
Abstract
High-throughput screening (HTS) is a widespread method in early drug discovery for identifying promising chemical matter that modulates a target or phenotype of interest. Because HTS campaigns involve screening millions of compounds, it is often desirable to initiate screening with a subset of the full collection. Subsequently, virtual screening methods prioritize likely active compounds in the remaining collection in an iterative process. With this approach, orthogonal virtual screening methods are often applied, necessitating the prioritization of hits from different approaches. Here, we introduce a novel method of fusing these prioritizations and benchmark it prospectively on 17 screening campaigns using virtual screening methods in three descriptor spaces. We found that the fusion approach retrieves 15% to 65% more active chemical series than any single machine-learning method and that appropriately weighting contributions of similarity and machine-learning scoring techniques can increase enrichment by 1% to 19%. We also use fusion scoring to evaluate the tradeoff between screening more chemical matter initially in lieu of replicate samples to prevent false-positives and find that the former option leads to the retrieval of more active chemical series. These results represent guidelines that can increase the rate of identification of promising active compounds in future iterative screens.
Introduction
Among the various approaches to early drug discovery, high-throughput screening (HTS) is often applied in the pharmaceutical industry and academic research centers. Numerous successful therapeutics discovered via HTS campaigns 1 and major advances in automation and miniaturization have made HTS an increasingly convenient strategy for lead discovery. A typical HTS campaign ( Fig. 1 ) involves screening a collection comprising millions of compounds in functional or binding assays at a single concentration. Frequently, however, screening the full deck is not the best course of action. Many assays of interest in industry can be run in only low throughput because of the limited availability of the relevant proteins and living cells they require, as these are often difficult or costly to produce at the scale of a typical HTS campaign. In addition, there are often costs and obstacles associated with the sourcing and disposal of reagents, which may lead to assays performed at very low volumes. Compounds present in the full deck that were produced for a program that is no longer supported represent ephemeral chemical space—once these compounds are depleted, they will likely not be synthesized any longer. Moreover, the secondary assay post-HTS is frequently low throughput. This requires both screening a smaller number of compounds and a hit list that a project team will be able to prosecute in the secondary assay in a reasonable amount of time. Pooling compounds into mixtures was proposed as a way to reduce the number of wells in a primary screen. 2 However, deconvolution efforts could be significant when the hit rate is high, especially in cell-based assays. Furthermore, pooling compounds into mixtures necessitates a significant compound logistics effort, especially when new compounds are added to the screening collection. Finally, because each sample is typically tested only once at a single concentration in traditional HTS, the false-positive and false-negative rates in this technology are not fully understood.
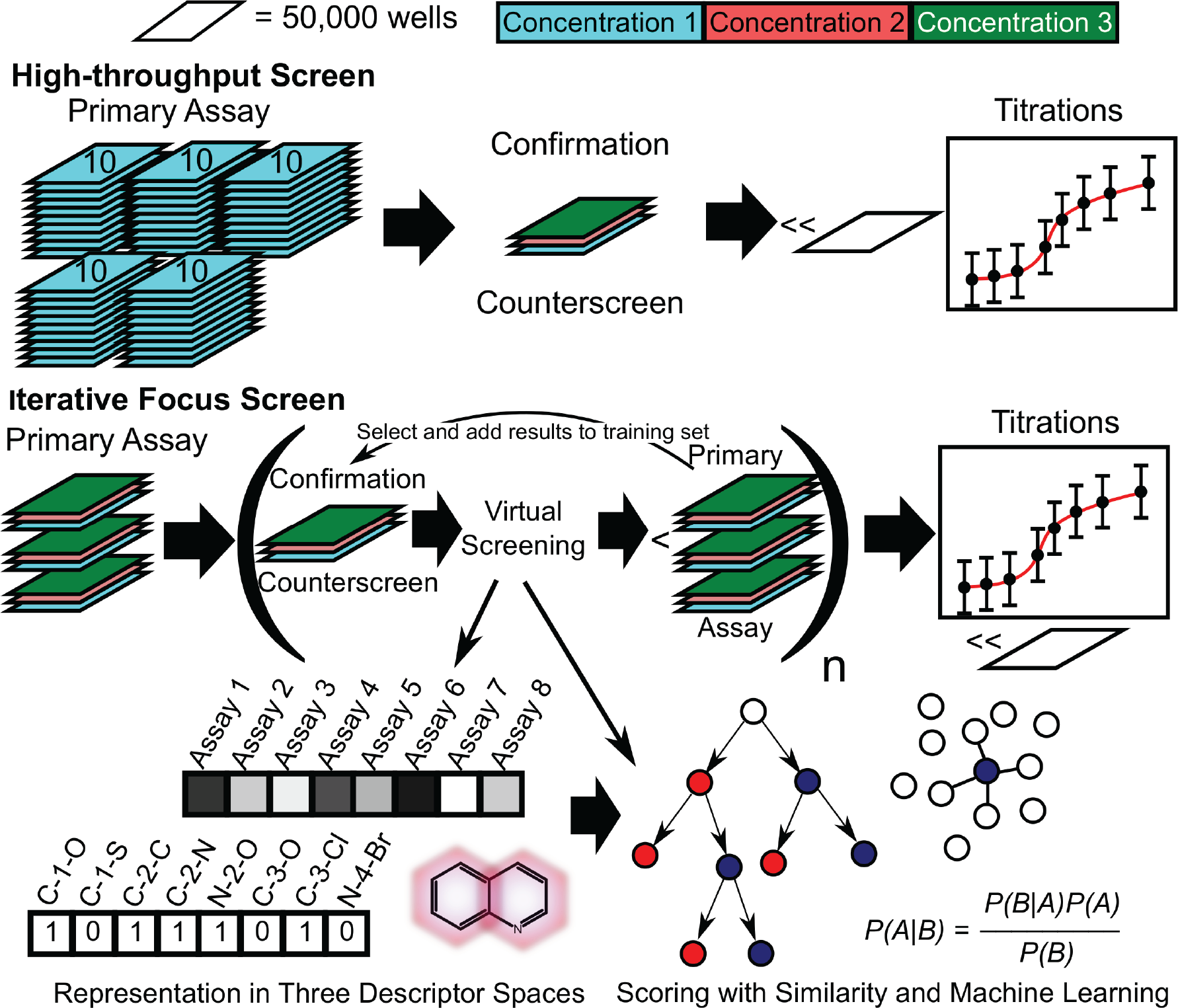
Comparison of a full-deck screen and iterative focus screening (IFS). IFS results in an order-of-magnitude reduction in the number of compounds screened. The current study screens diverse compounds of 50,000 compounds in triplicate at three concentrations, although the use of fewer wells is plausible. The remaining compounds in the collection at every iteration of IFS are scored, and a number of the top compounds (up to ~1% of the size of the full collection) are selected for screening in the next iteration.
