucei procyclic forms, the parasite stage found in the insect vector, has revealed that this isoform is essential for parasite survival, whereas TbMEc, along with the PPP, is responsible for NADPH production in the cytosol.
17
In T. cruzi, there is no information regarding the essentiality of the distinct ME isoforms. Although TcMEc and TbMEc catalyze the same reaction, both isozymes differ in their kinetic properties. TcMEc is allosterically activated by
Herein, we present the discovery of 262 novel TcMEc and TcMEm inhibitors identified using a biochemical high-throughput screen (HTS). Based on their structure, most (250) were placed into six distinct chemical classes, while the remainder represent structurally unrelated compounds (singletons). Different compounds specifically targeted TcMEc or TcMEm without significantly affecting the activity of human ME isoforms, with several displaying a growth inhibition effect against cultured T. cruzi epimastigotes and amastigotes.
Materials and Methods
Chemicals
The screening library DIVERSet and the resupply compounds were purchased from Chembridge (San Diego, CA). Clostridium kluyveri diaphorase was obtained from Worthington Biochemical (Lakewood, NJ), while resazurin, NADP+, NADPH,
HsME Expression Constructs
DNA fragments encoding for HsME1 (NM_002395), HsME2 (NP_002387), and HsME3 (NM_001161586) were amplified from MDA-mB-231 breast cancer (for HsME1 and HsME3) or human fetal brain (for HsME2) cDNA using the primers GC
Protein Expression and Purification
The TcMEs heterologous expression followed the protocol published elsewhere. 12 In the case of HsMEs, Escherichia coli Rossetta (DE3) pLysS containing the expression vector (pET28_HsME1, pET28_HsME2, or pET28_HsME3) was cultured in the autoinduction ZYM5052 medium, 20 in the presence of 30 µg/mL kanamycin and 34 µg/mL chloramphenicol, at 22 °C for 60 h. The cells were harvested and the pellets stored at −20 °C.
The purification steps were the same for TcMEs and HsMEs. Cells were resuspended in buffer A (50 mM Tris-HCl, pH 8.0, and 500 mM NaCl) containing 5 mM imidazole and lysozyme (1 mg/mL). Following sonication, the clarified lysates were passed through a HisTrap HP column (GE Healthcare Life Technologies, Pittsburgh, PA) and proteins eluted using an imidazole gradient (5–400 mM) in buffer A. ME-containing fractions were pooled and the proteins concentrated. The histidine tag was removed with thrombin (2 U/mg), for 16 h at 4 °C. Finally, the proteins were subjected to size exclusion chromatography on a Superdex 200 16/60 column (GE Healthcare Life Technologies) equilibrated with GF buffer (50 mM Tris-HCl, pH 8.0, and 150 mM NaCl). Purified proteins were concentrated using an Amicon Ultra-15 Centrifugal Filter device of a 30 kDa molecular weight cutoff (Millipore, Billerica, MA) and stored at −80 °C with 5% (v/v) glycerol.
Kinetic Characterization
The malate oxidative decarboxylation activities of recombinant MEs were assayed in a coupled reaction using diaphorase and resazurin ( Fig. 1A ). In this system, NADPH generated during the ME-catalyzed conversion of malate to pyruvate is utilized by diaphorase to drive reduction of resazurin to resorufin. Resorufin production can then be followed by monitoring the change in fluorescence using a plate reader set to an excitation λ = 570 nm and an emission λ = 590 nm. The apparent Michaelis–Menten constant (KmApp) for NADP+ or malate was determined by varying the concentration of one of the substrates while keeping the other at a saturating level. The activation constant (Ka) of aspartate for TcMEc was determined as previously described. 21 All the reactions were performed at 25 °C in reaction buffer (50 mM Tris-HCl, pH 7.5; 50 mM NaCl; and 0.01% [v/v] Triton X-100) supplemented with 2 mM MnCl2, 10 µM resazurin, and 2 U/mL diaphorase, in 384-well black v-bottom microplates and a final volume of 50 µL. The resorufin fluorescence intensity was measured using an EnVision plate reader (PerkinElmer, Waltham, MA).
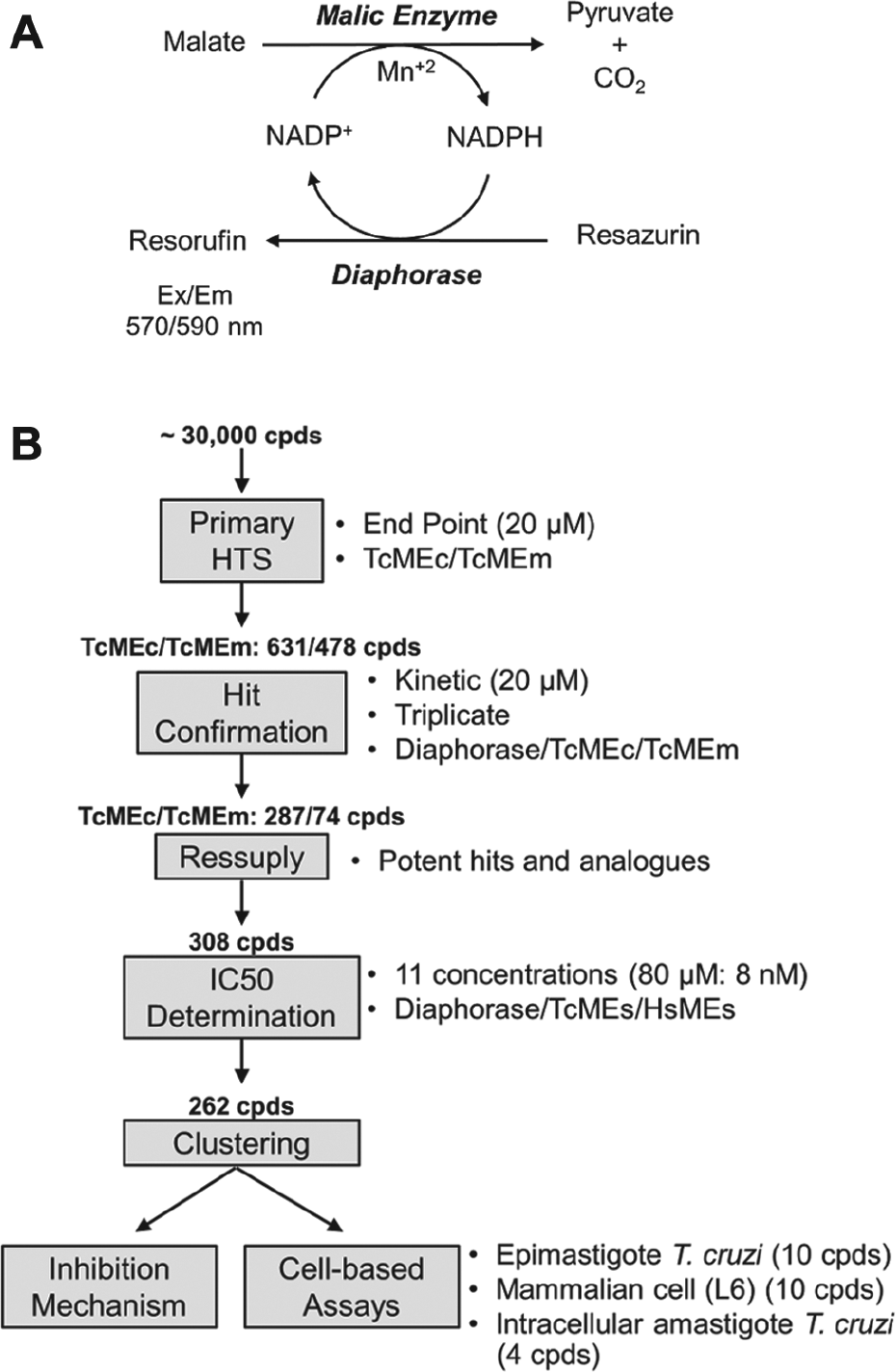
Schematic of HTS assay and workflow. (
Automated Primary HTS
The automated HTS assay for TcMEc and TcMEm was performed using the coupled system of diaphorase and resazurin, adapted from a previously reported dehydrogenase HTS assay.
