
Abstract
The human DEAD (Asp–Glu–Ala–Asp) box protein DDX41, a member of the DEXDc helicase family, has nucleic acid–dependent ATPase and RNA and DNA translocase and unwinding activities. DDX41 is affected by somatic mutations in sporadic cases of myeloid neoplasms as well as in a biallelic fashion in 50% of patients with germline DDX41 mutations. The R525H mutation in DDX41 is thought to play important roles in the development of hereditary myelodysplastic syndrome and acute myelocytic leukemia. In this study, human DDX41 and its R525H mutant (R525H) were expressed in Escherichia coli and purified. The ATPase activities of the recombinant DDX41 and R525H proteins were dependent on both ATP and double-stranded DNA (dsDNA), such as poly(dG–dC) and poly(dA–dT). High-throughput screening was performed with a dsDNA-dependent ATPase assay using the human R525H proteins. After hit confirmation and counterscreening, several small-molecule inhibitors were successfully identified. These compounds show DDX41-selective inhibitory activities.
Introduction
A comprehensive genetic analysis identified a somatic mutation in the DDX41 gene encoding a DEAD (Asp–Glu–Ala–Asp) box-type adenosine triphosphate (ATP)–dependent RNA helicase. The somatic mutations of DDX41 are dominated by c.G1574A (p.R525H). Germline mutations in DDX41 were recently isolated in a subset of familial acute myeloid leukemia (AML)/myelodysplastic syndrome (MDS) pedigrees. The R525H mutant (R525H) might also play an important role in tumorigenesis and therefore provides a drug target for cancer therapy.1–5
DDX41 is a member of the DEXDc family of ATP-dependent helicases.6,7 DEAD-box proteins, which contain the conserved sequence Asp–Glu–Ala–Asp, play essential roles in DNA and RNA metabolism, including in their replication, repair, recombination, transcription, translation, ribosome biogenesis, and splicing, which regulate growth and development.8,9 DDX41 has also been shown to sense microbial DNA, triggering the early induction of type I interferons (IFNs) in mouse splenic myeloid dendritic cells. 10 When it is stimulated with poly(dA–dT) or poly(dG–dC) in some cell-based assays, DDX41 combines with these polynucleotides physically and interacts with the downstream adaptor molecule stimulator of interferon genes (STING) and TANK-binding kinase 1 (TBK1), triggering the activation of interferon regulatory factor 3 (IRF3) and nuclear factor-kappa B (NF-κB), which are important transcription factors in the regulation of type I IFN expression.11,12 A recent study also determined the crystal structures of the DDX41 DEAD domain in the apo form. 2 However, very little is known about the enzymatic activities of DDX41.13,14
In the present study, we cloned the full-length cDNA of human DDX41 and R525H, characterized the proteins’ functions with an ATPase activity assay and used high-throughput screening (HTS) to identify R525H inhibitors. The purified human DDX41 and R525H enzymes showed ATPase activity against poly(dG–dC) or poly(dA–dT). Various compounds were found to inhibit R525H and DDX41. These results constitute significant advances in the biochemical research into DDX41.