Approaches that use subsets of compound collections designed to maximize chemical diversity have been investigated in the past.3–7 Broadly, these diversity sets fall into one of two categories: focused sets that are co-optimized for diversity and relevance to a particular target and compound sets that are constructed to capture the biological 5 or chemical diversity 8 in the screening deck. After the initial screen of a small, diverse compound (DC) set, subsequent hit expansion is performed to identify putative hits among compounds not yet screened. Hit expansion generally proceeds by enacting a similarity search using the initial hits as a starting point or training a machine-learning model in order to prioritize untested chemical matter. In the past, hit expansion efforts have also incorporated the fusion9,10 of similarity searching 11 and machine learning 12 predictions in disparate descriptor spaces to enhance the amount of chemical space covered.
In many cases, an iterative process has the potential to more effectively capture relevant portions of chemical space than a single hit expansion. 13 Three-dimensional (3D) methods 14 or biological descriptors 15 are used to hop into a new chemical space. In tandem, 2D descriptors are used to pinpoint the remaining analogs in the collection. In iterative focus screening (IFS; Fig. 1 ), active compounds identified in a hit expansion are subsequently screened, and the results are used in further hit expansions until a sufficient number of hits have been identified. Previously, colleagues at our company have investigated the utility of focused compound sets in a retrospective IFS study of a kinase inhibitor campaign carried out at our company 16 and concluded that such an approach was indeed able to identify novel starting points for lead optimization. We investigated the applicability of initiating an iterative screen with a DC collection across a number of different screening campaigns. We aimed to identify a virtual screening approach that could detect the largest number of active compounds between rounds of IFS, regardless of the target. To maximize the number of viable hits from this procedure, we carried out virtual screening in 2D and 3D descriptor spaces, in addition to the bioactivity space defined by our company’s historical assay descriptors.
In this study, we interrogate a unique data set generated at our company: in 17 different screening campaigns, a screen initialized with ~50,000 DCs in triplicate at three concentrations was run in parallel with a traditional full-deck screen to enable the direct comparison of the two methods. We introduce a novel method of fusing together scores from similarity searching and machine-learning models based on the hits found in the DC and demonstrate that it has the best active compound retrieval rate compared to any one virtual screening method alone across a broad range of assays. Using this fusion method, we are able to identify scenarios in which similarity searching alone may be more advantageous than machine learning and vice versa. This in turn enables strategic deployment of virtual screening methods in cases where screening is constrained to a subset of the full deck and maximizes high-probability active chemical matter that is moved down the pipeline.
Methods
Data Set
In an attempt to represent the diversity of the target biology and assay technologies present in our company’s HTS portfolio, a group of 17 different assays was chosen for this study. To initiate each IFS screening campaign, a diverse collection of ~50,000 DCs was screened in triplicate at three different concentrations: the full-deck screening concentration plus 3-fold and 10-fold dilutions (hereafter “high,” “medium,” and “low”). DCs were selected using an algorithm that allowed this set of compounds to approximate the chemical diversity of our company’s full screening deck (see Supplementary Methods ). The DC was screened in 17 different primary assays, with putative hits subsequently in secondary screens—either orthogonal confirmatory or counterscreening assays where available ( Suppl. Table S1 ). For the purposes of this study, hits were defined as compounds whose observed activity/inhibition both exceeded an expert-determined threshold in a primary screen and was below similar thresholds in any applicable counterscreens. To facilitate the training of subsequent machine-learning models, compounds were further assigned a pseudo-potency score based on the lowest concentration at which they were observed as a hit, with a score of “3” indicating the compound met the hit threshold at the lowest concentration, “2” if at the medium concentration, “1” if at the high concentration, and “0” if a compound was not a hit at any concentration. After these screens, the hits were expanded using a variety of similarity search and machine-learning methods in a number of descriptor spaces.
Metrics
Several metrics and visualizations in the current work are predicated on the notion of a chemical series. To derive these, all confirmed hits from a full-deck screen are clustered according to the fragment-based algorithm described by Kutchukian et al. 13 Each resulting cluster constitutes a separate chemical series. Therefore, a chemical series is considered to have been identified by a virtual screening method if any member of its corresponding cluster was retrieved. To evaluate which methods were most effective at prioritizing active chemical series, we employed the enrichment factor (EF; 1%) described by Truchon and Bayly 17 and substituted counts of chemical series for raw compound counts.
Similarity Searching
Similarity searches were conducted in three descriptor spaces: the 2D descriptor space spanned by atom pair-topological torsion descriptors18,19 (AP-TT), the biological activity space 20 described by our in-house high-throughput screening fingerprints (HTSFP), and 3D shape space. 21 For each hit in each assay, no more than the top 50 most similar compounds according to various similarity metrics were kept as the compounds most likely to be active in the next round of IFS. We selected this number to obtain approximately 20,000 compounds, since the average number of hits among the assays in this study was approximately 400. For searches in AP-TT space, descriptors were generated using components in Biovia PipelinePilot (version 16.5.0.143). Compounds represented as fingerprints x using these descriptors were prioritized according to their Tanimoto similarity (τ) to a first-round hit compound described by a bit-vector y (Eq. 1).
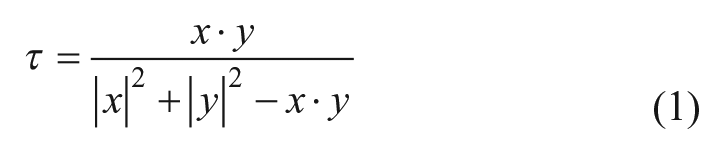
In HTSFP similarity searches, compounds are represented as vectors of transformed activities of that compound in historical assays at our company. 13 As has been the case in other applications of descriptors of this type,15,20 HTSFPs contain gaps corresponding to assays in which the compound in question has not been screened. To calculate similarity between two compounds in HTSFP space, we used the Pearson correlation metric reported by Petrone et al. 20 To avoid artifacts and simulate the effect of a prospective hit expansion, HTSFPs generated for similarity searching in each assay were adjusted to remove any descriptors corresponding to the assay for which the similarity search was being run. We also restricted our search to compounds sharing at least 50 available descriptors in common with the probes to avoid spurious, artificially large similarity scores.
Shape similarity calculations were performed using the FastROCS module from OpenEye Scientific. Molecules were standardized and converted to 3D coordinates in Biovia PipelinePilot and a minimized structure calculated using the Clean forcefield. 22 These energy-minimized structures were used as input into FastROCS, and the 50 nearest neighbors were prioritized according to the TanimotoCombo metric.