22
The activity of either trypanosomal enzyme in the presence of a diverse chemical library of 29,760 compounds was assessed using a homogeneous end-point assay in 384-well plate format and performed in an automated workstation (liquid handler, JANUS-MDT; plate reader, EnVision; both from PerkinElmer). First, 22 µL of reaction mix was added into the wells, followed by the transfer of 0.5 µL of sample compound (stock at 1 mM in 100% [v/v] DMSO). The reaction was then initiated by addition of 2.5 µL of malate (concentrations are described below). Following incubation at room temperature for 3 h, the end-point fluorescence intensity of resorufin was measured (excitation λ = 570 nm, emission λ = 590 nm). The final assay concentration (FAC) of reagents for assaying TcMEm activity was as follows: 10 µM NADP+, 2 mM MnCl2, 10 µM resazurin, 1 U/mL diaphorase, 63 pM TcMEm, and 1 mM malate. All plates contained 32 positive controls (reaction mix + DMSO + malate) and 32 negative controls (reaction mix + DMSO + reaction buffer). The fluorescence intensity readouts were normalized, considering the means of positive and negative controls in each plate as 100% and 0% of reminiscent activity (RA), respectively. The assay quality was evaluated by the Z factor.
23
The compounds with RA less than or equal to the cutoff RA were considered a hit candidate. The cutoff RA was defined as the average RA of all samples minus three standard deviations. For TcMEc-based reactions, the above assay was performed in the presence of
Confirmation Assay
The hit candidates were assayed against TcMEs and against diaphorase only, to exclude false positives and inhibitors of the coupled system. The hit candidates were transferred from the screening library plates to new assay plates and assayed in triplicate, monitoring the resorufin formation for 30 min. The calculated velocities were normalized using controls. For TcMEs, the assay conditions were the same for HTS assay, except 2× the amount of enzyme (TcMEs and diaphorase) was used. For diaphorase-only reactions, the assay concentrations of reagents were 15 µM NADPH, 2 mM MnCl2, 10 µM resazurin, and 0.035 U/mL diaphorase.
IC50 Determination
Initially, the effect of a single dose (80 µM) of hit compound against TcMEs, HsMEs, and diaphorase was assessed. For those structures that inhibited ME function by >50% without affecting diaphorase, the concentration of compound that inhibited ME activity (IC50) by 50% was determined. First, the selected compounds were serially diluted (2.5-fold dilution steps) in DMSO using a Versette Automated Liquid Handler (Thermo Fisher Scientific, Waltham, MA). Next, the reaction mix and compounds were dispensed in 384-well plates using the Versette Automated Liquid Handler and the reaction was initiated with malate using a Multidrop Combi Reagent Dispenser (Thermo Fischer Scientific). The FACs for each compound were 80, 32, 12.8, 5.12, 2.05, 0.82, 0.328, 0.13, 0.052, 0.021, 0.008, and 0 µM. The velocity of resorufin formation was followed using a Clariostar microplate reader (BMG Labtech, Ortenberg, Germany), following the manufacturer’s instructions for resorufin fluorescence (excitation λ = 545 nm, emission λ = 600 nm). The data were normalized with controls and analyzed in GraphPad Prism 5, using a sigmoidal curve.
For TcMEs, the reagent concentrations were the same as those used in the confirmation assay, while for human MEs, the above inhibition assays were performed using 6 µM NADP+ and 4.7 mM malate for the HsME1 (2.5 nM)-based assay; 1 mM NADP+, 0.7 mM fumarate, and 4.5 mM malate in reactions containing HsME2 (12.5 nM); and 10 µM NADP+ and 3.4 mM malate when testing HsME3 (1.25 nM). All the reaction mixtures contained 2 mM MnCl2, 10 µM resazurin, and 1 U/mL diaphorase.
Mechanism of Inhibition
The mechanism of inhibition was established for TcMEm, by determining the Michaelis–Menten saturation curves for malate or NADP+ in the presence of four different concentrations of inhibitor, and with DMSO as control. This enzyme was used because its activity is not effected by
Cell Culture
The L6 rat myoblast cell line was cultured at 37 °C under a 5% (v/v) CO2 atmosphere in RPMI-1640 supplemented with 5 g/L HEPES, pH 8.0; 0.34 g/L sodium glutamate; 0.22 g/L sodium pyruvate; 2500 U/L penicillin; 0.25 g/L streptomycin; and 10% (v/v) heat-inactivated fetal calf serum.
