
Abstract
BEAR (binding estimation after refinement) is a new virtual screening technology based on the conformational refinement of docking poses through molecular dynamics and prediction of binding free energies using accurate scoring functions. Here, the authors report the results of an extensive benchmark of the BEAR performance in identifying a smaller subset of known inhibitors seeded in a large (1.5 million) database of compounds. BEAR performance proved strikingly better if compared with standard docking screening methods. The validations performed so far showed that BEAR is a reliable tool for drug discovery. It is fast, modular, and automated, and it can be applied to virtual screenings against any biological target with known structure and any database of compounds.
Introduction
N
In the 1990s, combinatorial chemistry and high-throughput screening (HTS) emerged as useful and promising approaches for drug discovery. Although these techniques are successfully exploited to identify biologically active candidates in both industry and academia, some limitations remain, and several alternative approaches are also followed to drive the initial phases of the drug discovery process. One of these is virtual screening (VS), which emerged as the most interesting and straightforward computational tool for the rapid identification of bioactive compounds from large collections of chemicals. Typically, VS is performed with molecular docking, which is able to virtually screen and rank a large number of molecules into the active site of a target structure in a reasonable amount of time. This technique is powerful and cost-effective and allows one to prioritize and select best-hit compounds for biological assays based on the strength of interaction with the biological target.
Many VS tools, based on different algorithms for pose generation and ranking assessment, are now available. Moreover, the wide availability of computer resources makes VS applicable to big libraries, containing even millions of compounds, in relatively short times. Notwithstanding, drawbacks and limitations still exist. For example, docking techniques still lack reliable simulation of the flexibility of both ligands and target receptor, and scoring functions may fail to estimate ligand binding energies in reasonable agreement with experimental ones.
In light of these observations, we focused our efforts on the development, validation, and application of a new postdocking tool aimed at overcoming some of these limitations. The method, named BEAR (binding estimation after refinement), 1 is a new automated postdocking procedure based on the conformational refinement of docking poses through molecular dynamics (MD) followed by accurate predictions of protein binding free energies using molecular mechanics–Poisson Boltzmann surface area (MM-PBSA) and molecular mechanics–generalized Born surface area (MM-GBSA). The idea of applying MD and MM-PB(GB)SA to virtual screenings has been pioneered and extensively tested by our group and is proving to have significant impact and innovation potential in the drug discovery field.
Several validation studies, performed on different targets and using different conditions, demonstrated that BEAR scoring function is reliable. 2,3 In all cases, good correlations between experimental and calculated binding energies were achieved, confirming the BEAR scoring function reliability. Moreover, when applied to a huge virtual screening campaign aimed at identifying new plasmepsin inhibitors, BEAR discovered 26 novel and potent inhibitors out of the 30 compounds tested in the wet lab, thereby giving an impressive hit rate. 4 This evidence strongly confirmed the potential of BEAR in drug discovery.
In addition, other studies tested the ability of BEAR to distinguish among active and inactive compounds. This was initially achieved with 2 independent experiments performed against Plasmodium falciparum dihydrofolate reductase (Pf-DHFR) using 2 different sets of compounds. The first investigated the BEAR ability to enrich 14 known ligands seeded into the National Cancer Institute (NCI) diversity database, containing 1720 compounds with unknown activities on Pf-DHFR. 1 The second was performed using the 201 ligands and 7150 decoys contained in the DHFR data set present in the directory of useful decoys (DUD). 5,6 Both experiments revealed a striking performance of BEAR in comparison to standard dockings. In fact, BEAR was able to enrich the known inhibitors of pf-DHFR at the top of ranked lists despite decoys and unknown binders.
In this study, BEAR was applied to a new VS project to further benchmark the BEAR performance when very big libraries of compounds are used. The present screening was performed against Pf-DHFR using the lead-like subset of the ZINC database, containing 1.5 million commercial compounds. A detailed analysis of the BEAR performance in terms of enrichment factors of known active compounds is presented.
Materials and Methods
Docking screening
For this screening, we used the crystal structure of Pf-DHFR in complex with NADPH and the potent inhibitor WR99210 (PDB code 1J3I). 7 The structure was prepared for docking as already described in previous investigations. 5
The virtual screening was performed using the lead-like subset of the ZINC database 8 containing around 1.5 million commercial compounds. This set of molecules was seeded with the 201 known ligands of the DUD DHFR benchmarking data set developed by Huang et al. 6 The DUD DHFR ligand data set has been downloaded from the DUD at blaster.docking.org/dud.