Machine-Learning Models
To complement the similarity approaches to hit expansion, two machine-learning methods were used in this study to identify the most probable hit candidates among the unscreened compounds for subsequent testing. We deployed both Laplacian-corrected naïve Bayesian classification23–25 with HTSFPs and random forest regression26,27 with AP-TT fingerprint models to make these predictions. We selected the naïve Bayes for use with the HTSFPs since it is able to handle missing values in the fingerprint without imputation. 13
The naïve Bayesian classifier is a popular machine-learning method that poses the question of classifying a molecule represented by a vector of features
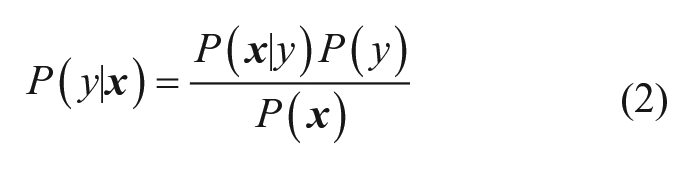
In the current study, the class labels were derived from the previously described pseudo-potency scores to enable a binary classifier: the “inactive” class corresponded to compounds with a pseudo-potency score of 0 and the “active” class to compounds with a nonzero pseudo-potency score. The class y that maximizes Eq. 2 becomes the predicted class for the input compound. To make the necessary calculations, compounds represented as HTSFPs were processed by feature binning: the values of each feature in each fingerprint were sorted into 10 bins of equal occupancy, and the corresponding feature was then transformed into a bit-vector of length 10, with a “1” in the position corresponding to the bin into which the feature in question was sorted and “0” elsewhere. In the case in which the feature was missing in the HTSFP, that feature was ignored. The required terms for a set of training compounds of size N containing Ny active compounds, Ni compounds containing feature i, and Nyi actives containing feature i are calculated as follows:

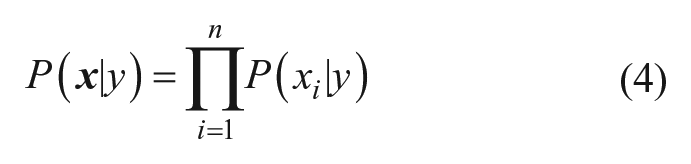
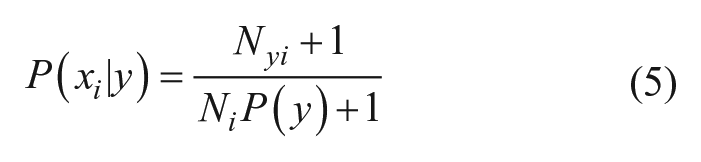
Since P(
To correct for the biased scores that result from the naïve Bayesian approach with HTSFPs (vide infra), the Bayesian scores were binned according to their HTSFP size and converted to Z-scores using the metrics of the distribution of scores in the bin. This prioritizes compounds that have high scores relative to their size, rather than globally high scores ( Suppl. Fig. S1 ). Compounds were ranked based on both their corrected and uncorrected scores, and compounds scoring in the top 5% of scores or the top-scoring 30,000 compounds were selected as suggestions for the next round of IFS, whichever was fewest.
Random forest regression models were also used to predict likely hits for the second round of IFS. Fingerprints with AP-TT descriptors used to train the random forest model, with the corresponding pseudo-potency scores
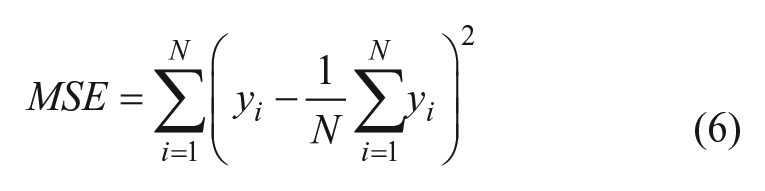
The splits consist of an inequality on one of the D descriptors, with compounds satisfying the inequality sorted into one of the leaves, and compounds not satisfying the inequality sorted into the other. This procedure is continued until N = 1 for each node, for a preset number of trees. Predictions for the pseudo-potencies of a given compound from each tree in the forest are averaged to yield the random forest prediction for the pseudo-potency. Compounds whose predicted pseudo-potency score was greater than the lowest self-predicted score corresponding to an active compound in the diversity set were selected as suggestions for the next round of IFS.
Both machine-learning models were cross-validated by training a model with 70% of the time-split three-point dose-response DC data set for each assay. Previously, accuracy metrics calculated using time-split cross-validation have been shown to more closely approximate prospective prediction accuracy than metrics calculated with randomly split or class-split cross-validation. 28 The remaining 30% of the data set was used to calculate the area under the receiver operating characteristic curve (AUC) score for each model ( Suppl. Table S2 ). AUC represents the probability that an active compound selected at random from a test will be scored more highly than a second randomly selected inactive compound by the model. 29 In the context of the DC, the AUC score is particularly meaningful, since it is a proxy for the ability of a model to correctly score compounds that by definition are highly dissimilar to the training set.
Model Fusion
To combine scores from both similarity search methods and machine-learning methods, we implemented a fusion score that combines approaches to similarity score fusion and model fusion. First, the similarity scores si from each method i (where i ranges from 1 to 3) for a given compound j are fused into a score φ j according to Eq. 7 (the SUM rule) 9 :
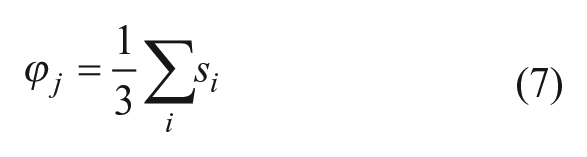
where similarity scores that are absent due to not meeting the corresponding threshold for the method are assumed to be zero. Then, scores from the k (where k ranges from 1 to 3) machine-learning methods Sk were combined using a modified weighted voting method 30 that uses the cross-validated AUC score to weight each model-generated score, as shown in Eq. 8. Scores from the machine-learning models are not necessarily constrained to a given range and so were normalized so that they fell into the interval [0, 1] before being used to calculate the machine-learning fusion score χ j . The exception to this was the corrected Bayesian score, which was first converted to a Z-score and expressed as the value of the normal cumulative distribution function at that Z-score. As in the calculation of φ j , missing values of Si were considered to be 0.