The T. cruzi (strain Y) epimastigote cells were grown at 27 °C in liver infusion tryptose medium 24 supplemented with 10% (v/v) heat-inactivated fetal calf serum. T. cruzi (strain Sylvio X10/6) epimastigotes engineered to express the thermostable red-shifted firefly luciferase (PpyRE9h) 25 were cultured at 27 °C in RPMI-1640 containing 5 g/L trypticase; 20 mg/L heamin; 5 g/L HEPES, pH 8.0; 0.34 g/L sodium glutamate; 0.22 g/L sodium pyruvate; 2500 U/L penicillin; 0.25 g/L streptomycin; and 10% (v/v) heat-inactivated fetal calf serum. Infection of L6 monolayers with recombinant T. cruzi and the culturing of amastigote parasites were carried out as previously described. 26
Antiproliferative Assays
All assays were performed in a 96-well plate format. To determine mammalian cytotoxicity, growth medium (200 µL) containing 1500 L6 cells and different concentrations of compound (80–0.82 µM) were incubated at 37 °C for 96 h. Resazurin (FAC = 50 µM) was added to each well and cultures incubated for a further 6 h. The fluorescence of each culture was determined using a Gemini fluorescent plate reader (Molecular Devices, Sunnyvale, CA) (excitation λ = 530 nm, emission λ = 585 nm, filter cutoff at 550), with the change in fluorescence resulting from the reduction of resazurin to resorufin being proportional to the number of live cells. The compound concentration that inhibits cell growth by 50% (EC50L6) was established using the nonlinear regression tool on GraphPad Prism 5 (GraphPad Software).
Growth inhibition assays against T. cruzi (strain Y) epimastigotes were performed using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega) as described. 22 Initial screens were performed in the presence of 80 µM compound, with the trypanocidal activity scored as active, moderate, or inactive when the percent of viable cells was <25%, between 25% and 75%, or >75%, respectively. For active structures, parasite growth was evaluated using different compound concentrations (80–0.82 µM), and from the resulting dose–response curve, a compound’s EC50Epi was established.
Growth inhibition assays against luciferase expressing T. cruzi amastigotes were performed as previously described. 26 Briefly, L6 (1500) cells in mammalian growth medium (100 µL) were allowed to adhere to the base of each well for 6 h. T. cruzi trypomastigotes (10,000 in 100 µL of mammalian growth medium) were then added to monolayer, and infections performed overnight at 37 °C under 5% (v/v) CO2. The cultures were washed in growth medium to remove noninternalized parasites, treated with compound-containing fresh growth medium, and subsequently incubated at 37 °C under 5% (v/v) CO2 for 72 h. Growth medium was removed, the cells lysed in cell culture lysis reagent (Promega), and reporter levels determined using the luciferase assay kit (Promega). Luminescence was then measured using a Fluostar Omega plate reader (BMG Labtech), with this activity being proportional to the number of live cells.
Data Analysis
The HTS output was analyzed using Microsoft Office 365. JChem for Office was used for chemical database access, search, and reporting (JChem for Office 16.8.2200). The kinetic data, inhibition mechanism, and IC50 and EC50 values were analyzed using GraphPad Prism 5 (GraphPad Software).
Results
Automated Primary HTS
Recombinant TcMEc and TcMEm were purified and their kinetic parameters determined under the HTS assay conditions (
Suppl. Fig. S1
and
Suppl. Table S1
). The
The hit candidates were transferred from the screening library plates to new assay plates and assayed in triplicate against ME coupled assay and against diaphorase, both in kinetic mode. Confirmed hits fulfilled the following criteria: ME inhibition (RAME ≤ 50%), inactive against diaphorase (RAdiaphorase ≥ 80%) and time-independent inhibition (ME activity linear along the assay time). Following these criteria, 287 and 74 hits were confirmed for TcMEc and TcMEm, respectively, with 27 common for both enzymes.
Kinetic Characterization of Hits
The most potent hits (RAME ≤ 30%), together with 99 commercially available analogues, totalizing 308 molecules, were resupplied. This compound collection was then assayed in triplicate against both TcMEs, the three HsMEs, and diaphorase, at 80 µM. The DNA fragments encoding for the catalytic regions of HsME1, HsME2, and HsME3 were heterologously expressed in E. coli, each enzyme purified, and their kinetic parameters determined ( Suppl. Table S1 ). These biochemical screens revealed that 262 compounds inhibited at least one of the T. cruzi MEs without affecting diaphorase activity: 7 molecules were eliminated due to their inhibitory effect on diaphorase, and 39 compounds did not inhibit TcMEs activity. For the ME-specific inhibitors, IC50 values of each compound toward the TcMEs and HsMEs were determined ( Fig. 2 and Suppl. Table S2 ). For TcMEm, the IC50 values ranged from 0.5 to 71 µM, while for TcMEc, this varied from 0.0032 to 64 µM ( Suppl. Fig. S2 ). Many compounds in the “resupply” library also inhibited at least one of the human MEs, although encouragingly, some were inactive against all three mammalian isoforms. Based on their chemical backbone and ability to target the trypanosomal enzyme, the compounds that constitute the resupply library could be clustered into six structurally related different groups, designated as ATR1 to ATR6, with a seventh assemblage consisting of 12 nonstructurally related singletons, designated as ATR7 ( Fig. 2 ).
