
Abstract
As an obligate step for picornaviruses to replicate their genome, the small viral peptide VPg must first be specifically conjugated with uridine nucleotides at a conserved tyrosine hydroxyl group. The resulting VPg-pUpU serves as the primer for genome replication. The uridylylation reaction requires the coordinated activity of many components, including the viral polymerase, a conserved internal RNA stem loop structure, and additional viral proteins. Formation of this complex and the resulting conjugation reaction catalyzed by the polymerase, offers a number of biochemical targets for inhibition of an essential process in the viral life cycle. Therefore, an assay recapitulating uridylylation would provide multiple opportunities for discovering potential antiviral agents. Our goal was to identify inhibitors of human rhinovirus (HRV) VPg uridylylation, which might ultimately be useful to reduce or prevent HRV-induced lower airway immunologic inflammatory responses, a major cause of asthma and chronic obstructive pulmonary disease exacerbations. We have reconstituted the complex uridylylation reaction in an AlphaScreen suitable for high-throughput screening, in which a rabbit polyclonal antiserum specific for uridylylated VPg serves as a key reagent. Assay results were validated by quantitative mass spectrometric detection of uridylylation.
Introduction
Human rhinoviruses (HRVs) are the primary causative agent of the common cold and play a critical role in triggering asthma and chronic obstructive pulmonary disease (COPD) exacerbations.1,2 HRVs are nonenveloped, single-stranded, positive-sense RNA viruses within the Enterovirus genus of the Picornaviridae family and comprise more than 133 genotypes classified into three species: HRV-A, HRV-B, and HRV-C.3,4 This large genetic diversity makes it practically impossible to develop natural immunity to subsequent infections and has also hampered the development of a vaccine. This diversity also poses a challenge for the development of a broadly active direct-acting antiviral, and no effective treatment of HRV infection is currently approved for use.
Similar to other Picornaviridae such as poliovirus, the rhinovirus genome contains ~7200 nucleotides and encodes a polyprotein divided into three regions that are posttranslationally processed by virally encoded proteases into 11 structural and nonstructural proteins. 5 The P1 region encodes the capsid structural proteins (VP4, VP2, VP3, VP1), whereas the nonstructural proteins are encoded by the P2 (2A protease, 2B, 2C ATPase) and the P3 (3A, 3B, 3C protease, 3D polymerase) regions. The P3-encoded proteins, both fully processed and some cleavage intermediates, directly participate in viral genome replication. The 3Dpol of picornaviruses is an RNA-dependent RNA polymerase composed of approximately 460 amino acids. It transcribes the positive-stranded RNA genome, and the resulting negative-stranded copy is then used by 3Dpol as a template for the production of progeny positive strands. 3Dpol activity is both primer and template dependent. In biochemical assays, the polymerase catalyzes the elongation of an oligonucleotide primer on an RNA template, but in vivo polymerization activity requires the addition of two UMP molecules onto the tyrosine hydroxyl at residue 3 of the short 3B protein (also known as VPg), which is catalyzed by 3Dpol. This uridylylated protein then serves as the natural primer for polymerization of the RNA genome. Biochemical and genetic studies of RNA genome replication of poliovirus, the most extensively studied picornavirus, have shown that the uridylylation reaction requires assembly of a ribonucleoprotein complex consisting of a VPg-containing precursor protein, probably 3BC or 3BCD 6 ; the precursor protein 3CDpro; 3Dpol; and a cis-acting replication element (creRNA), an RNA stem-loop structure.7–9 The cre-templated production of the protein primer VPg-pUpU is essential for poliovirus RNA replication. 10 The position of the cre element in the picornavirus genome is not conserved, but in all cases the loop contains a conserved A5A6 sequence. The first adenosine serves as the template for addition of both uridines to VPg, using a slide-back mechanism.11,12 3CDpro, an intermediate in the processing of the P3 region of the viral polyprotein, was shown to stimulate the uridylylation reaction and to interact with the creRNA stem during VPg uridylylation via the 3Cpro RNA binding site.6,13,14 It exhibits protease activity 15 but is devoid of polymerase activity. 16 Organization of the HRV uridylylation complex is similar to poliovirus, as shown by the in vitro reconstitution of HRV-A2 and HRV-B14 complexes able to synthesize VPg-pU and VPg-pUpU.17,18 The HRV cre element is found in the portions of the genome encoding 2Apro for HRV-A, VP1 for HRV-B, and VP2 for HRV-C.17,19,20
A few small-molecule inhibitors of poliovirus, coxsackievirus B3 (CVB3) and foot-and-mouth disease virus (FMDV) 3Dpol, have been reported.21–23 Also, the nonspecific nucleoside analogues 5-fluorouridine triphosphate and amiloride have been reported to inhibit FMDV and CVB3 RNA replication by affecting VPg uridylylation.22,24 However, the literature in this area is sparse, and furthermore, no rhinovirus replication inhibitors targeting the polymerase activity or the formation of the uridylylation complex have been reported to date. A robust high-throughput screen might be able to identify such inhibitors. The HRV 3Dpol is well conserved across genotypes, as are the N-terminal residues of VPg and the 3Cpro RNA binding residues, and therefore it may be possible to identify molecules exhibiting broad genotype coverage.
Uridylylation is a complex activity with multiple essential components, and all of the in vitro studies reported to date have been performed using micromolar concentrations of proteins and [α-32P]UTP for the detection of reaction products by gel electrophoresis. Our goal was to develop a nonradioactive assay that was sufficiently robust for high-throughput screening (HTS). In this report, we describe a cre-dependent uridylylation AlphaScreen in which the reaction is performed using reasonable concentrations of purified 3BC and 3Dpol proteins, cre(2A)RNA and UTP. The availability of an uridylylation AlphaScreen now makes it feasible to screen large compound collections to discover novel HRV replication inhibitors.
Materials and Methods
Materials
Bis-Tris was obtained from Hampton Research (Aliso Viejo, CA). MgCl2, DEPC-treated water and EDTA 0.5 M (pH 8.0) were purchased from Ambion (Carlsbad, CA). DMSO, glycerol, NaCl, Mg acetate, and MnCl2 were obtained from Sigma (St Louis, MO); HEPES and propidium iodide (PI) were from Invitrogen (Carlsbad, CA); RNasin and UTP were from Promega (Madison, WI); TCEP was from Pierce (Rockford, IL); Tween-20 was from Bio-Rad (Hercules, CA); and bovine serum albumin (BSA) was from New England BioLabs (Ipswich, MA). Streptavidin–horseradish peroxidase (HRP) conjugate and QuantaBlu fluorogenic peroxidase substrate were purchased from Thermo Scientific (Waltham, MA). All oligonucleotides used in this study were custom-synthesized at Integrated DNA Technologies (Coralville, IA) with the exception of poly(rA), which was purchased from GE Healthcare (Piscataway, NJ). AlphaScreen streptavidin donor beads, protein A acceptor beads, and [α-32P]UTP (3000 Ci/mmol) were obtained from PerkinElmer (Waltham, MA). The HRV-A16 N-terminal VPg decapeptide GPYSGEPKPK-aminohexanoic acid-[Cys]-amide and the corresponding mono- and di-uridylylated tyrosine peptides were synthesized at Cambridge Research Biochemicals (Billingham, Cleveland, UK).