Materials and Methods
Cloning
The genes encoding DDX41, R525H, and E345Q (ATPase defective mutant) were amplified with PCR using PrimeSTAR Max DNA Polymerase (Takara Bio, Kusatsu, Japan), with which BamHI and NotI restriction sites were inserted upstream and downstream from DDX41, respectively. The primer sequences for DDX41 were F1, 5′-GTACTTTCAGGGATCCATGGAGGAGTCGGAA CCCGA-3′, and R1, 5′-TGCTCGAGTGCGGCCGCTCA GAAGTCCATGGAGCTGTGGG- 3′. R525H and E345Q were constructed with overlap extension PCR. The first round of PCR to amplify R525H was performed with primers F1 and R2 (5′-TGTGTTTCCCGAGTGCCCGGTGCG GCCAAT-3′), and the open reading frame (Thermo Fisher Scientific, Waltham, MA) of the DDX41 gene was used as the template DNA. The second round of PCR was performed with primers F2 (5′-ATTGGCCGCACCGGGC ACTCGGGAAACACA-3′) and R1, and DDX41 was used as a template. In the third round of PCR, the products of the first- and second-round PCRs were used as the templates and amplified with the F1 and R1 primers. To construct E345Q, the first round of PCR was performed with primers F1 and R3 (5′-GATCATGCGGTCAGCCTGGTCCAGG GCCAGGTAGCGACA-3′), and DDX41 was used as the template. The second round of PCR was performed with primers F3 (5′-CCCTGGACCAGGCTGACCGCATGATC GACATG- 3′) and R1, and DDX41 was used as the template. The third round of PCR was performed in the same way as that for R525H. The amplified gene was inserted into the pET21HH(V) vector using the In-Fusion HD Cloning Kit (Takara Bio) and introduced into Escherichia coli JM109 to isolate the plasmid. dnaK was amplified with PrimeSTAR GXL DNA polymerase (Takara Bio) and restricted with the NdeI and NheI enzymes. The genomic DNA of E. coli BL21(DE3) was used as the template. The first round of PCR was performed with primers F4 (5′-AAGGAGATATACATATGGGTAAAATAATTGGTA TCG-3′) and R4 (5′-TCCCTGAAAGTACAGGTTCTCT TTTTTGTCTTTGACTTCTTCAAATTC-3′). The second round of PCR was performed with primers F5 (5′-AAGGA GATATACATATGGGTAAAATAATTGGTATCG-3′) and R5 (5′-CGTTCAGTCCGCTAGCTCCCT GA AAGTACAG GTTCTC-3′), and the PCR product amplified with the F4 and R4 primers was used as the template. After its digestion with restriction enzymes, the amplified gene was inserted into the pET21HH/hTARS-Avi-His vector using the In-Fusion HD Cloning Kit (Takara Bio) and introduced into E. coli JM109 for plasmid isolation. The isolated recombinant plasmids carrying DDX41, R525H, E345Q, and dnaK were introduced into E. coli BL21(DE3) (Nippongene, Toyama, Japan).
Protein Expression and Purification
E. coli BL21(DE3) clones were grown at 37 °C in Luria–Bertani broth to an optical density of 0.4 at 600 nm (OD600). Isopropyl β-
The presence of the recombinant protein in the collected fractions was checked by staining with Coomassie Brilliant Blue (Thermo Fisher Scientific) and immunoblotting with a DDX41-specific antibody (Abnova, Taipei, Taiwan), a His-tag–specific antibody (Wako Pure Chemical Industries, Ltd., Osaka, Japan) or a DnaK-specific antibody (in-house antibody).
Mass Spectrometric Analysis
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) of the purified enzymes was performed with SYPRO Ruby Protein Gel Stain (Thermo Fisher Scientific). The protein bands were excised and digested with trypsin (Promega, Madison, WI) in a MultiScreen 96-well filter plate (Merck Millipore, Darmstadt, Germany). The tryptic digests were analyzed with nano-LC (Shimadzu, Kyoto, Japan) and an LTQ OrbitrapXL ETD Hybrid Ion Trap-Orbitrap Mass Spectrometer (Thermo Fisher Scientific) and identified using the Mascot database search system (Matrix Science, Boston, MA) and the SProt database. Data processing was performed by Proteome discoverer software version 1.4 (Thermo Fisher Scientific). The precursor mass tolerance was set at 20 ppm, the fragment ion mass tolerance was 0.2 Da, and the false discovery rate was 0.01.
DDX41 Enzymatic Activity Assay
ATPase assays were performed with the ADP-Glo Kinase Assay (Promega), all of which were performed within the linear portion of the standard curve for the conversion of ATP to ADP.
The kinetic parameters for ATP, poly(dG–dC), and poly(dA–dT) were determined by incubating various concentrations of each substrate with DDX41 or R525H for 30 min in 6 µL reaction volume in Greiner 784075 384-well microplates. The initial rate data were fitted to the Michaelis–Menten equation with GraphPad Prism v.5 (GraphPad Software Inc., San Diego, CA).