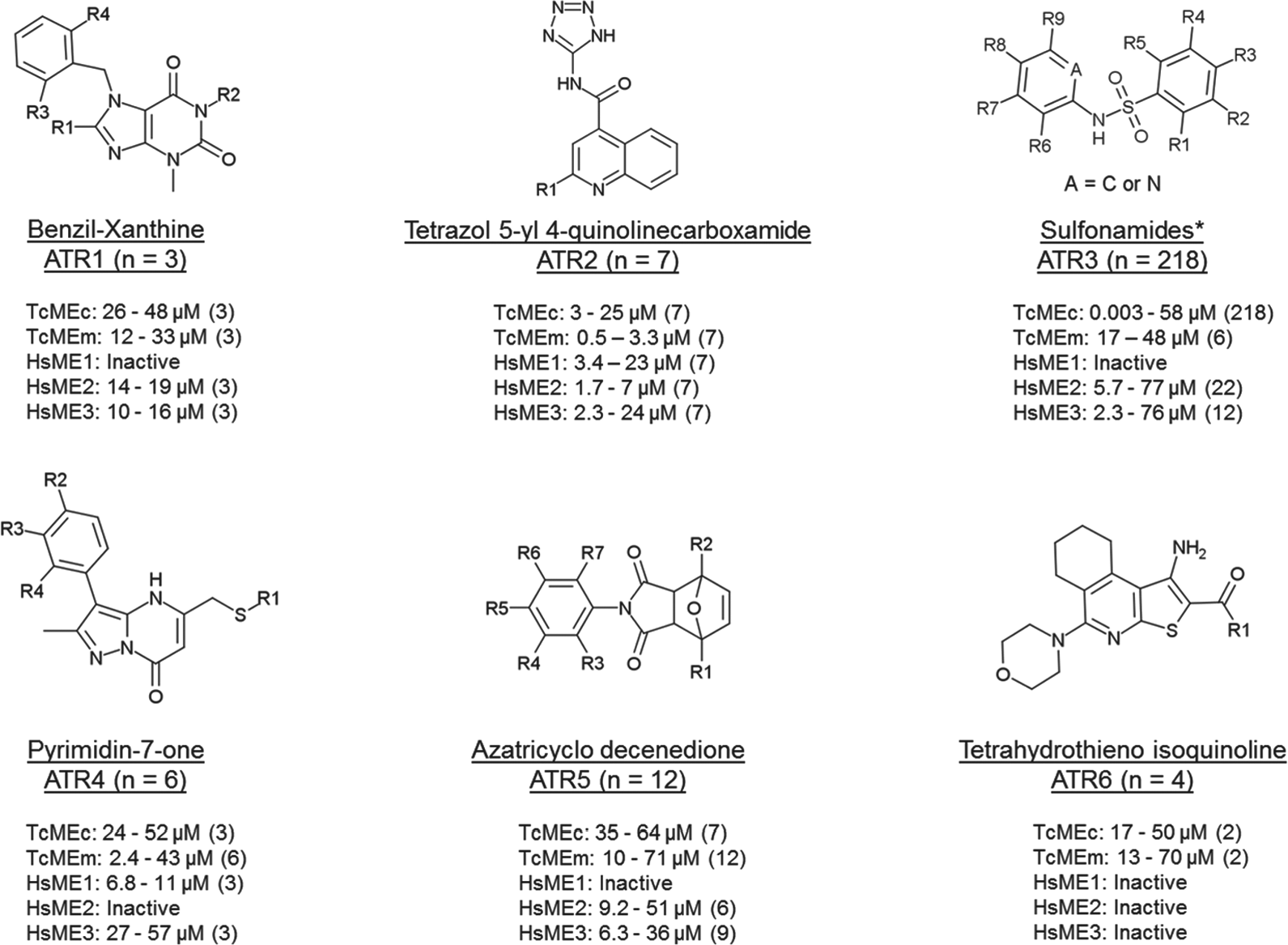
Scaffold of TcMEs inhibitor groups. TcME inhibitors were clustered into six structurally related groups (ATR1 to ATR6). The distinguishing chemical motifs and number (n) of compounds for each grouping are shown. The potency range (as judge by IC50) and number of compounds (in parentheses) targeting the trypanosomal (TcMEc and TcMEm) and/or human (HSME1, HsME2, and HsME3) are given. Compounds were deemed inactive if no IC50 could be determined over the concentration range used (the highest assayed concentration was 80 µM). * Scaffold representative of 80% of ATR3 compounds. Within this group, compounds are also found with substituted five-membered heterocycles attached at the nitrogen of sulfonamide (10% of compounds), besides other substituents connected to sulfonamide, as aliphatic groups.
To determine their mechanism of inhibition, a representative member from each structural group that generated an IC50 value of ≤10 µM (ATR2-001, ATR3-001, ATR4-001, and ATR5-001) was tested against TcMEm; unlike TcMEc, this isoform was used, as it does not undergo allosteric activation, thereby simplifying interpretation of the inhibition data. For ATR3-001 and ATR5-001, a mixed-type inhibition with respect to both substrates was noted ( Suppl. Fig. S3 ). In contrast, TcMEm activity in the presence of ATR2-001 resulted in a competitive inhibitory mechanism toward malate and a mixed type to NADP+, whereas ATR4-001-treated enzyme was competitive to malate and uncompetitive to NADP+ ( Suppl. Fig. S4 ).
In Vitro T. cruzi Antiproliferative Assays
To determine whether any of the TcME inhibitors displayed antiparasitic properties, a selected subset of compounds were screened for trypanocidal activity against cultured T. cruzi epimastigotes. The compounds used were selected based on their IC50 values toward the trypanosomal enzymes. The structures tested generated IC50 values of <15 µM against one or both TcMEs (ATR1, ATR2, and ATR4 to ATR7), while a range (20 out of 183) of ATR3 compounds that elicited IC50 values ranging from 3 nM to 10 µM were screened. Out of the 44 agents examined, 13 displayed a moderate trypanocidal effect at 80 µM, with 21 having no significant effect on parasite growth at this concentration ( Suppl. Table S2 ). For the remaining 10 active compounds ( Fig. 3 ), growth inhibition assays were conducted to determine their potency against epimastigote parasites, yielding EC50Epi values ranging from 14 to 56 µM ( Table 1 ). In parallel, benznidazole, used as a control treatment, generated an EC50Epi of 12 ± 1 µM. The biological activity of the 10 active compounds was extended to evaluate their toxicity toward mammalian cells and T. cruzi amastigotes. Against the rat skeletal L6 line, two compounds (ATR5-001 and ATR7-005) had no effect on mammalian cell growth at concentrations up to 80 µM, while the remaining eight structures yielded EC50L6 values ranging from 14 to 55 µM ( Table 1 ). When tested against T. cruzi amastigotes and using a single compound dosage at a concentration that did not affect host cell growth, four molecules (ATR3-128, ATR6-001, ATR7-005, and ATR7-008) displayed antiparasitic activity, preventing growth of the intracellular pathogen by >90% relative to untreated controls ( Table 1 ).