Genetic Constructs
The HRV-A16 3Dpol coding region (GenBank accession no. L24917) with a C-terminal hexahistidine tag was obtained from DNA 2.0 (Menlo Park, CA) and subcloned into the pET11a bacterial expression vector. We found later that this HRV-A16 isolate encodes a rare polymorphism at amino acid 149 of 3Dpol, with the consensus Gly residue replaced by Asp. Preliminary experiments were performed with the Asp-149 variant, but for assay development, the aspartic acid residue was changed to the glycine residue using the QuickChange II site-directed mutagenesis kit according to the manufacturer’s instructions (Stratagene, La Jolla, CA).
HRV-A16 3BC was cloned from HRV-A16 RNA isolated from a viral stock (ATCC no. VR-283; ATCC, Manassas, VA) produced in H1-HeLa cells. Isolated RNA was reverse-transcribed into complementary DNA (cDNA) using the QIAGEN OneStep RT-PCR kit (QIAGEN, Valencia, CA) with HRV-specific primers corresponding to the 3B N-terminal and 3Cpro C-terminal with a hexahistidine tag sequence (forward primer: 5′-AATTACCGCGGTG-GAGGCCCTTACTCGGGGGAGCCCAAACCTAAG-3′; reverse primer: 5′-TTGGGCTCG AGTTAATGGTGATGG-TGATGGTGTTGTTGTTCAGTGAAGTATGATCTCAATAG-3′). To the purified reverse transcription (RT)–PCR product, the amino acid sequence GLNDIFEAQKIEWHE (avitag) was added by PCR 5′ to the hexahistidine tag (forward primer: 5′-CTATTGAGATCATACTTCACTGAAC-AACAAAGCGGAGGTGGTCTCAACGACATCTTTGAGGCCCAAAG-3′; reverse primer: 5′-TTGGGCTCGAG-TTAGTGATGGTGATGGTGATGACCTCCGCTTTCAT GCCATTCGATCTTTTGGGCCTCAAAGATGTCG-3′). The resulting PCR product was ligated into pCR-Blunt, and the 3Cpro active site cysteine was mutated to alanine (C147A) using the QuickChange II site-directed mutagenesis kit. Finally, the purified HRV-A16 3BC(C147A)-Avitag-His6 PCR product was subcloned into the pET26-Ubi bacterial expression vector.
Expression and Purification of HRV-A16 3Dpol
The HRV-A16 3Dpol-His6 was expressed in Escherichia coli BL21(DE3). Bacteria were grown at 37 °C in CircleGrow medium (MP Biomedicals, Santa Ana, CA) supplemented with 100 µg/mL ampicillin. At an OD of 0.8 to 0.9 at 600 nm, the culture was cooled to 25 °C and the protein expression induced with 0.4 mM isopropyl-β-D-thiogalactoside for 16 h. Cells were harvested by centrifugation, and the cell paste was frozen at −80 °C. The cell paste was resuspended in 3 mL of lysis buffer (50 mM sodium phosphate [pH 7.5], 10% glycerol, 0.3 M NaCl, 1 mM TCEP, 20 U/mL benzonase, 500 µg/mL lysozyme) per gram of cells. The suspension obtained was sonicated and incubated at 4 °C for 1 h with shaking. The extract was then clarified by a 30-min centrifugation at 14,500 g at 4 °C. Imidazole was added to the supernatant to a final concentration of 20 mM followed by a filtration through a 0.8-µm filter. The solution was then applied to a Hi-Trap Ni+2-chelating column (GE Healthcare). The HRV-A16 3Dpol was eluted in 50 mM sodium phosphate (pH 7.5), 0.3 M NaCl, and 10% glycerol using a 20- to 500-mM imidazole gradient. The enzyme-enriched fractions were pooled and dialyzed twice against 50 mM sodium phosphate (pH 7.5), 0.15 M NaCl, and 10% glycerol. A final dialysis was performed in 50 mM sodium phosphate (pH 7.5), 0.15 M NaCl, and 50% glycerol. The purified enzyme was stored at −80 °C. HRV-A16 3Dpol was obtained with >95% purity, as judged by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Introduction of the polymerase active site substitutions D327A/D328A into the HRV-A16 3Dpol coding region was done using the QuickChange II site-directed mutagenesis kit. This active site variant of 3Dpol was expressed and purified as described above and found to be inactive at concentrations up to 100 nM, ruling out the copurification of a contaminating polymerase.
Expression, Purification, and Biotinylation of HRV-A16 3BC
HRV-A16 3BC(C147A)-Avitag-His6 was expressed as an ubiquitin fusion protein in E. coli BL21(DE3) pCG1 transformed with the pET26-Ubi expression vector. 25 Bacteria were grown at 37 °C in CircleGrow medium supplemented with 20 µg/mL kanamycin and 30 µg/mL chloramphenicol. At an OD of ~0.6 at 600 nm, the culture was cooled to 25 °C and the protein expression induced with 1 mM isopropyl-β-D-thiogalactoside for 4.5 h. Cells were harvested by centrifugation, and the cell paste was frozen at −80 °C. The cell paste was resuspended in 5 mL of lysis buffer (40 mM HEPES [pH 8.0], 5% glycerol, 0.1 M NaCl, 1 mM TCEP, protease inhibitors cocktail) per gram of cells. The suspension obtained was processed in a Dounce homogenizer (Kimble Chase-Kontes Glass, Vineland, NJ) and the lysate further homogenized using a microfluidizer. The extract was clarified by a 30-min centrifugation at 14,500 g at 4 °C followed by passage of the supernatant through a 0.8-µm filter. Imidazole was added to the supernatant to a final concentration of 5 mM and the solution applied to a Hi-Trap Ni+2-chelating column. HRV-A16 3BC was eluted in 40 mM HEPES (pH 8.0), 0.1 M NaCl, 5% glycerol, and 1 mM TCEP using a 5- to 500-mM imidazole gradient. The protein-enriched fractions were pooled, applied to a Superdex 200 size exclusion column (GE Healthcare), and equilibrated in 40 mM HEPES (pH 8.0), 0.1 M NaCl, 10% glycerol, 2 mM EDTA, and 1 mM TCEP. The 3BC-enriched fractions were pooled and biotinylated using an in vitro biotinylation kit, which provides the biotin-protein ligase, according to the manufacturer’s instructions (Avidity, Aurora, CO). Finally, the protein was repurified on a Hi-Trap Ni+2-chelating column according to the procedure described above. Biotinylated 3BC was >95% pure as judged by SDS-PAGE and was 100% biotinylated as verified by mass spectrometry analysis.