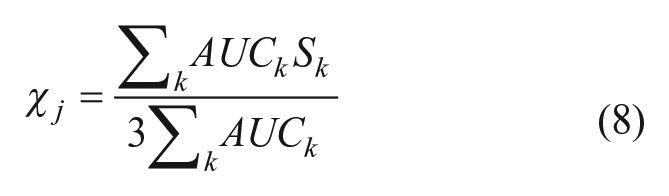
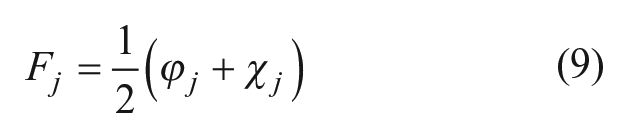
The combined fusion score Fj is the arithmetic mean (Eq. 9) of the fusion score for the similarity methods φ
j
and the fusion score for the machine-learning methods χ
j
. Compounds were then ranked according to their combined fusion score. To perform optimization on the weight of similarity scores and machine-learning scores in Eq. 9, we introduce a free parameter c to yield a weighted fusion score
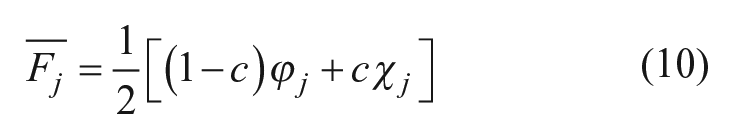
Replicate Strategies
To investigate the effect of different replicate strategies on virtual screening, we selected a subset of compounds screened in each campaign and reassigned pseudo-potency scores to align with three different approaches designed to keep the number of screening wells constant at 45,000. The first scheme is represented by a set of compounds that we refer to in this text as “45,000/9,” composed of the first ~5000 compounds of the DC screened in triplicate at all three concentrations. The second set of compounds, “45,000/ 3,” consisted of the first ~15,000 compounds in the DC set and were rescored as though they had been screened in triplicate at only the highest concentration, meaning compounds were scored as “1” if their median performance surpassed the hit threshold and “0” otherwise. The final set of compounds, “45,000/1” represented a situation in which compounds were screened once at the highest concentration. This set of compounds consisted of the first ~45,000 compounds from the DC and were scored as “1” if a randomly selected measurement at the screening concentration surpassed the hit threshold and “0” otherwise.
Results
Identification of Hits Occupying Different Regions of Chemical Space
To identify additional bioactive compounds in each screening campaign, hits from the initial screen with the DC were used to run three similarity searches and train two different machine-learning models, which in total yield six different scores (see the Methods section). Each method was then used to score each compound in the full screening collection. To determine if each method was capable of retrieving different confirmed hits, we first ranked the compounds according to the score assigned by each method. We then generated nonlinear maps (NLM) of the chemical series found in confirmation for three screening campaigns with low, moderate, and high initial hit rates. The chemical series found using each method were projected onto the maps to visualize the chemical space covered by each method’s predictions ( Suppl. Fig. S2–S4 ). Because each of the iterations in focused screening involves selecting a subset of the predicted compounds, we concerned ourselves only with the chemical series recovered in the top 20,000 compounds ranked by each method. The NLMs demonstrate that the machine-learning methods capture more active chemical series than the similarity methods, although both identify chemical series not apparent in the initial DC screen. Furthermore, while overlap exists, each method identifies a number of unique chemical series ( Fig. 2A–C ); across all 17 campaigns, up to 40% of the active chemical series identified by each method were found uniquely by that method ( Suppl. Fig. S5 ). This is also apparent when broken down to individual active compounds, where we observed that the confirmed actives captured varied depending on the descriptor set used alongside the virtual screening method ( Suppl. Fig. S6 ). We subsequently hypothesized that a fusion score that could combine the scores from the disparate virtual screening methods could improve the retrospective retrieval rate of active clusters of compounds in the screening campaigns in this study.