All the ligands were docked into the Pf-DHFR structure using AutoDock 4. 9 The Lamarckian genetic algorithm (LGA) was applied to model ligand-protein interactions. The docking area was constrained to a 22.5 Å cubic space centered on the inhibitor WR99210, using a grid point spacing of 0.375 Å. Before docking screenings, the Gasteiger partial atomic charges of each ligand were calculated and used for docking analyses. For each molecule, 10 docking runs were carried out with 150 individuals in the first population and 2.5 million energy evaluations. After docking, all the ligand orientations were clustered using root mean square tolerance of 2.0 Å. The orientation with the best binding energy and belonging to the most populated cluster was selected and postprocessed with BEAR.
BEAR postprocessing
Bond charge correction (BCC) partial atomic charges were calculated and assigned to the ligands. Each protein-ligand complex was postprocessed with the same BEAR settings described in previous studies. Briefly, each complex was submitted to energy minimization using Amber 9 10,11 through a multistep MM/MD/MM simulation consisting of 2000 steps of minimization on the entire complex, followed by 100-ps molecular dynamics in which only the ligand was allowed to move, plus a final minimization of the complex (2000 steps). The MD simulation was performed at 300 K, with SHAKE turned on for bonds involving hydrogen atoms, allowing a time step of 2.0 fs. Then, the single structure of each refined complex was used to estimate the free energy of binding through MM-PBSA and MM-GBSA algorithms. 12-15
Results and Discussion
With the aim of comparing the BEAR performance with that of traditional docking methods, a wide virtual screening based on the lead-like subset of the ZINC database (1.5 million compounds) was performed against Pf-DHFR. The choice of this subset is related to the compound molecular properties and their commercial availability. In fact, the ZINC database contains commercially available compounds in a ready-to-screen format. Moreover, the screening of lead-like compounds will allow the further optimization of the best-ranked compounds. To test the BEAR performance, the ZINC lead-like subset was seeded with 201 known DHFR inhibitors taken from the DUD database. 6 In this experiment, the ratio between the number of known inhibitors and the number of compounds in the database was around 1.3 × 10−4 (i.e., 50- and 200-fold lower than those used in previous validations). 1,5 Therefore, the present screening represents the most intensive benchmark ever performed with BEAR.
After the screening, the results were evaluated with an enrichment factor analysis. The enrichment curves were calculated as a function of rank position, according to equation (1), where I x is the number of known inhibitors found within the first x positions of the ranked database and I tot the total number of known inhibitors contained within the data set. Figure 1 reports the enrichment factor results for AutoDock and BEAR scoring functions (MM-PBSA and MM-GBSA). In this plot, the higher the percentage of known ligands found at a given percentage of the ranked database, the better the enrichment performance of the virtual screen. Ideally, all the known ligands should be grouped at the top of the ranked list, leading to a perfect enrichment despite a random choice where ligands are equally distributed along the ranked list.
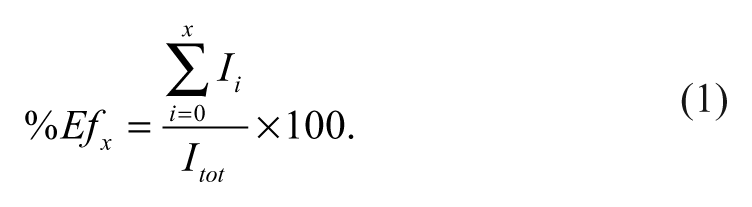
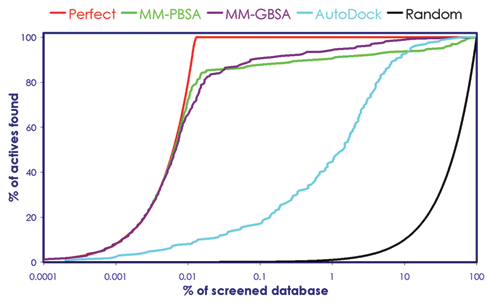
Enrichment factor plot according to AutoDock and BEAR (binding estimation after refinement) scoring functions. The random and perfect curves are also shown. MM-GBSA, molecular mechanics–generalized Born surface area; MM-PBSA, molecular mechanics–Poisson Boltzmann surface area.
By comparing AutoDock and BEAR, enrichments we found that BEAR performed strikingly better than AutoDock ( Table 1 and Fig. 1 ). In addition, BEAR enrichments resemble the perfect trend with both MM-PBSA and MM-GBSA scoring functions. This gives evidence that the application of BEAR resulted in a significant improvement of the ranking even in a case in which the inhibitors were seeded into a huge database of 1.5 million compounds.