Anti-VPg-pU(pU) Evaluation
Rabbit polyclonal anti–VPg-pU(pU) antisera were raised against the VPg peptide GP[Tyr(pU)]SGEPKPK-aminohexanoic acid-[Cys]-amide. Antisera were first passed through a column derivatized with the antigen peptide to deplete antibody able to bind to nonuridylylated peptide and then purified on a column derivatized with the VPg peptide. To evaluate specificity of the purified antiserum for uridylylated VPg, the in vitro uridylylation reaction was performed in the presence of 3 µM 3BC, 1 µM cre(2A)RNA, 1 µM 3Dpol (wild-type or inactive mutant as negative control), and 3 µM UTP in 50 mM HEPES (pH 7.5), 5 mM Mg acetate, 10 mM β-mercaptoethanol, 10% (v/v) glycerol, 80 U/mL RNasin, and 5% DMSO. After an incubation of 90 min at 30 °C, the products of the uridylylation reaction were separated on SDS-PAGE (12% polyacrylamide) and an immunoblot analysis was performed using the anti–VPg-pU(pU) diluted 1000-fold. Proteins were visualized using the ECL Western Blot Detection Reagents (Thermo Fisher). To monitor uridylylation with ELISA, biotinylated 3BC was used as substrate, 0.01% BSA added to the assay buffer, and the uridylylated 3BC complex captured with immobilized anti–VPg-pU(pU). For antibody immobilization, 100 µL per well of a 0.28-µg/mL anti–VPg-pU(pU) solution prepared in a 50-mM bicarbonate/carbonate buffer (pH 9.6) was added to a 96-well Microfluor-2 white plate and incubated overnight at 4 °C. After washing the plate with 3 × 100 µL of phosphate-buffered saline (PBS) containing 0.1% Tween-20, 150 µL of PBS containing 0.1% Tween-20 and 2% BSA was added. After 90 min at 4 °C, the plate was washed as described above. Then, 100 µL of the uridylylation reaction was added, followed by a 2-h incubation at 23 °C. The plate was then washed and 100 µL of streptavidin-HRP (1:40,000 dilution) was added. Following a 2-h incubation at 23 °C, the plate was washed again and 100 µL of the QuantaBlu substrate added. After a 60-min incubation at 23 °C, the peroxidase reaction was stopped by the addition of 100 µL of the QuantaBlu stop solution. The fluorescence was monitored on a Tecan GENios Pro plate reader (Tecan, Männedorf, Switzerland) equipped with a 340-nm excitation filter and a 430-nm emission filter.
HRV-A16 3Dpol Scintillation Proximity Assay
The HRV-A16 polymerase activity is quantified by measuring the incorporation of 3H-UTP during the elongation of the biotin-U12 RNA primer annealed to the homotemplate poly(rA) by the enzyme. The polymerase reactions were performed in a 384-well polystyrene plate (OptiPlate-384; PerkinElmer). To 5 µL of the primer/template solution (25 and 2.5 µg/mL final concentrations, respectively) was added 5 µL of the test compound diluted in 20% DMSO/assay buffer (20 mM Bis-Tris [pH 6.8], 0.75 mM MgCl2, 0.5 mM KCl, 80 U/mL RNasin, 0.01% BSA, and 1 mM TCEP) and 5 µL HRV-A16 3Dpol (4.5 nM final concentration). Following a 15-min incubation at 23 °C, the reaction was initiated by adding 5 µL of the UTP/[3H]UTP solution (7 µM and 500 nM final concentrations, respectively). The reaction was stopped after a 30-min incubation at 23 °C with the addition of 30 µL of the streptavidin-coated PS imaging beads (5 mg/mL in 0.5 M EDTA [pH 8.0]). The plate was shaken gently for 5 min and left for 30 min at 23 °C to allow for the capture of the double-stranded RNA product. Then, 35 µL of 5 M cesium chloride solution was added and the plate left at 23 °C for at least 2 h in the dark before counting for 2 s per well on an EnVision multilabel reader (PerkinElmer) under enhanced luminescence mode.
HRV-A16 VPg Uridylylation AlphaScreen Optimization
The 3BC and 3Dpol titrations were performed in 20 mM Bis-Tris (pH 6.8), 5% (v/v) glycerol, 0.5 mM Mg acetate, 0.01% BSA, 80 U/mL RNasin, 1 mM TCEP, and 5% DMSO in a 384-well AlphaPlate (PerkinElmer). To 10 µL of 3Dpol diluted in assay buffer (7.8–500 nM) was added 10 µL of 3BC (7.8–500 nM). After a 15-min incubation at 23 °C, the uridylylation reaction was initiated with the addition of 10 µL of a solution containing 400 µM UTP (buffer only for negative control) and 31.25 or 125 nM 48-nt cre(2A)RNA (5′-GAUUUAAUCAUAUACCGAACAAGCACAC- AAGGUGAUGGUUAUAUUCCA-3′). The reaction was stopped after 30 min by adding 10 µL of the Alpha bead–antibody solution, prepared in 25 mM HEPES (pH 7.5), 100 mM NaCl, 0.25% Tween-20, and 80 mM EDTA and containing 40 µg/mL streptavidin donor beads, 20 µg/mL protein A acceptor beads, and 1.56 nM anti–VPg-pU(pU) incubated for 60 min at 23 °C in the dark. The plate was incubated for at least 1 h at 23 °C in the dark and was read on an EnVision multilabel reader. A second 3BC/3Dpol cross-titration was performed using 15.6 to 250 nM 3Dpol, 15.6 to 250 nM 3BC, 125 nM cre(2A)RNA, and 10 µM UTP.
For titration of UTP, 10 µL of 3BC (125 nM final) was added to 10 µL of a solution containing 3Dpol and cre(2A)RNA (125 nM final for each). After a 15-min incubation at 23 °C, the uridylylation reaction was initiated by adding 10 µL of UTP (0.625–50 µM final; buffer only for negative control) and the reaction stopped after 5, 10, 20, 30, or 60 min by adding 2 µL of EDTA (20 mM final). Capture of the uridylylated products was performed as described above. Km was determined by nonlinear regression analysis of data points best corresponding to initial velocity of uridylylation calculated at each UTP concentration (Prism version 4.02; GraphPad Software, La Jolla, CA).