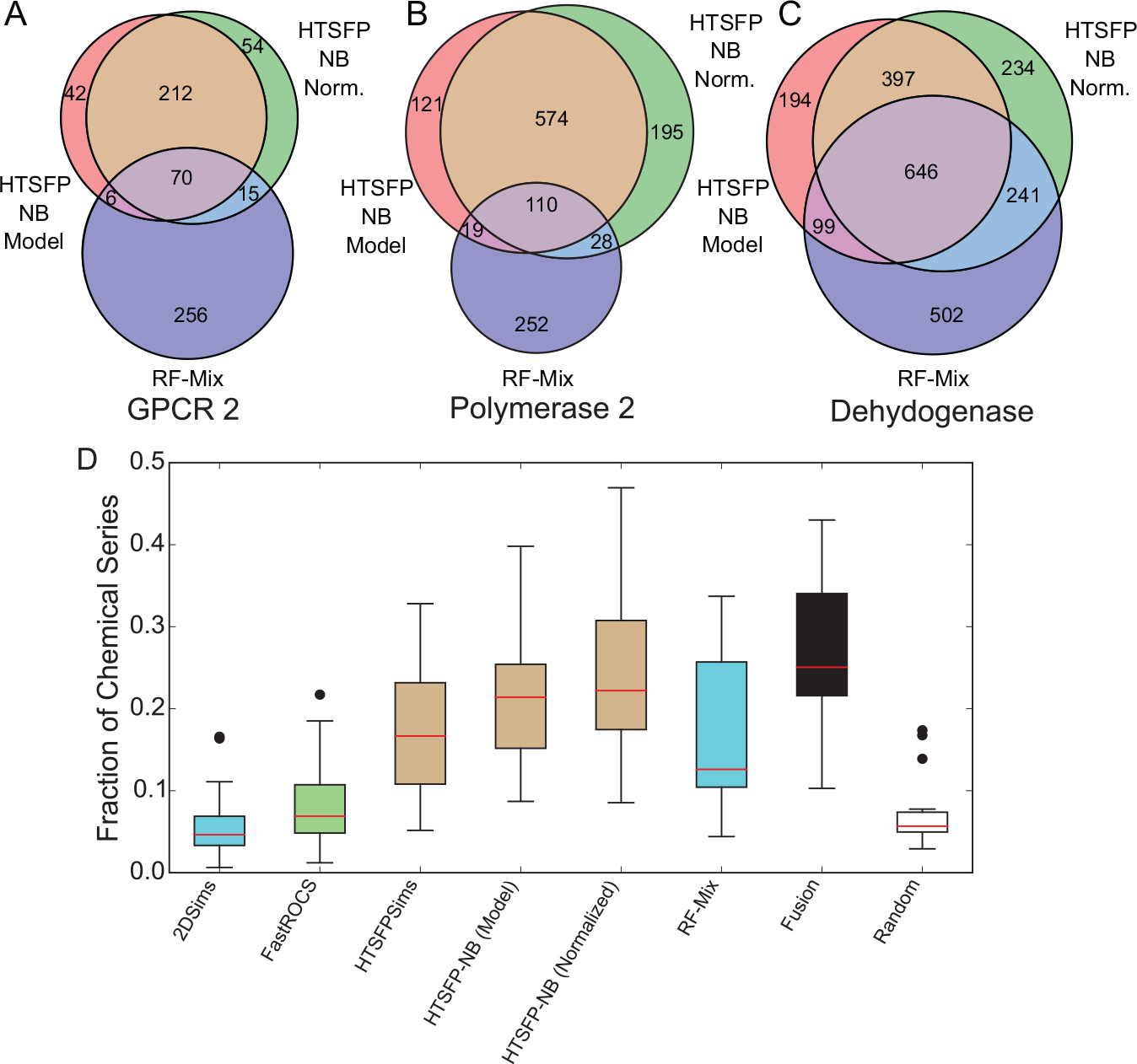
While there is overlap between chemical series identified by individual machine-learning methods (random forest [RF-Mix], naïve Bayesian [HTSFP-NB Model/Normalized]) in virtual screening, each method also retrieves unique chemical series in each campaign. Shown here are GPCR 2 (
Fusion Increases Enrichment of Active Chemical Series
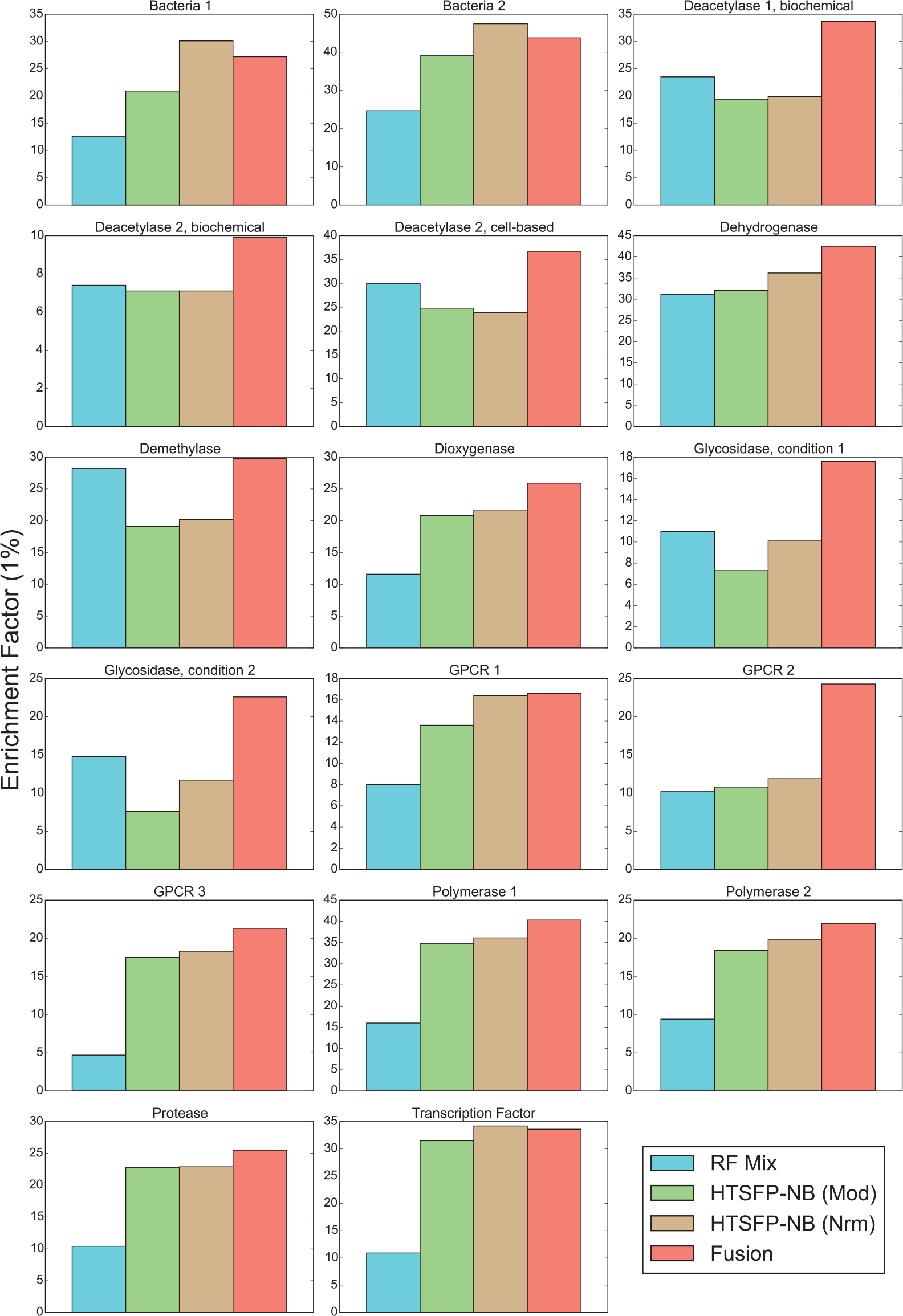
To combine the scores given to compounds in the collection by each of the six virtual screening methods used in this study, we applied Eqs. 7 to 9 (see the Methods section) to the collected scores given to each compound. To assess the benefit of the fusion metric, we embarked on a retrospective analysis of the 17 campaigns in this study. Our analysis focused on the number of unique chemical series identified by a method, rather than the gross number of active compounds required, since a subsequent step of focused screening using similarity searching could identify additional members of a chemical series found in the initial round. Hence, we favor chemical diversity over quantity of hits in our analysis. First, we ranked compounds in our internal screening collection for the 17 campaigns analyzed in this study. A list of confirmed active compounds clustered according to chemical series was then joined with this rank-ordered list. In most campaigns, we found that the fusion scoring method could retrospectively identify more active chemical series on average than any individual method in 14 of 17 campaigns ( Fig. 2D ), particularly in the first 1% of the screening collection ( Fig. 3 ; Suppl. Table S3 ). Importantly, fusing the scores assigned by the individual virtual screening methods resulted in a retrieval of an average of nearly 75% of chemical series with >20 members ( Suppl. Fig. S7 ). While fusion collectively outperformed the individual methods in general over the course of screening the entire collection virtually ( Fig. 4 ), we noted that Bayesian models trained with HTSFPs contributed the most to the overall performance of the fusion method. Because HTSFPs are not structure based per se, virtual screening methods based upon them are able to find compounds that, while structurally dissimilar (i.e., occupying disparate clusters in topological space), are related in their activity profiles. Looking past the first 1% of the collection, approximately 80% of all active chemical series can be retrieved by the fusion of virtual screening scores within the first 20% of the collection ( Fig. 4 ; Suppl. Fig. S8 ).
Initial enrichment is greatest when applying the fusion method in 14 of the 17 screening campaigns included in this study when compared with scoring approaches from individual machine-learning methods. Enrichment factors were not calculated for similarity methods because the entire collection was not scored for each hit using these methods.