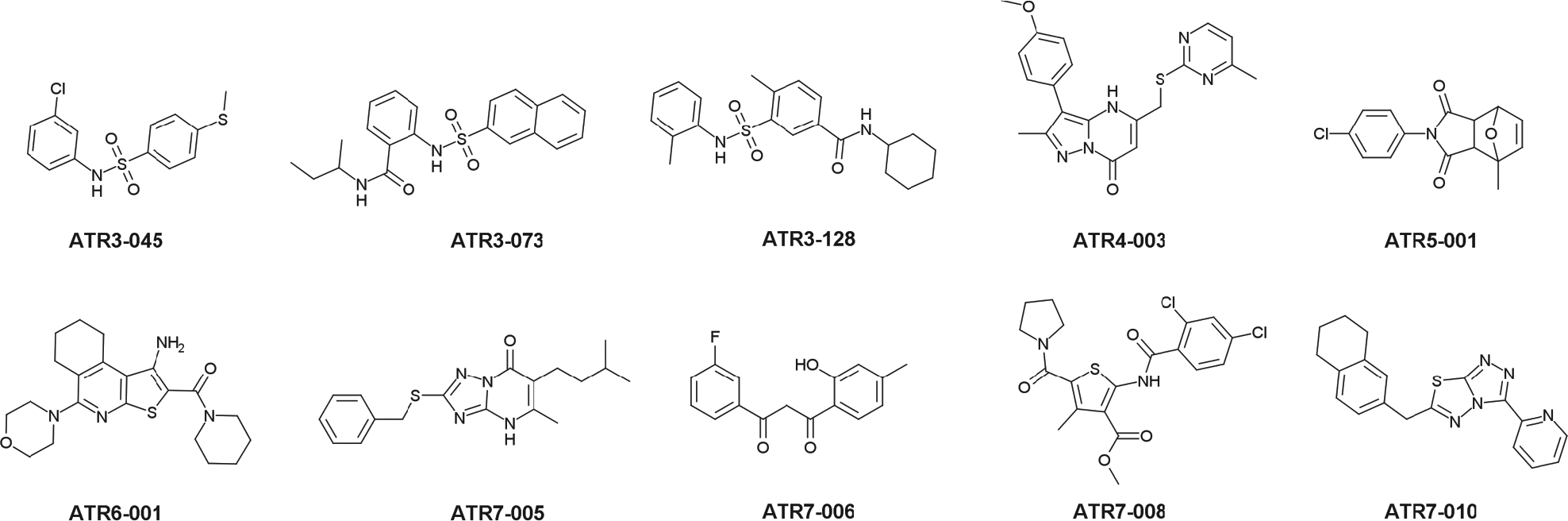
Structure of TcME inhibitors used in phenotypic screens. Selected compounds were assayed against T. cruzi epimastigote and amastigote forms (see Materials and Methods). Values of EC50 are presented in Table 1.
Compounds with Trypanocidal Effect.
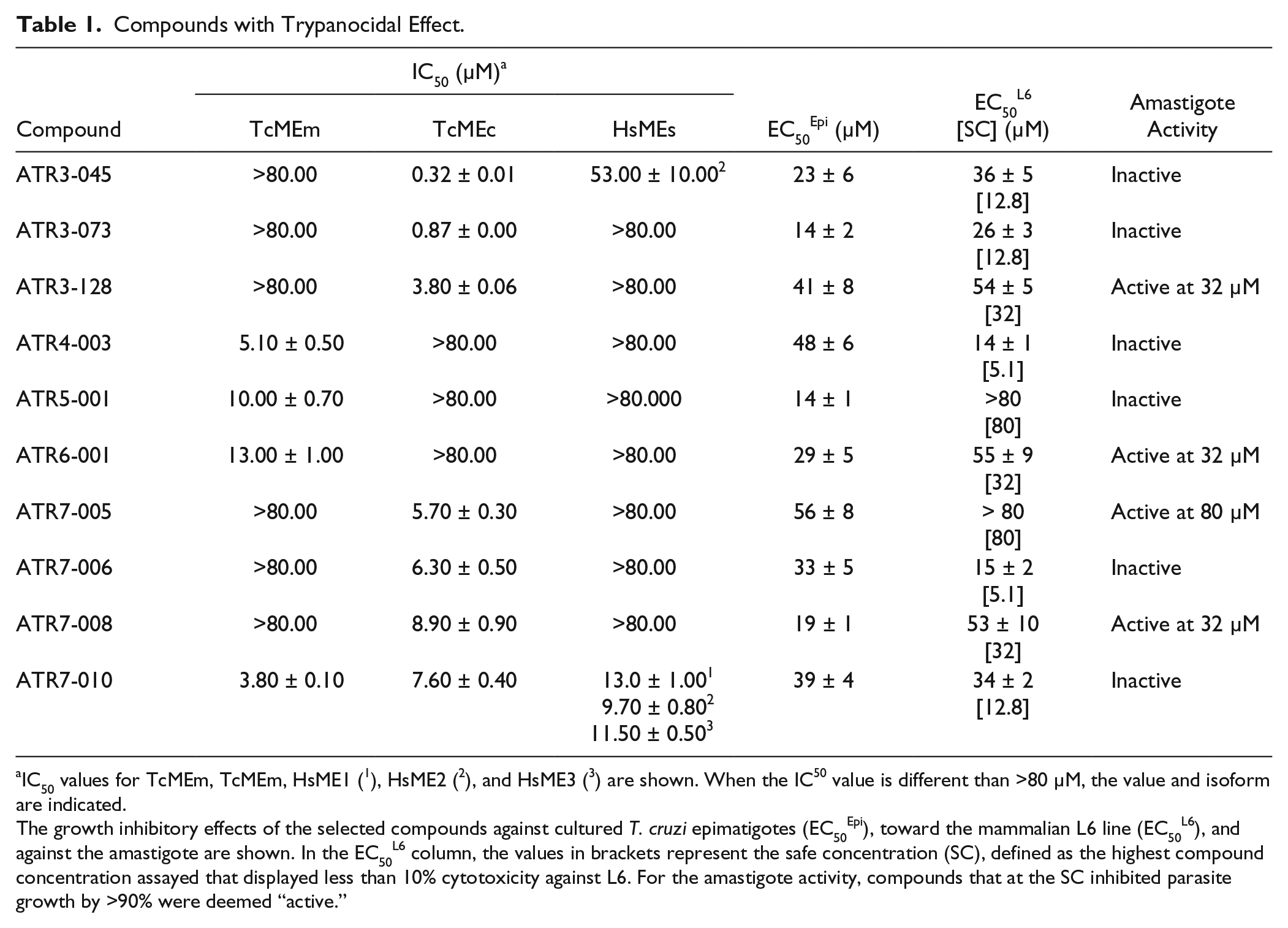
IC50 values for TcMEm, TcMEm, HsME1 ( 1 ), HsME2 ( 2 ), and HsME3 ( 3 ) are shown. When the IC50 value is different than >80 µM, the value and isoform are indicated.
The growth inhibitory effects of the selected compounds against cultured T. cruzi epimatigotes (EC50Epi), toward the mammalian L6 line (EC50L6), and against the amastigote are shown. In the EC50L6 column, the values in brackets represent the safe concentration (SC), defined as the highest compound concentration assayed that displayed less than 10% cytotoxicity against L6. For the amastigote activity, compounds that at the SC inhibited parasite growth by >90% were deemed “active.”
Discussion
The TcMEs are a promising drug target for Chagas disease treatment, due to their importance in helping maintain NADPH levels in the T. cruzi parasite. In this study, using a biochemical HTS we evaluated approximately 30,000 compounds as potential TcMEs inhibitors. A total of 262 compounds were identified and confirmed as blocking this activity, with the hits falling into six groups of structurally related molecules and a group with 12 singletons. The results obtained for each group are discussed below.