To determine the optimal concentration of anti–VPg-pU(pU) required to capture the biotinylated 3BC-pU and 3BC-pUpU products, the uridylylation reaction was performed with 125 nM 3BC, 125 nM 3Dpol, 125 nM cre(2A)RNA, and 10 µM UTP. The reaction was stopped by adding 10 µL of the Alpha bead–antibody solution, in which the final antibody concentration was varied from 0.006 to 6.25 nM.
To summarize the optimized screening conditions, the uridylylation reactions are performed in a 384-well AlphaPlate in 20 mM Bis-Tris (pH 6.8), 5% (v/v) glycerol, 0.5 mM Mg acetate, 0.01% BSA, 80 U/mL RNasin, 0.05% Tween-20, and 1 mM TCEP. To 5 µL of the test compound diluted in 15% DMSO/assay buffer (5% DMSO final content) is added 5 µL of the 3Dpol/3BC/cre(2A)RNA solution (125 nM each final concentration). Following a 15-min incubation at 23 °C, the reaction is initiated by adding 5 µL of UTP (10 µM final concentration). The reaction is stopped after 30 min by adding 5 µL of the Alpha bead–antibody solution, prepared in 25 mM HEPES (pH 7.5), 100 mM NaCl, 0.25% Tween-20, and 80 mM EDTA and containing streptavidin donor beads, protein A acceptor beads, and anti–VPg-pU(pU) (10 µg/mL, 5 µg/mL, and 0.39 nM final concentrations, respectively) incubated for 60 min at 23 °C in the dark. The plate is incubated for at least 1 h at 23 °C in the dark and is read on an EnVision multilabel reader.
Mass Spectrometry Analysis
Aliquots from the uridylylation reaction were spiked with 1 M ammonium bicarbonate for a final concentration of 50 mM and digested with trypsin (Promega) overnight at 37 °C. In samples from duplicate experiments, tryptic peptides were analyzed by liquid chromatography–mass spectrometry (LC-MS) using a Dionex Ultimate 3000 RSLCnano (Thermo Fisher) connected to a hybrid linear ion trap LTQ-Orbitrap XL (Thermo Fisher). Liquid chromatography separation of peptides was performed with a Dionex 5-µm Acclaim PepMap100 C18 100-µm × 2-cm precolumn and a Dionex 2-µm Acclaim PepMap C18 75-µm × 15-cm column using a linear gradient of 96% solvent A (100% water, 0.1% formic acid) and 4% solvent B (20% water, 80% acetonitrile, 0.1% formic acid) to 35% solvent A and 65% solvent B over 30 min with a 0.25-µL/min flow rate. The mass spectrometer was operated in the data-dependent mode. Data acquisition was set to acquire one high-resolution scan in the Orbitrap with a 60,000 resolution at m/z 400, followed by five collision-induced dissociation (CID) MS/MS scans in the linear ion trap. Target values were 1 × 106 ions for the survey scan over a maximum time of 500 ms and 3 × 105 ions for the MS/MS scan over a maximum time of 50 ms. Peptide identification was performed with Proteome Discoverer 1.3 embedded with SEQUEST (Thermo Fisher). The raw data were searched against the tryptic peptide database containing the amino acid sequences of 3BC and 3Dpol with one missed cleavage site tolerated. The mass tolerance for precursor ion masses was 10 ppm and 0.5 Da for fragment ion masses. The following variable modifications were considered: methionine oxidation (+15.995) and tyrosine mono-uridylylation (+306.025) or di-uridylylation (+612.050). For each of the identified peptides with biochemical modification, a manual examination of the fragmentation pattern was performed to eliminate search artifacts.
To calculate the stoichiometries of the modified peptides, the extracted ion current chromatograms (XICs) as well as the area under the curve (AUC) of the precursors were used for relative quantification. The relative abundance of a modified peptide was calculated using the following formula: 100% × [(sum of AUCs of the modified peptide under all available charge states)/(sum of AUCs under all charge states of all different forms of the peptide in the sample)]. Nonuridylylated, mono-uridylylated, and di-uridylylated N-terminal VPg decapeptides were used to evaluate potential changes in peptide ionization and ion current intensities associated with the addition of the uridyl group. The percentage of uridylylated VPg was estimated from the AUCs corresponding to the 3BC, 3BC-pU, and 3BC-pUpU peptides as follows: [(3BC-pU + 3BC-pUpU)/3BC] *100. Calculated percent inhibition at each PI concentration was used to determine the median effective concentration (IC50) using Prism.
Results and Discussion
3BC as Uridylylation Substrate
It has been shown that the creRNA-dependent synthesis of VPg-pUpU can be reconstituted in vitro using purified VPg, 3CD, 3Dpol, and creRNA. Detection of the uridylylated product has previously been performed using [α-32P]UTP and SDS-PAGE analysis. The VPg precursor form used for initiation of genome replication in infected cells is not known for certain, but it has been suggested for poliovirus that it is most likely the precursor protein 3BC(D). Both 3BC and 3BCD have been reported to be efficient uridylylation substrates in vitro. For practical reasons, we chose to use 3BC to serve as both the uridylylation substrate and the source of 3Cpro, with the 3Cpro active site cysteine mutated to alanine to prevent autocleavage at the 3B-3C junction. Furthermore, it has been reported that in vitro poliovirus uridylylation of 3BC is at least 3-fold more efficient than uridylylation of VPg, with optimal 3BC uridylylation achieved at a 3BC:3Dpol:creRNA stoichiometry of 3:1:1.
6
We initially confirmed by gel analysis that uridylylated 3BC was produced upon performing the uridylylation reaction with 3 µM HRV-A16 3BC, 1 µM HRV-A16 3Dpol, 1 µM cre(2A)RNA, and 1 µM UTP/32P-labeled UTP, whereas no product could be observed when we used an inactive variant of 3Dpol (
We sought to identify an antibody specifically recognizing the uridylylated product as a starting point for the development of a nonradioactive assay. Rabbit polyclonal antiserum was raised against the VPg N-terminal decapeptide GP[Tyr(pU)]SGEPKPK-aminohexanoic acid-[Cys]-amide and depleted of antibodies that could also bind to unmodified VPg. This purified serum was evaluated for its ability to discriminate uridylylated and nonuridylylated 3BC by immunoblot analysis. As shown in Figure 1A , the anti–VPg-pU(pU) detected 3BC (molecular weight of 26 kDa) after an uridylylation reaction performed with wild-type 3Dpol, but no band was visible when the uridylylation reaction was carried out with inactive 3Dpol, indicating that the anti–VPg-pU(pU) is specific for uridylylated 3BC. Surface plasmon resonance (SPR) analysis demonstrated that the anti–VPg-pU(pU) recognized both mono- and di-uridylylated VPg N-terminal decapeptides, with ~10-fold higher affinity for the mono-uridylylated peptide, whereas no binding was observed to the nonuridylylated VPg decapeptide (data not shown).