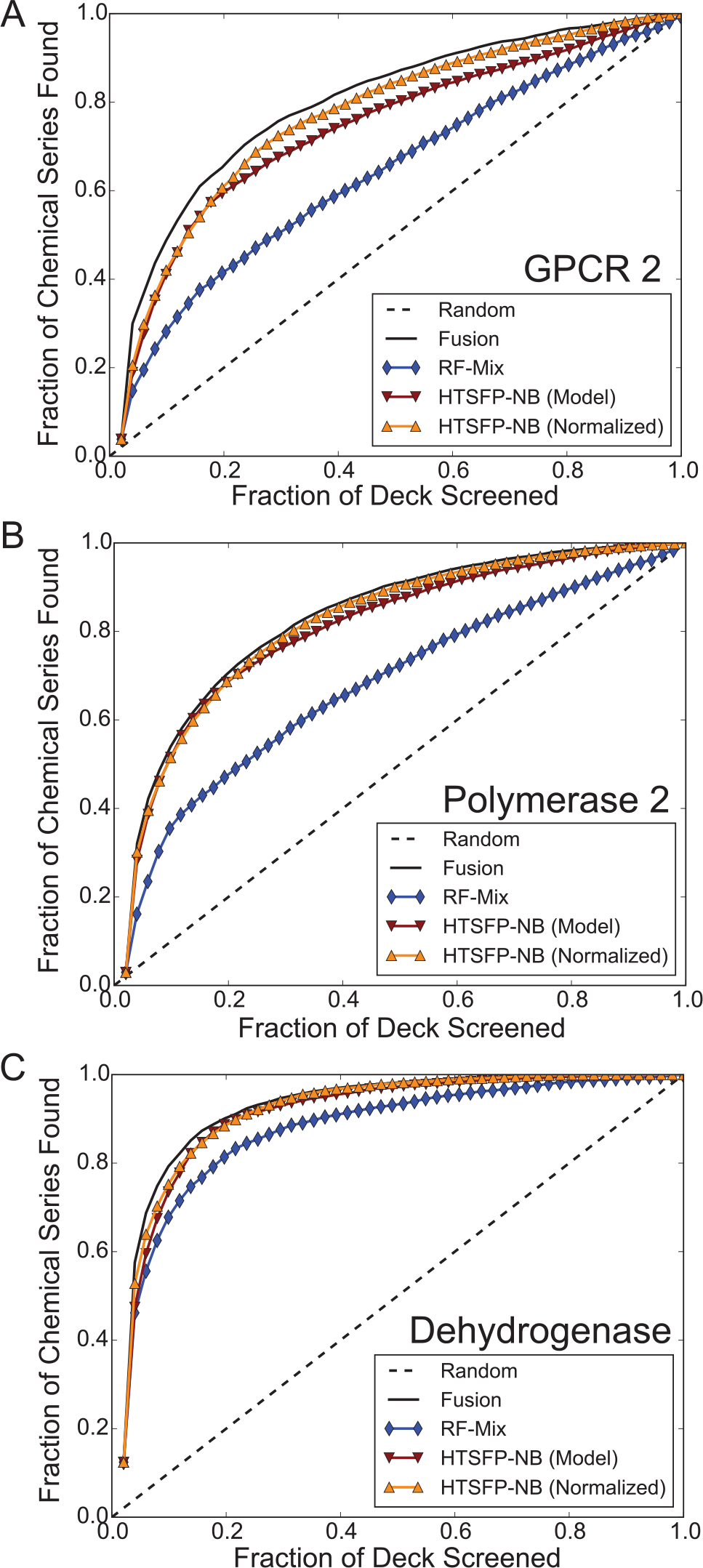
Enrichment from a fusion scoring method consistently outperforms single machine-learning method scoring in the GPCR 2 (
Optimizing the Weight of Similarity versus Machine Learning Splits Campaigns by Initial Hit Rate
We applied an unweighted scoring scheme Eq. 9 to fuse the predictions from the six virtual screening methods we used in this study. To assess whether improvement in fusion could be realized by emphasizing either similarity scores or machine-learning scores more heavily in the fusion, we performed an optimization on the coefficient c in the weighted fusion scoring function Eq. 10 to obtain a ranking of the compounds in the collection that maximized the value of EF (1%). Interestingly, EFs improve with increased weight on similarity methods in roughly half of the campaigns ( Table 1 ). When the campaigns are split according to which set of approaches receives more weight (similarity vs. machine learning), we observe that the campaigns whose enrichment benefits most from giving greater weight to similarity scores in the fusion metric have on average a lower hit rate in the DC. Conversely, those campaigns that benefit most from applying more weight to the machine-learning scores have a significantly higher average hit rate.
Improvement in fusion enrichment factor (EF; 1%) after optimization of the weighting of machine-learning scores. a
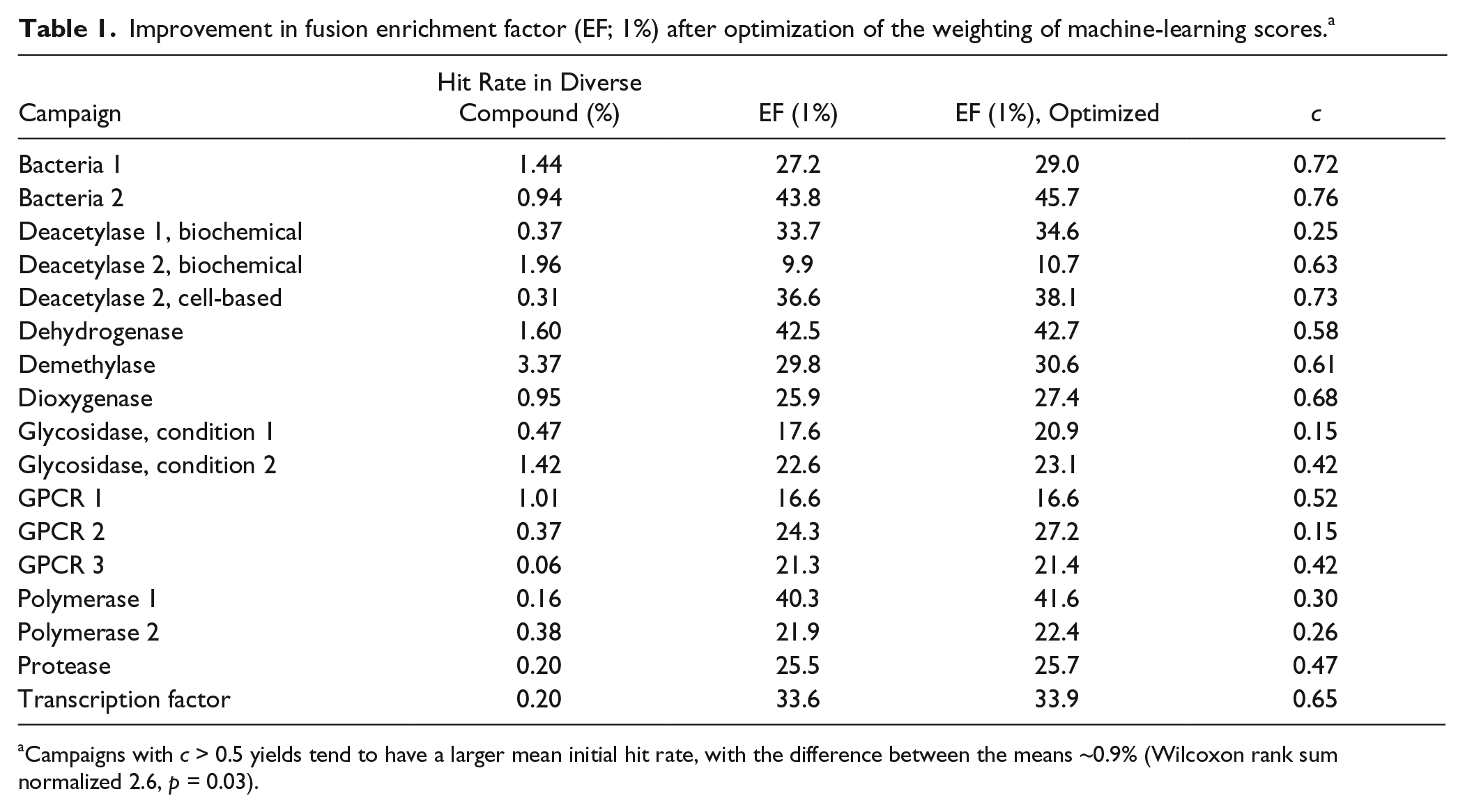
Campaigns with c > 0.5 yields tend to have a larger mean initial hit rate, with the difference between the means ~0.9% (Wilcoxon rank sum normalized 2.6, p = 0.03).
This result reflects the amount of data available for model building in the initial DC screen: lower hit rates observed in the campaign imply there will be fewer positive examples to be found in the DC on which to base machine-learning models. Consequently, similarity scoring methods are more desirable initially to amass sufficient chemical matter in the data set. Furthermore, because the similarity methods employed in this study span several descriptor spaces (topological, shape, bioactivity), they can be effective at identifying hits belonging to chemical series that are not well represented in the DC, allowing for the retrieval of diverse chemical matter, which will in turn lead to the favorability of machine-learning methods. In a prospective application, it is likely that the best default approach—one that eliminates any guesswork—would be to give greater weight to similarity scores in the first iteration and then resort to machine-learning–only scoring methods in subsequent iterations after having built up