Enrichment Factors at Several % of Ranked Database According to AutoDock and BEAR Scoring Functions (MM-PBSA and MM-GBSA)
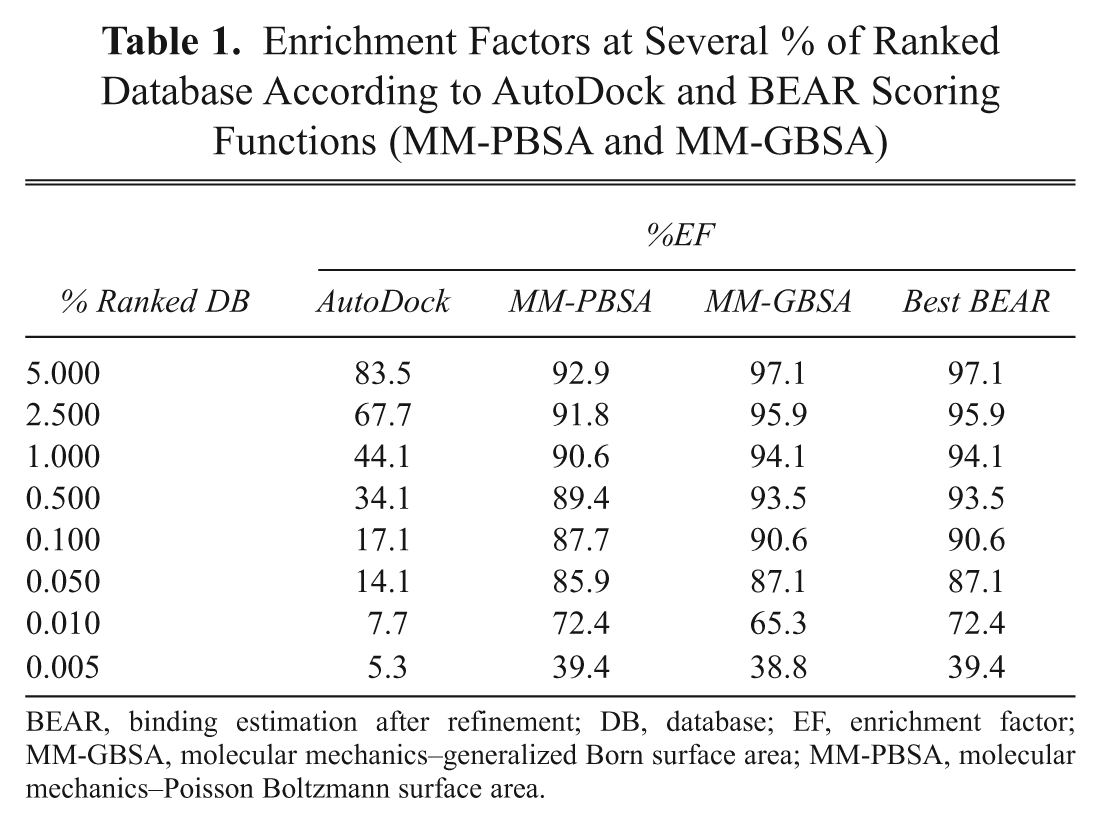
BEAR, binding estimation after refinement; DB, database; EF, enrichment factor; MM-GBSA, molecular mechanics–generalized Born surface area; MM-PBSA, molecular mechanics–Poisson Boltzmann surface area.
An analytical summary of the enrichment performances is reported in Table 1 . Taking into account the first 5% of the ranked database (corresponding to around the first 75,000 positions), the percentage of known ligands enriched is around 90% for both BEAR and AutoDock scoring functions. However, at lower percentages, the enrichment factors measured for BEAR remain clearly superior. In fact, at 0.1% of the ranked database (corresponding to the first 1500 positions), BEAR enrichments were still around 90%, whereas AutoDock values lowered to around 17%. Focusing on the first 0.01% of the ligand database (first 150 positions), BEAR enrichment was around 9-fold better than AutoDock, with 70% of the ligands still retrieved by BEAR and only 8% by AutoDock. Finally, taking into account only the compounds at the topmost position of the ranked database (0.005% of the database, 75 compounds), BEAR and AutoDock enrichments were 40% and 5.3%, respectively. Considering that after a VS, only the topmost (typically 40-100) compounds are tested experimentally, most of the active compounds would have been discarded by AutoDock but successfully retrieved by BEAR.
Finally, a structural analysis and a comparison of ligand orientations assigned by docking and postprocessed with BEAR were performed. First, visual inspection of the orientations of known inhibitors before and after BEAR refinement was performed. Figure 2A reports the docking pose of an active compound and its refined orientation. The orientations are quite similar, and the interactions established with the target involve residues such as Asp54, Ile14, and Ile164, known to be crucial for binding to the protein. This compound is ranked at the 8th position according to MM-PBSA but at more than the 40,000th position according to AutoDock, indicating a failure in the standard docking scoring function to assign a favorable score. On the contrary, the BEAR scoring function was able to assign a favorable score and to rank this inhibitor at the topmost position on the list. On the other hand, Figure 2B reports an example of an unknown ligand that was ranked in the first position by AutoDock (25th) and in the last position by BEAR (around 1,000,000th). In this case, the ligand orientation was more perturbed by the BEAR refinement procedure than by the known inhibitor of Figure 2A , and less favorable interactions are established with binding site residues.