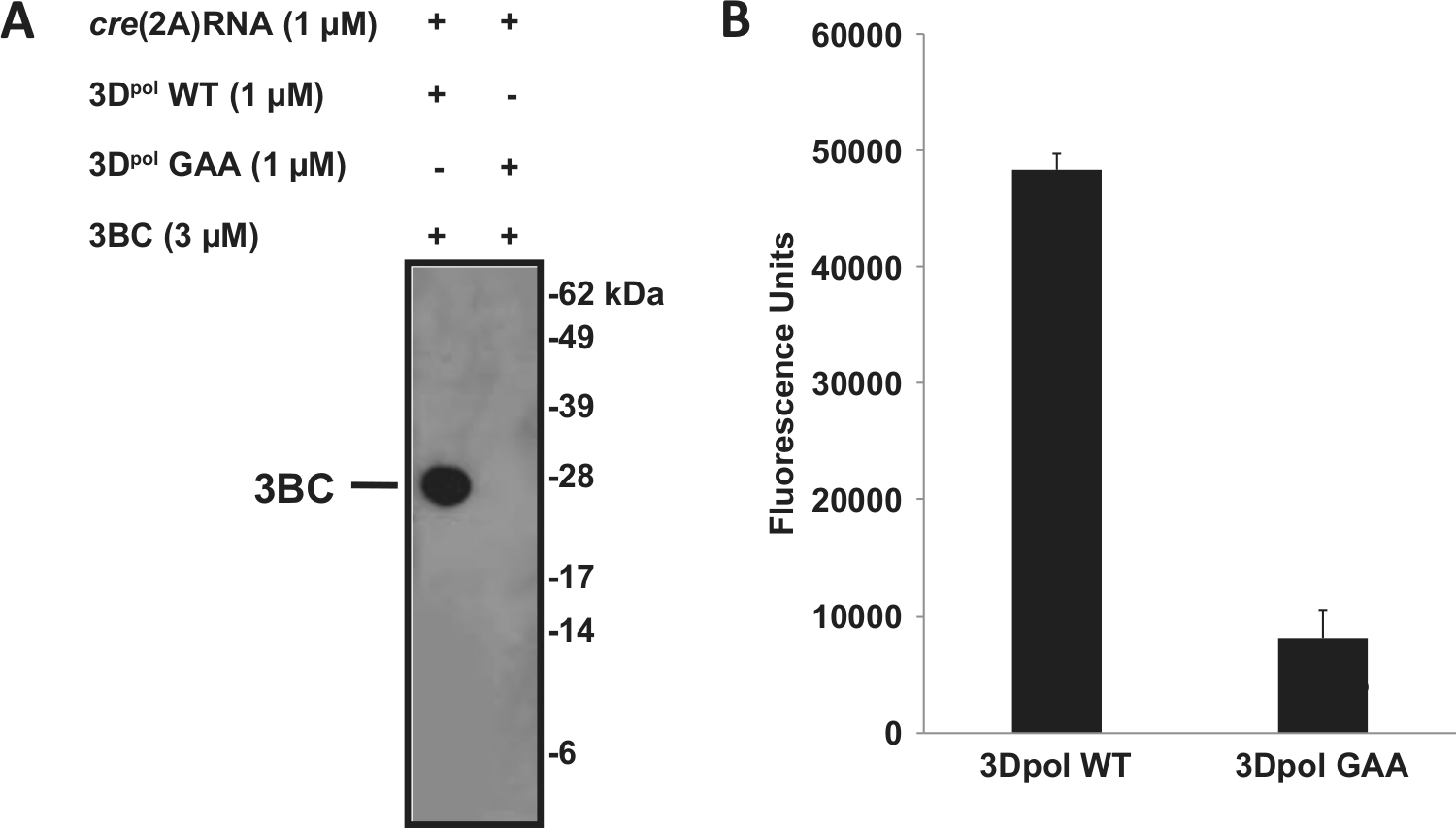
Anti–VPg-pU(pU) purified rabbit polyclonal antiserum specificity. The uridylylation reaction was performed with wild-type or inactive (GAA mutant) HRV-A16 3Dpol, as described in Materials and Methods. (
Plate-based detection of the uridylylation reaction was performed with biotinylated 3BC. The biotin-3BC uridylylated product was captured by the anti–VPg-pU(pU) serum immobilized on a 96-well plate and detected with streptavidin-HRP. A 6-fold increase in the enzyme-linked immunosorbent assay (ELISA) signal was obtained in the presence of wild-type 3Dpol in comparison to the inactive enzyme, consistent with the immunoblot data ( Fig. 1B ).
These initial experiments were performed with a relatively rare 3Dpol Asp-149 variant. For assay development, the more common Gly-149 variant was used. Although it is a surface-exposed residue, the identity of amino acid 149 did appear to affect activity, with the Gly-149 form having a lower apparent Km for UTP under uridylylation (but not simple elongation) assay conditions (data not shown). Interestingly, a recent publication has reported a possible role for the adjacent Tyr-148 in poliovirus uridylylation. 26
HRV VPg Uridylylation Assay Optimization
The ELISA format described above is not ideal for robust high-throughput detection of uridylylated products. AlphaScreen, a bead-based homogeneous method involving signal amplification to provide high sensitivity, was selected for further assay development, and conditions were optimized to yield a robust signal with the lowest possible protein concentrations. A schematic description of the assay is shown in
A cross-titration of 3BC and 3Dpol was performed at protein concentrations of each ranging from 7.8 to 500 nM in the presence of 125 nM creRNA and of an excess of UTP (400 µM). As shown in Figure 2A , the highest AlphaScreen signals (>8000 cps) were observed when the uridylylation reaction was performed with 62.5, 125, or 250 nM 3BC and 125 or 250 nM 3Dpol. A maximal signal-to-background (S/B) value of 117 was obtained in the presence of 125 nM 3BC, 125 nM 3Dpol, and 125 nM creRNA. At a lower creRNA concentration of 31.25 nM, we observed a 2-fold decrease in the maximum AlphaScreen signal. At this lower creRNA concentration, a slightly lower 3Dpol concentration (62.5 nM) but slightly higher 3BC concentration (250 nM) were optimal ( Fig. 2B ). In both experiments, a decrease in signal was observed at the highest 3Dpol concentrations, independently of the 3BC concentration. Altogether, and based on the observation that HRV 3Dpol exhibits a 10-fold higher affinity for creRNA than 3BC (data not shown), these data suggest that formation of a maximally productive 3BC/3Dpol/creRNA uridylylation complex is dependent on maintaining the optimum 3Dpol:creRNA ratio. For experiments described below, the uridylylation reaction was performed with 125 nM each of 3BC, 3Dpol, and creRNA.