a sufficient corpus of chemical matter. Alternatively, an initial focus on scaffold hopping methods that make use of biological activity or 3D/shape descriptors can enable an increased rate of analog finding in subsequent iterations using 2D methods. Another option for simplifying the fusion procedure could be to increase the number of different compounds included in the initially screened DC. This approach would frontload the screen with a larger amount of chemical matter that would be suitable for the favorability of machine-learning scoring methods and therefore could reduce the number of cherry-picked compounds downstream.
Screening DCs Yields More Information per Well
To this point, our analysis has hinged on the results of screening the compounds in the DC in triplicate at three concentrations, but it is possible to fill the same number of sample wells using a different strategy. For example, fewer concentrations could be used to accommodate more diverse chemical matter in the same number of sample wells. To investigate which replicate strategy was able to contribute the most enrichment of active chemical series, we rescored the hits from the initial DC screen according to one of three replicate strategies (see the Methods section). Because 45,000/1, 45,000/3, and 45,000/9 are subsets of the original DC, we retrained models for each of the 17 campaigns in this study under each simulated replicate strategy, resulting in scores from 103 distinct machine-learning models. Furthermore, because compounds’ pseudo-potency scores may have changed, which in turn would change the set of probes used in similarity searching, we used the newly defined hits to extract only the relevant nearest neighbors for each assay under each of the scenarios. We then used the previously defined unweighted fusion scoring function in Eq. 9 to score and rank order screening collection compounds. Interestingly, we found that virtual screening performed under the 45,000/1 scenario was consistently the most effective at retrospectively retrieving active chemical series among all the 45,000/N schemes ( Fig. 5 ; Suppl. Fig. S9 ), significantly outperforming 45,000/3 and 45,000/9 in every campaign ( Suppl. Table S4 ). The 45,000/3 scenario was generally the second best case on average, with 45,000/9 surpassing it in select campaigns. Although most campaigns realized a modest variation in the EF between the three scenarios—including variation between EF (1%) values between the base fusion method and 45,000/9 fusion, which differed only in the number of compounds—others showed stark differences (Suppl. Fig. S10; Suppl. Table S4).