The three compounds that comprise the ATR1 group all contain a benzyl-xanthine core within their backbone ( Fig. 2 ), with this type of scaffold already described as an inhibitor of human BRAF protein kinase. 27 Here, members of this family presented moderate inhibitory activity toward the TcMEs (IC50 values of 12–48 µM) and HsME2 and HsME3 (IC50 around 18 µM), with no effect on HsME1. For one of the ATR1s, ATR1-003, the presence of a sulfhydryl group attached to an imidazole ring on the xanthine backbone appears to underlie its mode of TcME inhibition; the ATR1-003 related compounds, ATR1-004 and ATR1-005, that contain a hydrogen or thiomethyl side chain at R1, have no affected enzyme function ( Fig. 4A ). Unfortunately, none of the ATR1s screened against T. cruzi epimastigotes had any effect on parasite growth.
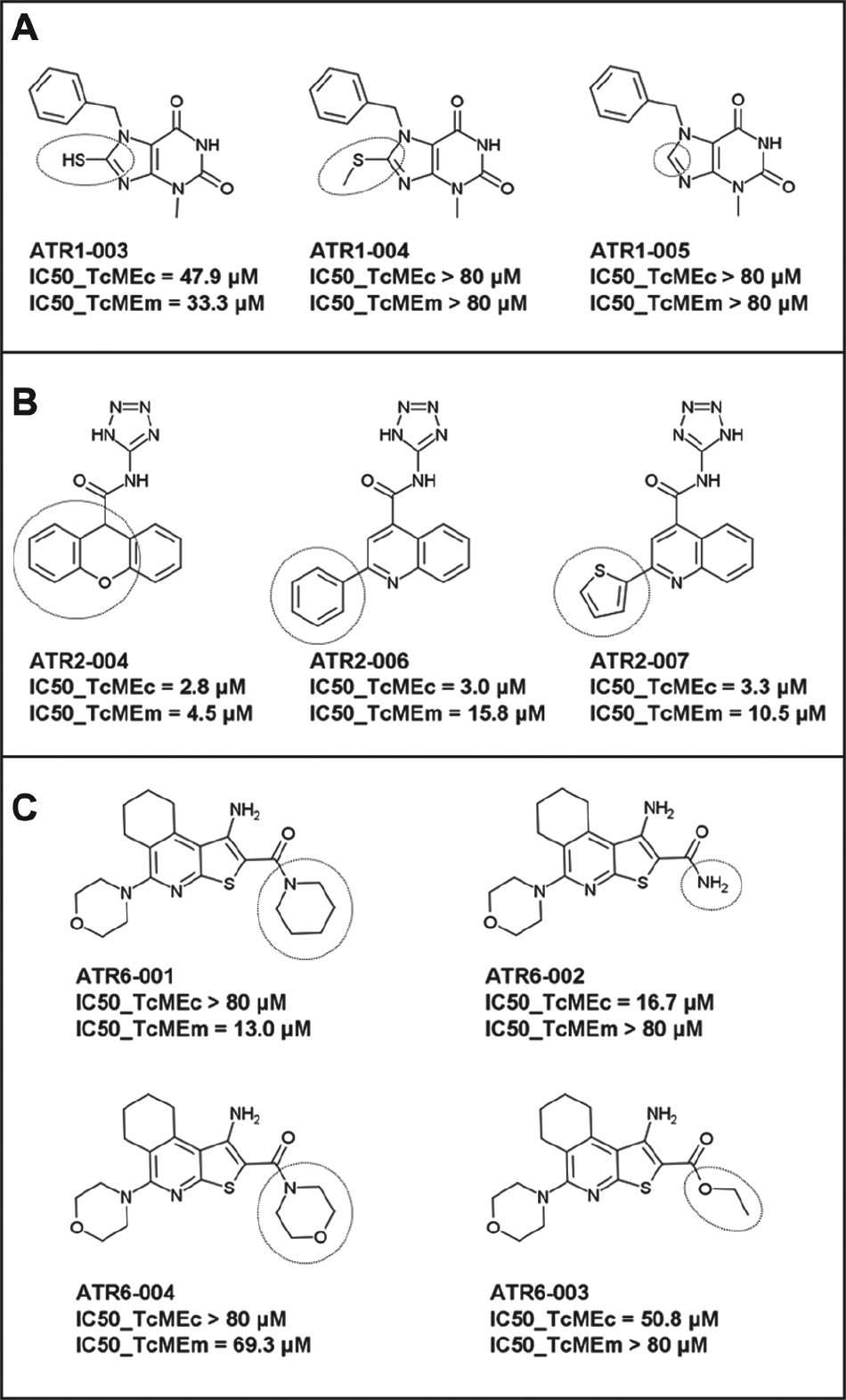
SAR of groups ATR1, ATR2, and ATR6. (
All seven ATR2s, which contain a tetrazol-yl 4 quinolinecarboxamide or N-1H-tetrazol-5-yl-9H-xanthene-9-carboxamide core, proved to be general ME inhibitors targeting each parasite and mammalian isoform tested. Intriguingly, some of these compounds were up to ~5-fold more selective toward TcMEm relative to the other enzymes. Despite only determining the IC50 values of a limited number of ATR2s, it is clear that having different ring-based substituents at the R1 position on the conserved quinoline does not affect a compound’s inhibition potency ( Fig. 4B ): ATR2-004, ATR2-006, and ATR2-007 all exhibit similar IC50 values toward TcMEc or TcMEm even though the former contains a tricyclic xanthene structure, whereas the remaining two compounds possess a benzyl or thiophene moiety linked to the bicyclic quinolone ring. As with the ATR1 structure samples, no ATR2 compound displayed trypanocidal activity.
The ATR3 compounds are related in that they possess a sulfonamide linker that connects two aromatic rings (five or six membered). A total of 218 structures were identified as inhibiting the trypanosomal MEs, with most (212) only targeting TcMEc. Of these, many generated an IC50 below 100 nM, with ATR3-007 yielding a value of 3.2 nM. When the potency of these compounds toward the human ME isoforms was examined, 189 had no effect on any of the mammalian enzymes, further demonstrating the specificity of these inhibitors toward TcMEc. This way, a preliminary structure–activity relationship (SAR) analysis is presented for the most potent compounds, with IC50 values below 100 nM ( Fig. 5 ).