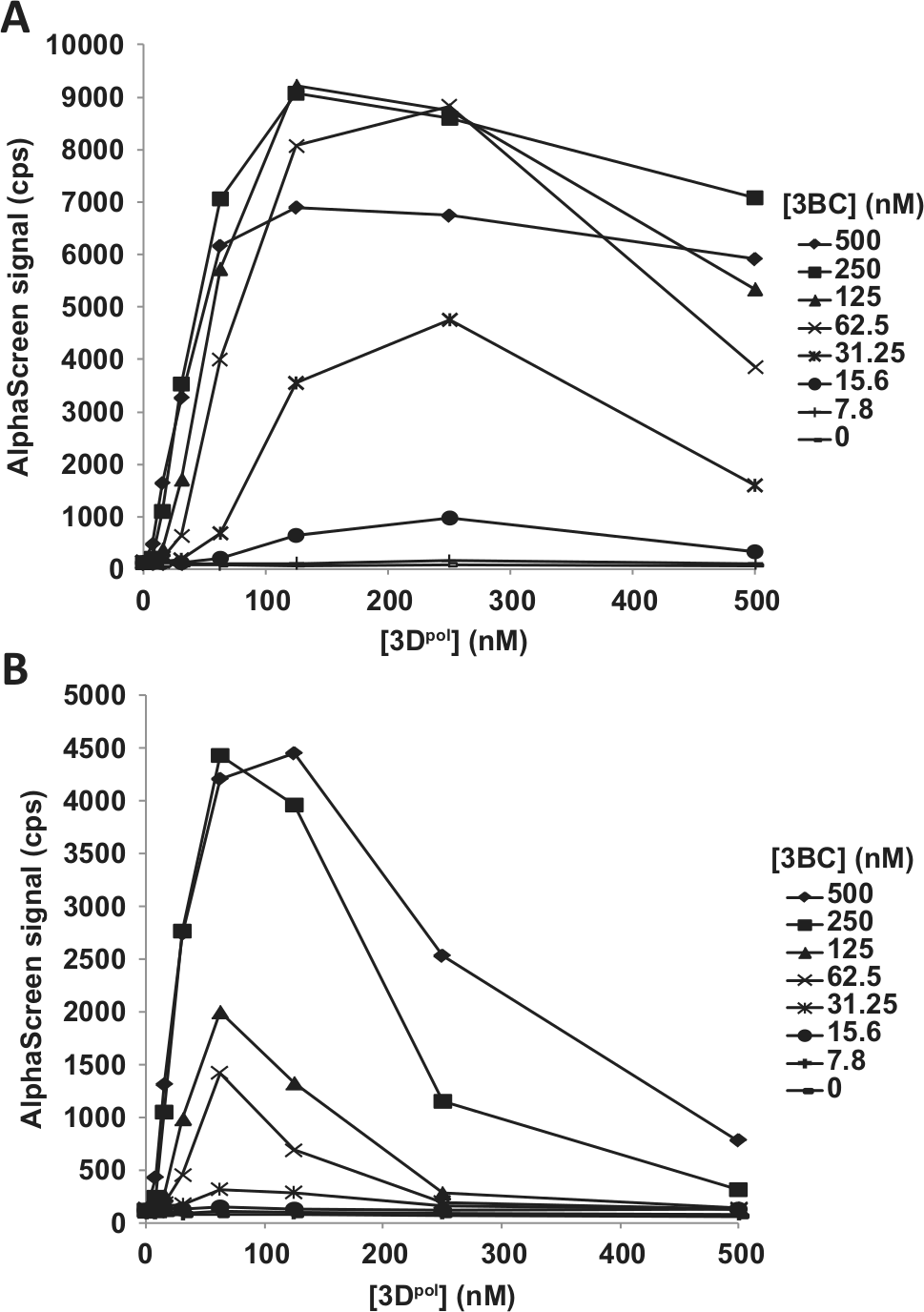
Cross-titration of HRV-A16 3BC and 3Dpol. Reactions (30 µL) were performed for 30 min at 23 °C in 20 mM Bis-Tris (pH 6.8), 5% (v/v) glycerol, 0.5 mM Mg acetate, 0.01% bovine serum albumin, 80 U/mL RNasin, 1 mM TCEP, and 5% DMSO in a 384-well AlphaPlate and initiated with the addition of 400 µM UTP. The AlphaScreen bead–anti–VPg-pU(pU) solution (10 µL), prepared as described in Materials and Methods, was added to the assay mixture to capture uridylylated biotin-3BC. Following a 60-min incubation in the dark at 23 °C, the AlphaScreen signal was measured. (
During initial assay optimization, the uridylylation reaction was carried out in the presence of 400 µM UTP. Time-course experiments at UTP concentrations ranging from 0.6 to 50 µM were performed to determine the Km for UTP in our assay system. As shown in Figure 3A , the signal was approximately linear over 60 min at UTP concentrations less than 10 µM. However, at higher UTP concentrations, the signal was not linear. Loss of linearity at higher substrate concentrations is unusual, and based on the mass spectrometry experiments described below, it is probably not related to substrate depletion but rather to the decreased affinity of the antibody for the final di-uridylylated product versus the initial mono-uridylylated product, which is more abundant at later times. Using the 5-min time point for lower UTP concentrations, an apparent Km for UTP of 20 µM was obtained ( Fig. 3B ). This result is generally consistent with the apparent Km values of 3.9 µM and 16.6 µM reported for poliovirus and FMDV VPg uridylylation, respectively.24,27 We obtained a similar Km value of 5 µM in the HRV-A16 3Dpol elongation activity assay (data not shown). The purpose of this experiment was to establish a screening concentration for UTP near its Km, and for further work, we selected a concentration of 10 µM. Additional experiments would be needed to definitively establish whether the nucleotide substrate Km is different in uridylylation and elongation reactions. A cross-titration of 3BC and 3Dpol (15.6–250 nM each) in the presence of 125 nM creRNA and 10 µM UTP was performed ( Fig. 3C ). The highest signals were observed when the uridylylation reaction was performed with 125 or 250 nM 3BC and 125 or 250 nM 3Dpol. Reduction of the UTP concentration from 400 µM to 10 µM had no impact on the 3BC and 3Dpol concentrations yielding the maximum signal (~9000 cps for both UTP concentrations) (cf. Fig. 2A with Fig. 3C ). However, performance of the assay at the lower UTP concentration improved the signal linearity as a function of the 3Dpol concentration, especially at lower 3BC concentrations.