The reactions for HTS were performed in a total volume of 1 µL in Greiner 782075 1536-well microplates and included 25 nM R525H enzyme, 70 µM ATP, and 0.2 µg/mL poly(dG–dC) (Sigma, Darmstadt, Germany) or poly(dA–dT; Sigma) diluted with assay buffer (20 mM Tris-HCl [pH 7.5], 100 mM KCl, 2.5 mM MgCl2, 1 mM DTT, 0.01% Tween 20, and 0.01% bovine serum albumin [Sigma]). After incubation for 120 min at room temperature, the reactions were terminated by the addition of 0.5 µL of ADP-Glo reagent (Promega). The samples were incubated for 40 min at room temperature before the addition of 1 µL of Kinase Detection Reagent (Promega). After 40 min at room temperature, the luminescence was read with an EnVision Multiplate Plate Reader (PerkinElmer, Waltham, MA). The assay quality of HTS was analyzed with the Z′ factor, which was calculated from the means and standard deviations of the 100% and 0% inhibitory activities. We considered the luminescent signals of the reaction without poly(dG–dC) to indicate 100% inhibitory activity and those of the complete reaction mixture to indicate 0% inhibitory activity. Curve fitting and calculation of the 50% inhibitory concentrations (IC50) were performed with GraphPad Prism v.5 (GraphPad Software).
The substrate competition assays using R525H enzyme were performed under the following four conditions: 70 µM or 700 µM ATP in the presence of 0.2 µg/mL poly(dG–dC) and 0.2 µg/mL or 2 µg/mL poly(dG–dC) in the presence of 70 µM ATP. The time-dependent inhibition was then confirmed by setting the preincubation between the R525H enzyme and the inhibitor to 0 min or 120 min before the reaction was initiated with 2 µg/mL poly(dG–dC) and 70 µM ATP. The reaction time was 5 min to confirm the change in the IC50 by setting different preincubation times of the enzyme with the inhibitor.
EIF4A3 Enzymatic Activity Assay
An RNA-dependent ATPase assay for eIF4A3 was also performed using the ADP-Glo assay system, and proteins were prepared by the method described by Ito et al. 15 To enhance the ATPase activity of eIF4A3, 150 nM MLN51 was added to 150 nM eIF4A3 enzyme, and 35 µM ATP and 1.5 µg/mL poly-U (MP Biomedicals, Santa Ana, CA) were used as the substrates. The reagents were incubated at room temperature for 30 min. Detection with ADP-Glo was performed as for DDX41. We considered the luminescent signals of the reaction without enzyme to indicate 100% inhibitory activity and those of the complete reaction mixture to indicate 0% inhibitory activity. Curve fitting and the calculation of the IC50 values were performed with GraphPad Prism v.5.
DnaK Enzymatic Activity Assay
ATPase activity of DnaK was also confirmed using the ADP-Glo assay system. The reaction was performed with 24 µM DnaK, 0.5 µM ATP, and test compounds diluted with the same assay buffer as DDX41 at room temperature for 40 min. The detection with ADP-Glo, the calculation of activity, and curve fitting were performed as for EIF4A3.
Fluorescent Intercalator Displacement Assay
Poly(dG–dC; 1 µg/mL) and 30 µM candidate hit compound were incubated with 0.1 µM TO-PRO-3 stain (Thermo Fisher Scientific) in DDX41 assay buffer for 120 min in Greiner 784075 384-well microplates. The displacement activities were detected as the fluorescence at an excitation wavelength of 512 nm and an emission wavelength of 533 nm. We considered the fluorescent signals of the reaction without TO-PRO-3 to indicate 100% intercalating activity and those of the complete reaction mixture to indicate 0% intercalating activity.
Results
Activities of DDX41 and Mutants
The recombinant DDX41, R525H, and E345Q proteins were expressed in E. coli and purified through NiNTA and Superdex 200 columns. On SDS-PAGE and Western blotting, the mutant proteins appeared around the DDX41 band at 83 kDa ( Fig. 1A , B ). A protein mass analysis confirmed the types of proteins contained in the purified DDX41, R525H, and E345Q bands. A protein mass analysis revealed that the mass spectrometry–identified peptides covered human DDX41 ( Suppl. Table S1 ), and DnaK from E. coli was one of the proteins included with the purified DDX41 protein ( Suppl. Table S2 ). A Western blotting analysis of the purified DDX41 protein detected the DnaK protein with a DnaK-specific antibody ( Fig. 1C ).