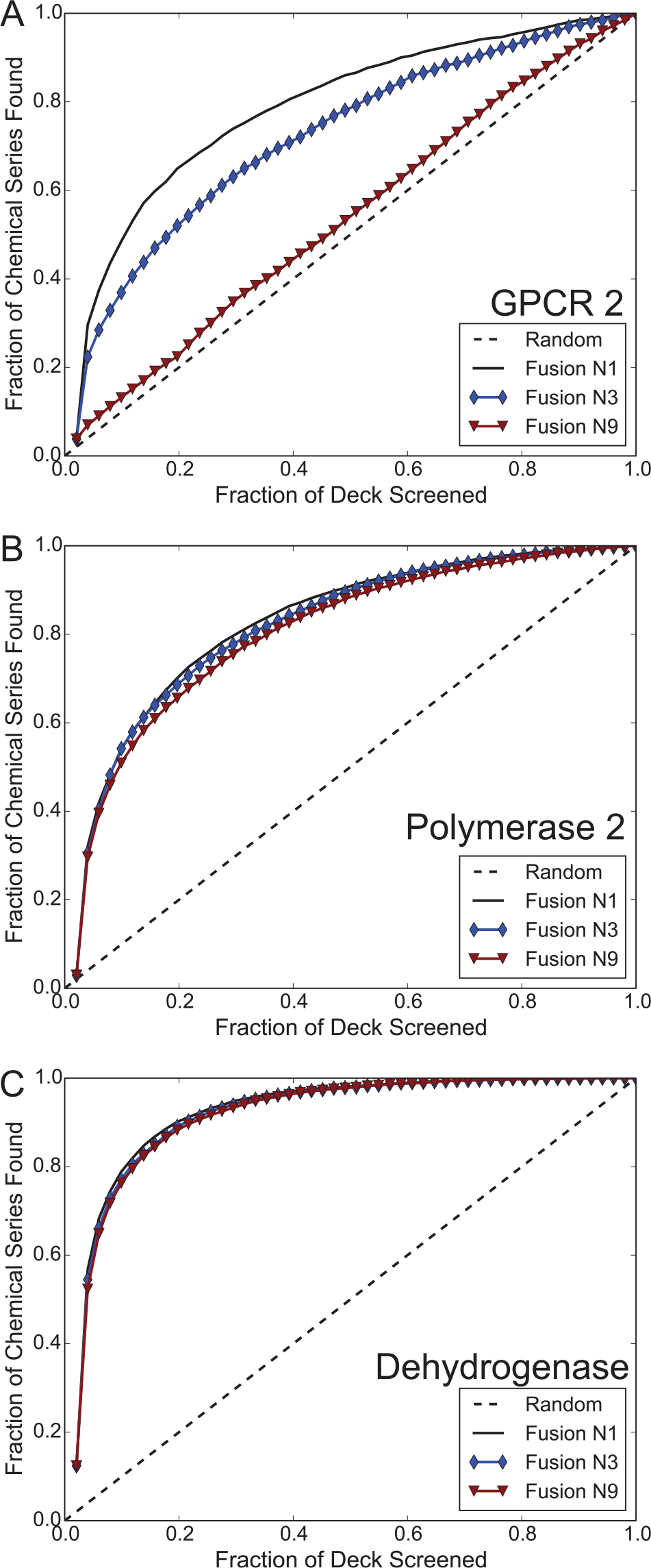
Among all the 45,000/N campaigns, the 45,000/1 strategy consistently outperforms the 45,000/3 and 45,000/9 replicate strategy in the GPCR 2 (
Discussion
The identification of bioactive compounds is a core activity in drug discovery that is frequently addressed by HTS and necessitates the expenditure of large amounts of time and resources. To reduce this burden, methods that combine small, rationally designed compound libraries with virtual screening approaches have been described to reduce the need to screen entire compound collections. In this study, we outline a strategy of iterative focused screening that couples a DC library to a hybrid virtual screening method that is applied to 17 different historical campaigns undertaken at our company to demonstrate its efficacy in a wide variety of assays. By initiating screening with a DC, we are able to leverage virtual screening methods to retrieve up to 80% of active chemical series while assaying approximately 20% of the deck ( Fig. 4 ). We observed that the best performance was achieved when fusing a combination of six mixed similarity and machine-learning scoring methods operating in three distinct descriptor spaces. An assessment of the performance of in silico approaches revealed that enrichment is boosted primarily by methods that make use of our company’s HTS fingerprint descriptors. Those are used to retrieve compounds with a similar activity profile in a similarity search or are used to learn desirable and undesirable activity profiles in machine-learning methods. Should circumstances dictate it, alternative machine-learning approaches such as support vector machines and neural networks or novel descriptor spaces can easily be added to the fusion approach. Moreover, structure-based methods such as docking can also be incorporated as an additional dimension. The fusion of docking scores has proved efficacious in the past,10,31 albeit with the caveat that such approaches frequently require large amounts of crystallographic data.
The base fusion method weighted scores given by similarity methods and machine-learning methods equally; when we allowed the weights to vary, a significant trend emerged. We noted that the campaigns in which optimal enrichment was achieved by increasing the weight given to similarity scores had a lower average hit rate than the rest of the campaigns. This result is consistent with expectations: scores generated from models with extreme class imbalance are less reliable. Therefore, similarity methods have the potential to surpass machine-learning methods in initial enrichment. It is likely that a DC co-optimized for diversity in several descriptor spaces rather than just topological space may more uniformly cover druglike chemical matter and generate sufficient data ab initio for a broad range of assays and targets. In the absence of this, similarity methods—particularly those reliant on shape and bioactivity descriptors—can aggregate chemical matter that can provide links to more hits in chemical space by escaping local pockets of chemical space that topological-based methods may become trapped in. We anticipate that this enhancement is similar to what has been observed with weak reinforcement strategies in solely topological space 32 and that upon reaching a critical mass of confirmed hits in the data set, topological methods can serve to amass structural analogues of a now-diverse corpus of compounds.
With the fusion strategy we developed and the screening data in this study, we sought to understand if there was a systematic advantage to filling a fixed number of sample wells in a given screen with single instances of a larger number of DCs versus using the sample wells to sample several replicates of fewer compounds at a number of different concentrations. The analysis demonstrates that screening more compounds once is preferable to screening fewer compounds multiple times. The implication is that false-negatives that inevitably occur in 45,000/1 screening are outweighed by the volume of true-positive and -negative data points collected—subsequent virtual screening methods can still score hits in such a way that provides increased enrichment over 45,000/3 and 45,000/9 screening. As the chemical space represented by the DC becomes more densely packed (i.e., progressing from 45,000/9 to 45,000/3 to 45,000/1), the effect of a single false-negative is ameliorated by other compounds in the set that can successfully provide sufficient information to identify active compounds that may otherwise have been lost. This is concomitant with the results of other studies, 33 with the scaffold hopping enabled by our methodology likely potentiating this effect despite the wide diversity of the DC. It should be noted that this analysis was performed assuming only 45,000 wells, which was the limit given the number of compounds in the DC screened in this study. However, given a larger number of wells with which to work, the difference in EF (1%) values among the replicate strategies could become more pronounced, particularly because screening decks are frequently enriched with chemical matter for specific targets or disease areas. In addition, if a more diverse screening at a single concentration is prescribed, the choice of concentration should take into account the possibility of identifying weak hits that may reveal novel modes of action. 34
In total, this study has highlighted three recommendations for enhancing the efficacy of virtual screening. Although the main focus of this study was HTS, the conclusions derived herein may be equally applicable to other screening methods. First, scoring the expansion compounds for screening using a fusion of several methods in orthogonal descriptor spaces has advantages over the use of any single method in retrieving active chemical series, especially ones with many members. Second, the parameters of the fusion method are related to the initial hit rate of the campaign under investigation and should be tuned accordingly. Extremely low hit rate campaigns appear to benefit most from emphasizing similarity methods rooted in descriptors that enable scaffold hopping, whereas campaigns with a high hit rate can retrieve more chemical series by placing a greater emphasis on machine-learning methods. Finally, fusion methods similar to what we have described herein are able to have improved enrichment if used with a more diverse initial screening library instead of one with many replicates. In situations in which resources are limited, this approach will allow for more parsimony in the allocation of sample wells in a screening campaign while maximizing the amount of active chemical matter retrieved.
Footnotes
Acknowledgements
The authors are extremely grateful to the late Frank K. Brown for enthusiastic support and advice regarding this work.
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.