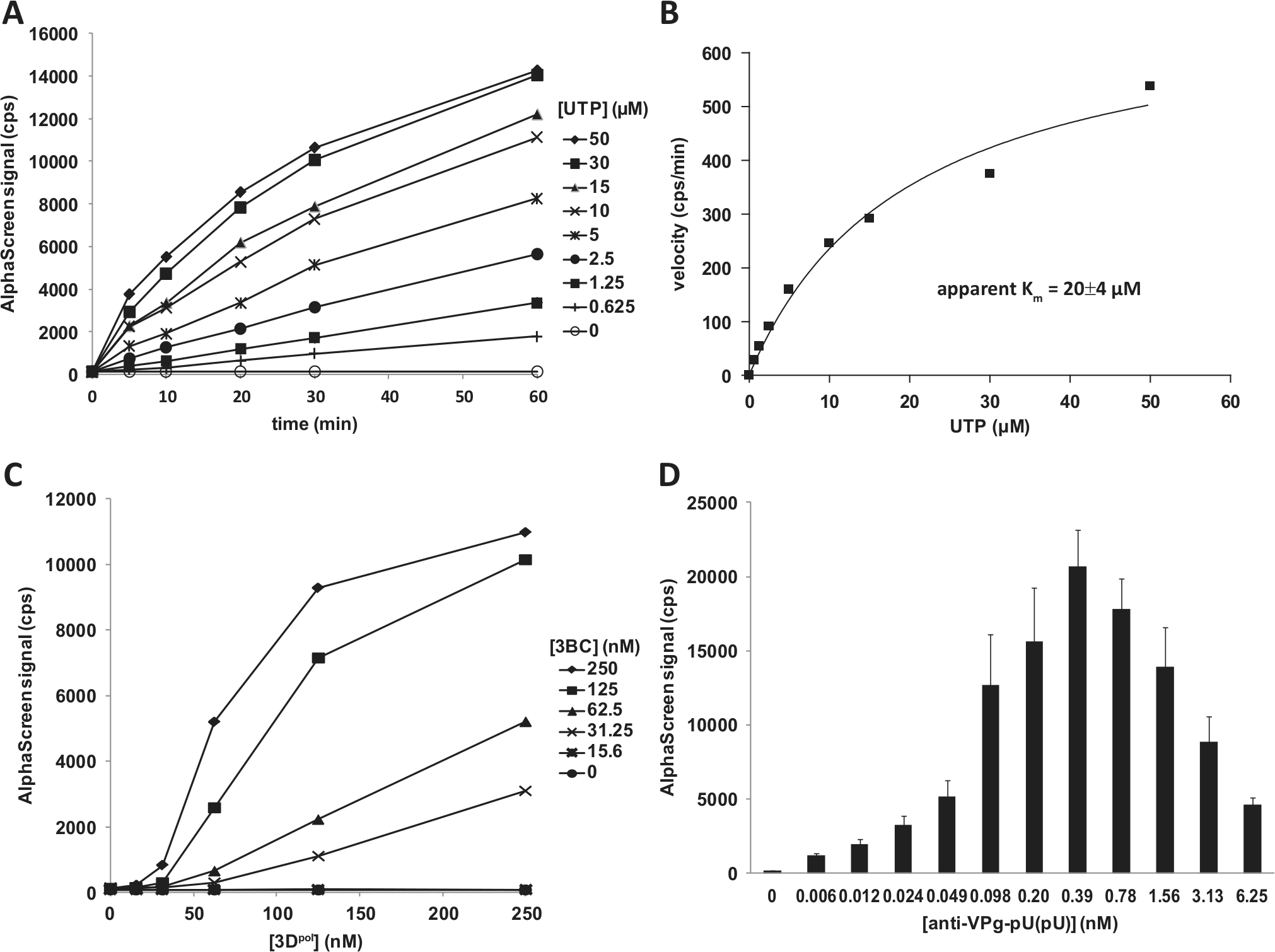
Optimization of HRV-A16 VPg uridylylation reaction. The uridylylation reactions (30 µL) were performed for 30 min at 23 °C in 20 mM Bis-Tris (pH 6.8), 5% (v/v) glycerol, 0.5 mM Mg acetate, 0.01% bovine serum albumin, 80 U/mL RNasin, 1 mM TCEP, and 5% DMSO in a 384-well AlphaPlate and initiated with the addition of UTP. Biotinylated product capture was performed as described in Materials and Methods. (
The anti–VPg-pU(pU) used to capture the uridylylated products was also titrated (
Fig. 3D
). The bell-shaped curve obtained is characteristic of the “hook effect” observed in a saturable bimolecular detection system such as AlphaScreen. From the range tested, the antibody concentration yielding the maximum signal was 0.39 nM, confirming that the concentration used in previous experiments is optimal. To obtain a preliminary assessment of assay performance, a control plate consisting of 336 positive control wells and 48 negative control wells was tested (
Use of poly(rA) in place of creRNA as a template during uridylylation was investigated and resulted in a 8- and 2.4-fold decrease in signal for the reaction performed in the presence of Mg+2 and Mn+2, respectively (
Monitoring of the HRV VPg Uridylylation Reaction by Mass Spectrometry
The VPg uridylylation AlphaScreen detection procedure does not allow us to differentiate the mono- and di-uridylylated 3BC products. Furthermore, the relative binding affinity of anti–VPg-pU(pU) for 3BC-pU versus 3BC-pUpU under assay conditions is not known, although SPR experiments with uridylylated peptides suggested that the antibody exhibits a ~10-fold higher affinity for the mono-uridylylated VPg peptide (data not shown). Mass spectrometry analysis was used to quantify the level of 3B, 3B-pU, and 3B-pUpU peptides present over the course of the uridylylation reaction. For MS detection, the uridylylation reaction was performed under the optimized assay conditions at UTP concentrations of 1, 10, and 50 µM for 0, 5, or 40 min. As shown in Figure 4A , an increase in mono- and di-uridylylated peptides was observed over time at all UTP concentrations, concomitant with a decrease in nonuridylylated 3B peptide, as exemplified for the reaction performed with 10 µM UTP (see inset in Fig. 4A ). As well, more uridylylated products and an increase in the relative abundance of 3BC-pUpU over 3BC-pU were observed with increasing UTP concentrations. Using the AUC value for the input 3BC (5.6 × 107) and the sum of AUCs for both uridylylated peptides, the percentage of uridylylation was calculated to estimate the amount of modified 3BC. After a 40-min incubation, 3.6%, 25%, and 67% of the input 3BC was mono- or di-uridylylated at 1, 10, and 50 µM UTP, respectively. On the basis of these results, we estimate that under our optimized assay conditions (125 nM each of 3BC, 3Dpol, creRNA, and 10 µM UTP incubated for 30 min), approximately 20% 3BC is uridylylated. This is comparable to results for poliovirus, for which at least 75% of the input 3BC was uridylylated after 20 min in reactions with 1 µM each of 3BC, 3Dpol, and creRNA. 6 Also consistent with these results, the same laboratory reported a direct comparison of HRV-B14 and poliovirus VPg uridylylation, suggesting that uridylylation is ~4-fold less efficient for rhinovirus. 18 The uridylylation reaction mixture submitted to MS analysis was also evaluated in the AlphaScreen assay. The relative levels of AlphaScreen signal were comparable to the results observed by MS (cf. Fig. 4B with Fig. 4A ) and therefore validate the use of the AlphaScreen format to measure uridylylation.