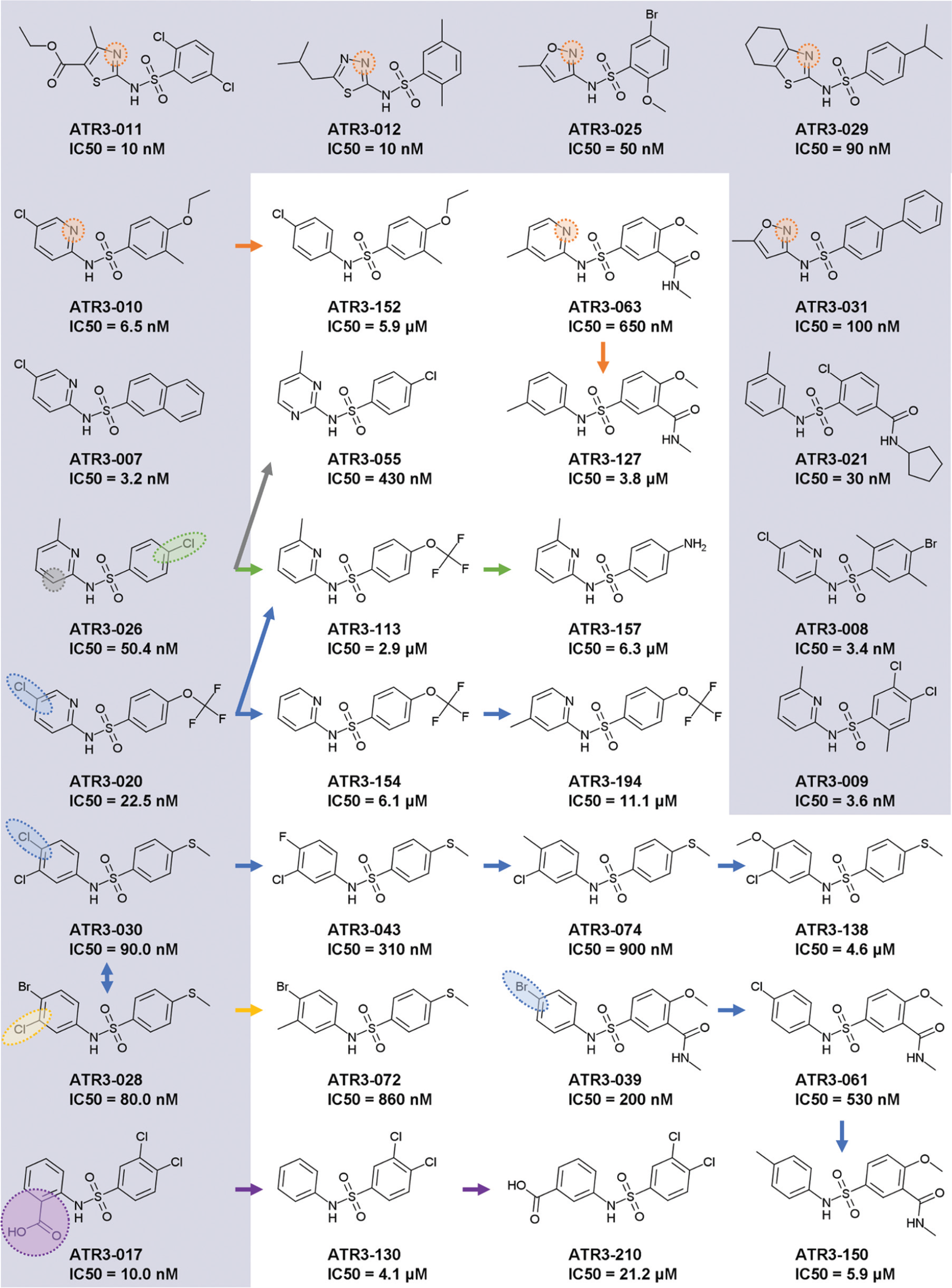
SAR of ATR3 group. In the grey background, the most potent compounds (IC50 < 10 nM), together with other representatives of inhibitors with IC50 values below 100 nM. The IC50 presented is for TcMEc. The arrows indicate the related compounds from a SAR series. The variable position among the analogues is highlighted for each series.
Despite the variety of chemical structures of the 27 ATR3 compounds yielding an IC50 below 100 nM, 19 contain a nitrogen atom within a six- or five-membered ring, two ligations apart from N-sulfonamide. The addition of this nitrogen to ATR3-152, rendering ATR3-010, was sufficient to improve compounds’ potency by 912-fold. Less dramatic, but following the same behavior, was the 5.8-fold gain in potency of compound ATR3-063, compared with ATR3-127. The addition of another nitrogen atom in the ring has a deleterious effect for inhibitory activity, as seen in compound ATR3-055, which is 8.5-fold less potent than ATR3-026. Other orientations of the N-pyridinyl ring were found, as in ATR3-170 and ATR3-188 (nitrogen located three ligations apart from N-sulfonamide), and in ATR3-003, ATR3-004, and ATR3-153 (nitrogen four ligations apart from N-sulfonamide); all these inhibitors exhibited IC50 values in the micromolar range ( Suppl. Table S2 ). Interestingly, ATR3-003 and ATR3-004 inhibited TcMEm as well. In the subgroup of N-(2-pyridinyl)-benzenesulfonamides (A = N, Fig. 2 ), the following substituents in the pyridinyl ring were found: methyl at R7, R8, and R9 (also concomitantly at R7 and R9), and halogens (chlorine and bromine) at R8. However, no compound with a substituent at R6 was identified. Analyzing analogues of ATR3-020, the higher activity provided by the addition of chlorine at R8 is noteworthy. Likewise, from the 14 compounds of this subclass with an IC50 value below 100 nM, 10 have a halogen atom at R8. Regarding the benzene region, different substituents were found at all the positions of the ring. For instance, halogens, alkyl, methoxy, and ethoxy, including a fused ring (ATR3-007), as well as another ring connected by a carboxamide (ATR3-002), were recognized. The presence of a chlorine at R3 (ATR3-026) improved activity at least 50-fold compared with trifluoromethoxy (ATR3-113) and amine (ATR3-157) ( Fig. 5 ).
Another representative ATR3 subgroup, presented among the most potent inhibitors, was formed by 4-(methylsulfanyl)-N-phenylbenzenesulfonamides (R3 = methylsulfanyl). Comparison of ATR3-028 analogues revealed the trend Br = Cl > F > methyl > methoxy for inhibitory activity, showing a preference for halogens at R8. The same occurs at position R7, where the replacement of methyl (ATR3-072) for chlorine (ATR3-028) improved potency by 10.8-fold. In the screened library, no compounds from this subgroup had substituents at positions R1, R2, R4, and R5. In contrast, within the subgroup of 4-(methoxy)-N-phenylbenzenesulfonamides (R3 = methoxy), inhibitors with halogens (chlorine, fluorine, and iodine), methyl, or amide at R2 were identified. Again, as seen for the other subgroups, the presence of a halogen atom at R8 (ATR3-039 and ATR3-061 as compared with ATR3-150) increased the inhibitory activity in one order of magnitude. Besides these subgroups, three compounds with a benzoic acid connected to N-sulfonamide were active in concentrations in the nanomolar range (<100 nM). The substitution of an acid group at R6 enhances the potency 40-fold (ATR3-130–ATR3-017). The acid position in the ring dramatically affected the activity, as seen in compound ATR3-210, which was 2000-fold less potent than ATR3-017 ( Fig. 5 ).