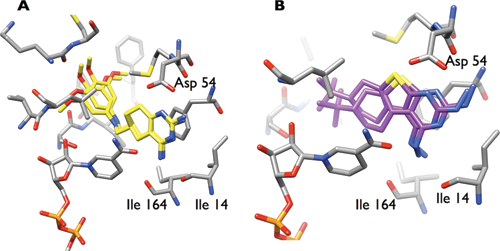
Two examples of docked ligands before and after BEAR structural refinement. (
In light of these results, the root mean squared deviation (RMSD) between docking and BEAR refined orientations was computed for the entire database and for the known inhibitors alone. Respectively, averaged RMSD values of 1.4 ± 1.2 and 1.1 ± 0.5 for the database and the inhibitors were obtained. The lower RMSD value measured for the inhibitors suggests that docking poses of active compounds are less perturbed by the BEAR structural refinement than those of the compound database. This finding indicates that BEAR recognizes correct orientations assigned by docking (as is the case of known inhibitors) but tries to explore different orientations when unlikely binders or decoys are evaluated. Thanks to the more accurate scoring functions applied by BEAR, the slightly refined docking orientations of the known ligands are enriched at the topmost position of the ranked database, and many false-positive compounds retrieved by docking are excluded from the top of the ranked list.
The BEAR procedure was included in a completely automated workflow ( Fig. 3 ); starting from ligand-receptor complexes obtained from docking, the procedure iteratively processes each compound of the database and provides the user with a final summary report that includes a list of interesting molecules in terms of predicted binding affinity and occurrence of key interactions with active site residues defined by the user. The workflow parameters were optimized to reduce the calculation time and to be suitable for submission to high-performance computing systems. According to the parameter set reported in the Materials and Methods section, the average time required to analyze a single molecule with BEAR was around 8 min on a single core of a 2.4-GHz AMD Opteron CPU. Additional validation studies still in progress show that the computing time per molecule can be further reduced without loss of accuracy. These features, together with its accuracy and reliability, enable BEAR to be applied on large databases of compounds, with timings comparable to traditional HTS systems.
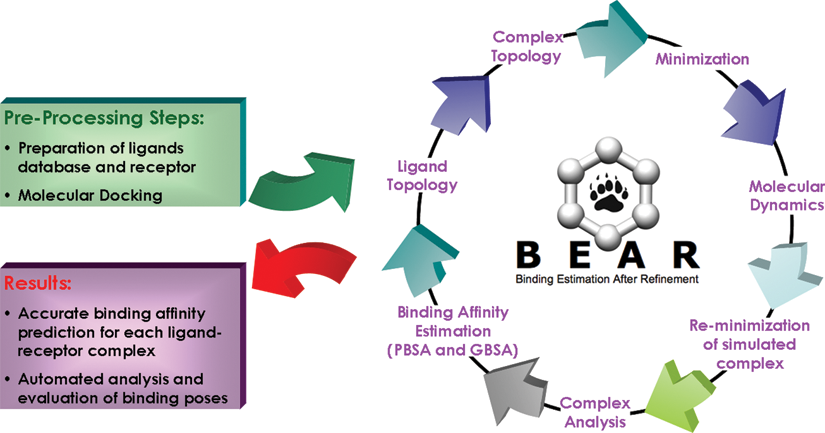
The BEAR (binding estimation after refinement) workflow. GBSA, generalized Born surface area; PBSA, Poisson Boltzmann surface area.
Conclusions
We performed a wide virtual screening against Pf- DHFR using our BEAR technology. An accurate analysis of the enrichment factors and molecular orientations revealed a low enrichment of known inhibitors using standard docking schemes but a strikingly better performance using BEAR. Although the docking method was generally able to predict the correct binding orientation of the known inhibitors, it proved inaccurate in ranking them among the best compounds. On the contrary, the BEAR scoring function was more reliable and able to recognize and discriminate known ligands among compounds with unknown activity.
BEAR provides a list of molecules prioritized according to predicted affinity for the biological targets considered and proved able to correctly identify known ligands (validation studies) as well as entirely new classes of inhibitors (drug discovery studies). Moreover, in our experience, BEAR screenings result in lists of best hot compounds that usually differ in structure from those obtained with standard docking techniques. This aspect represents a further benefit of BEAR because the identification of innovative and original scaffolds is required to allow intellectual property protection.
In conclusion, BEAR demonstrated to be a reliable tool for drug discovery. It is fast, modular, and automated, and it can be applied to virtual screenings against any biological target with known structure and any database of compounds, such as small organic compounds and small peptides.