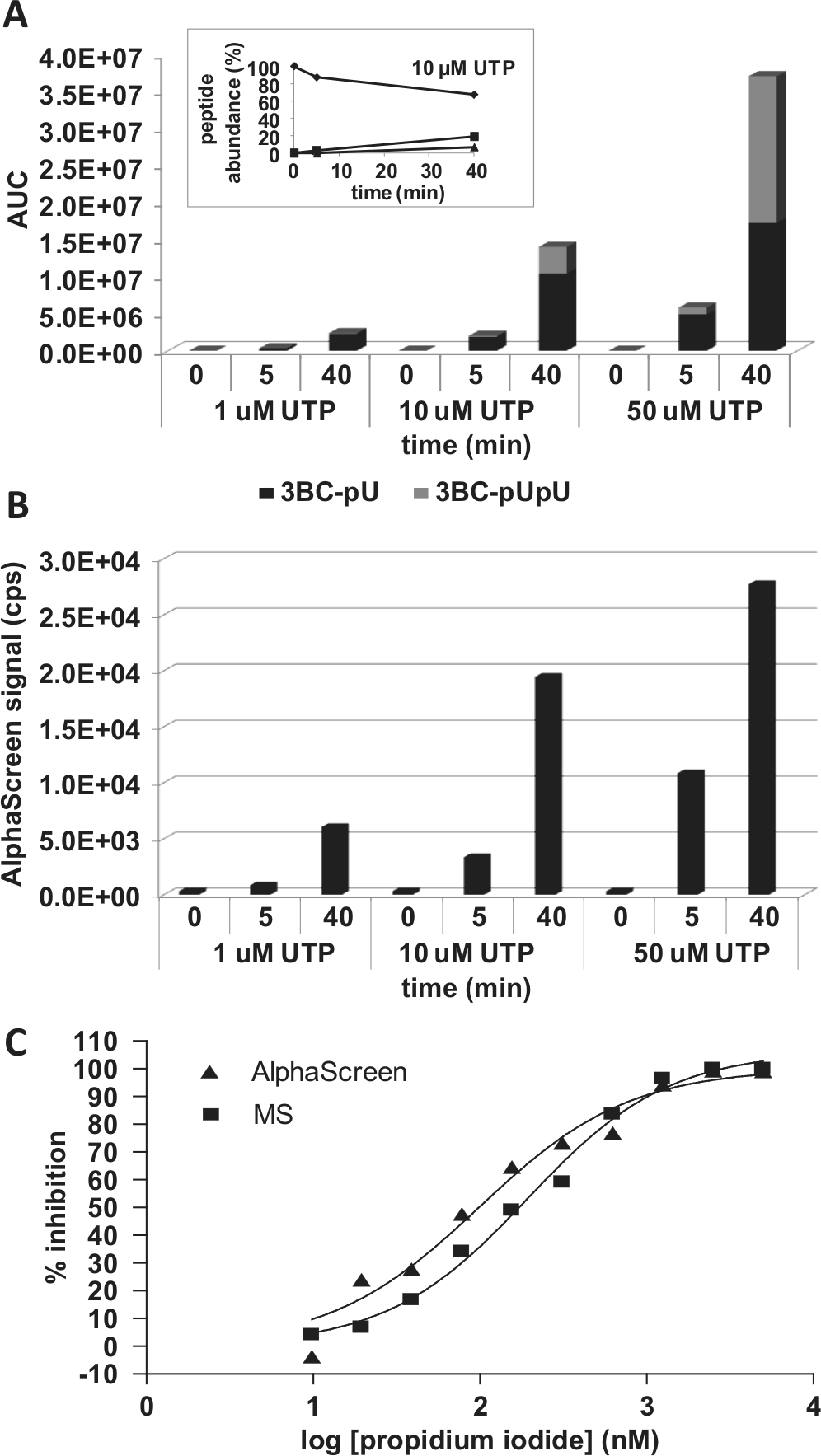
Mass spectrometry (MS) and AlphaScreen detection of uridylylated products. The uridylylation reactions were performed with 125 nM biotinylated 3BC, 125 nM 3Dpol, and 125 nM cre(2A)RNA in 20 mM Bis-Tris (pH 6.8), 5% (v/v) glycerol, 0.5 mM Mg acetate, 80 U/mL RNasin, 1 mM TCEP, and 5% DMSO and were initiated with the addition of 5, 10, or 50 µM UTP. For MS detection, reactions were stopped after 0, 5, or 40 min by the addition of 20 mM EDTA. Sample processing and MS analysis were done as described in Materials and Methods. For AlphaScreen, the biotinylated products were captured and detected as described in Materials and Methods. (
In the absence of specific small-molecule inhibitors of HRV VPg uridylylation, the effect of the intercalator PI on activity was evaluated. PI preferentially binds to double-stranded nucleic acids and is therefore expected to interfere with the formation of the uridylylation complex by altering the secondary structure of creRNA. Representative inhibition dose-response curves for PI using both AlphaScreen and MS detection are shown in Figure 4C . Both detection methods yielded similar IC50 values for PI: 100 nM and 190 nM for AlphaScreen and MS, respectively. This demonstrates that the lower-throughput MS method provides an antibody-independent method for confirmation of inhibitor activity.
In conclusion, a novel homogeneous and nonradioactive HRV-A16 VPg uridylylation AlphaScreen assay was developed and shown to be suitable for screening large compound collections to identify HRV replication inhibitors. Such compounds could then be confirmed as inhibitors of rhinovirus replication in cell culture assays. Furthermore, this assay might be used as a biochemical tool to further study the interplay between the different components involved in the rhinovirus uridylylation complex.
Footnotes
Acknowledgements
We thank Jessica Tamura-Wells for 3BC production, Robert S. McCollum for SPR experiments, Dr. Mohammed Kashem and John Wolak for helpful discussions, and Dr. Frédéric Vaillancourt for critical review of the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.