In addition to being able to block TcMEc activity, ATR3 compounds also displayed antitrypanosomal activity against epimastigote (ATR3-045, ATR3-073, and ATR3-128) and amastigote (ATR3-128) forms of T. cruzi ( Table 1 ). Sulfonamide-based drugs are frequently used to treat various bacterial and protozoal infections, blocking dihydropteroate synthetase (DHPS) activity, and therefore preventing folate synthesis. It is unlikely that the trypanocidal mode of action of the ATR3 structures identified here involves DHPS inhibition, as T. cruzi is auxotrophic for folates and pterins. 28 Interestingly, sulfonamides similar to the ATR3 TcMEc inhibitors are also present in the reported GSK Chagas Box, a set of 222 leadlike molecules that came out of a phenotypic HTS using a library of 1.8 million compounds. 29 A preliminary assay of TcMEs against the GSK Chagas Box confirmed 3 sulfonamides as potent inhibitors of TcMEc (data not shown). These data, together with the results reported here, encourage further phenotypic studies with the ATR3 dataset to evaluate possible correlation between TcMEc inhibition and parasite death.
Out of all the TcMEs inhibitor hits, the pyrimidinone-based ATR4 compounds demonstrated the highest selectivity toward TcMEm. Two of these specifically blocked the activity of this trypanosomal isoform, including one (ATR4-003) that displayed trypanocidal properties against T. cruzi epimastigotes (
Table 1
and
Fig. 3
). Unfortunately, ATR4-003 also exhibited toxicity to mammalian cells and did not affect T. cruzi amastigote growth. This group displayed the best behavior regarding the inhibition mechanism, which was competitive with respect to malate and uncompetitive with respect to NADP+. This feature indicates a sequential binding of NADP+, followed by the inhibitor, which occupies the malate site. Interestingly, ATR4-like compounds are reported to inhibit the 1-deoxy-
The ATR5 group comprises 12 structures distinguished by the presence of an azatricyclo decenedione motif. Generally, they function as moderate TcMEm, TcMEc, HsME2, and HsME3 inhibitors, with none affecting HsME1 activity. Some compounds that affect both trypanosomal enzymes appear to show a degree of selectivity (~5- to 8-fold) toward TcMEm. One of the compounds (ATR5-001) that specifically targets TcMEm displays growth inhibitory activity against T. cruzi epimastigotes (EC50Epi of 14.1 µM) ( Table 1 ) while having no cytotoxic effect on mammalian cells. However, ATR5-001 was inactive against intracellular parasites. Compounds related to ATR5 have been reported to be inhibitors of thioredoxin glutathione reductase expressed by Schistosoma mansoni, with such structures being particularly effective against this parasite’s larva stage. 33
The four active ATR6 molecules are of interest, as they have no activity toward any of the HsMEs and displayed specific inhibition against TcMEm (ATR6-001 and ATR6-004) or TcMEc (ATR6-002 and ATR6-003) ( Fig. 2 ). The TcMEm preference appears to be due to the presence of a heterocyclic ring (pyridine or morpholine) at position R1, while smaller side chains (amine or ethoxy) confer selectivity toward TcMEc ( Fig. 4C ). Encouragingly, ATR6-001 displays activity against T. cruzi epimastigotes and amastigotes. Given that very few members of the compound group were tested, the generation of ATR6 derivatives will aid in understanding how this class of chemicals interacts with TcMEm or TcMEc. This, coupled with further phenotypic screens, may lead to the identification of more potent trypanocidal ATR6 structures that exhibit low toxicity to mammalian cells.
In addition to the structurally related compounds, a total of 12 unrelated chemicals (singletons) were identified from the HTS as inhibiting TcMEm and/or TcMEc, with these being assigned to the ATR7 group. Most showed TcME inhibition specificity (nine targeted only TcMEc, while two were specific for TcMEm), with these structures having no effect on mammalian enzyme activity. Only one compound (ATR7-010) blocked the activity of both trypanosomal enzymes, with this effect extending across to all mammalian counterparts. Out of the 12 ATR7 variants, the most promising were ATR7-005 and ATR7-008. They both showed activity against T. cruzi epimastigotes and amastigotes, with the former having displayed no cytotoxic effect against mammalian cells at the highest concentration assayed (80 µM).
The variety of compounds discovered here demonstrates that TcMEs are amenable to druglike compound inhibition. This is an essential feature of a drug target, and the T. cruzi MEs were assessed for the first time in this context. An intriguing feature to emerge from this work stems from the high number of potent (IC50 values in the nanomolar range) TcMEc-specific inhibitors identified from the screens relative to those that target TcMEm, indicating that the cytosolic isoform may be more druggable than the mitochondrial variant. It is tempting to speculate that the higher availability of inhibitors targeting TcMEc may correlate with the presence of the allosteric site for aspartate. The fact that specific inhibitors for the two isoforms display a trypanocidal effect suggests that both TcMEs may be essential for the parasite survival. Further experiments to assess the target specificity of the identified molecules are required to confirm the essentiality of both TcME isoforms.
This work reports the first set of inhibitors against TcMEs, in addition to the trypanocidal activity of these compounds toward the intracellular stage of this pathogen. This medically relevant developmental stage represents the most important target for antiparasite therapies. Furthermore, new efforts in structural studies are required to identify the binding sites of the inhibitors on TcMEs molecules, especially regarding the compounds of the ATR3 class. Besides, an expansion of the in vitro antitrypanosomal studies is also important to optimize the best trypanocidal drug. Lastly, further studies to evaluate if the observed effects are due to an off-target contribution will also represent significant progress to defeat the neglected disease caused by this pathogen.
Footnotes
Acknowledgements
We thank Paul A. M. Michels (University of Edinburgh, UK) for reviewing the manuscript and GlaxoSmithKline for the transfer of GSK Chagas Box compounds.
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Sao Paulo Research Foundation (FAPESP) (process numbers 2013/03983-5, 2012/23682-7, and 2015/03336-5).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.