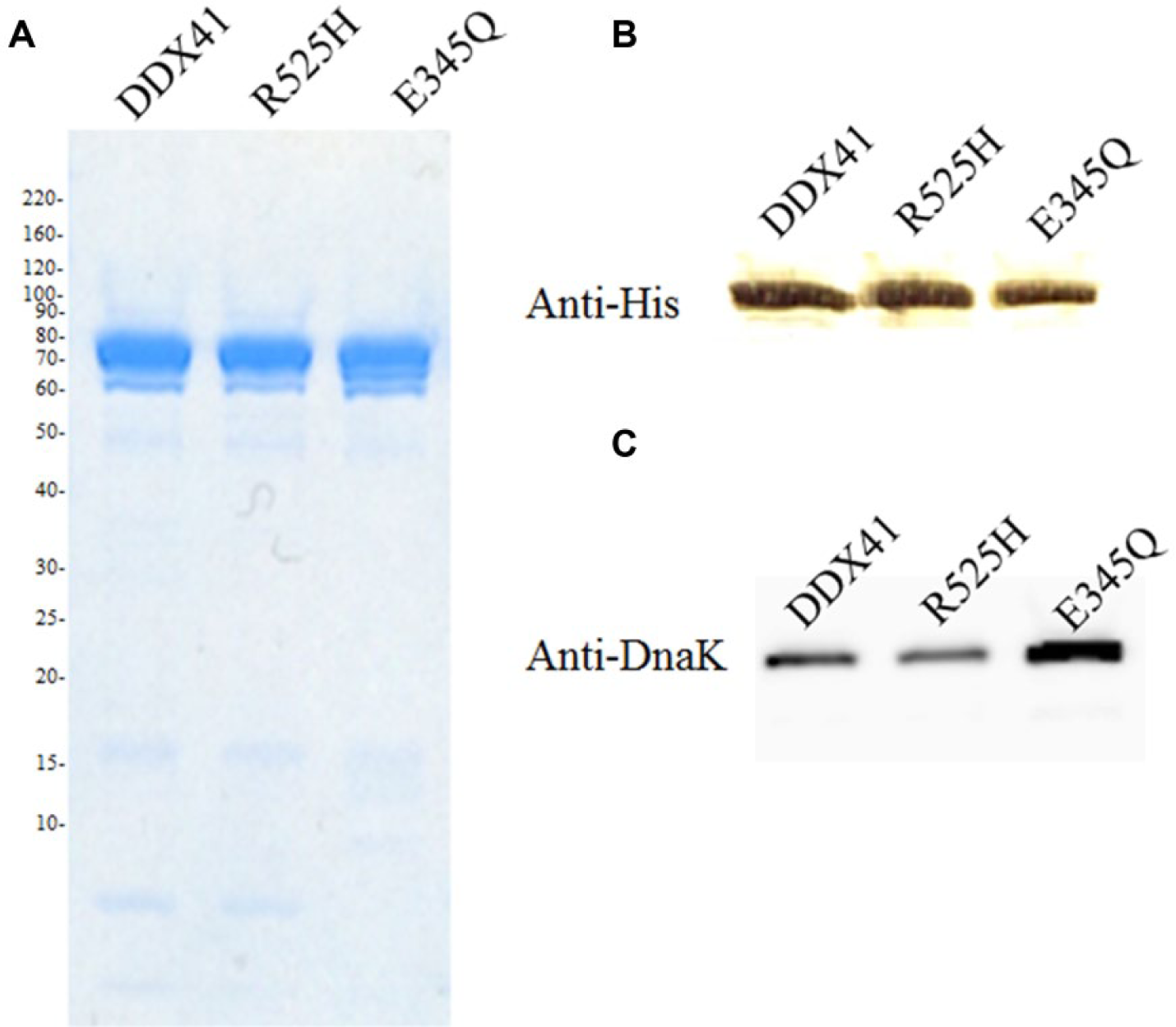
Analysis of purified enzymes. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis of DDX41, R525H, and E345Q stained with Coomassie Blue (
We constructed E345Q, which was expected to be defective in ATP hydrolysis activity based on an analysis of eIF4A3, 16 to confirm the difference in the ATPase activities of DDX41 and E345Q and rule out the possibility that the dsDNA-dependent ATPase activity observed with purified DDX41 was actually due to a contaminant, particularly DnaK. The results of the ATPase activity assay are shown in Figure 2 . Both DDX41 and R525H displayed double-stranded DNA (dsDNA)-dependent ATPase activity, whereas E345Q did not. Analysis of the substrate specificity of DnaK revealed that DnaK did not use dsDNA as a substrate. We defined the luminescent signal of the reaction without dsDNA as indicating 100% inhibitory activity and that of the complete reaction mixture as indicating 0% inhibitory activity and measured the dsDNA-dependent activities of DDX41 and R525H accordingly.
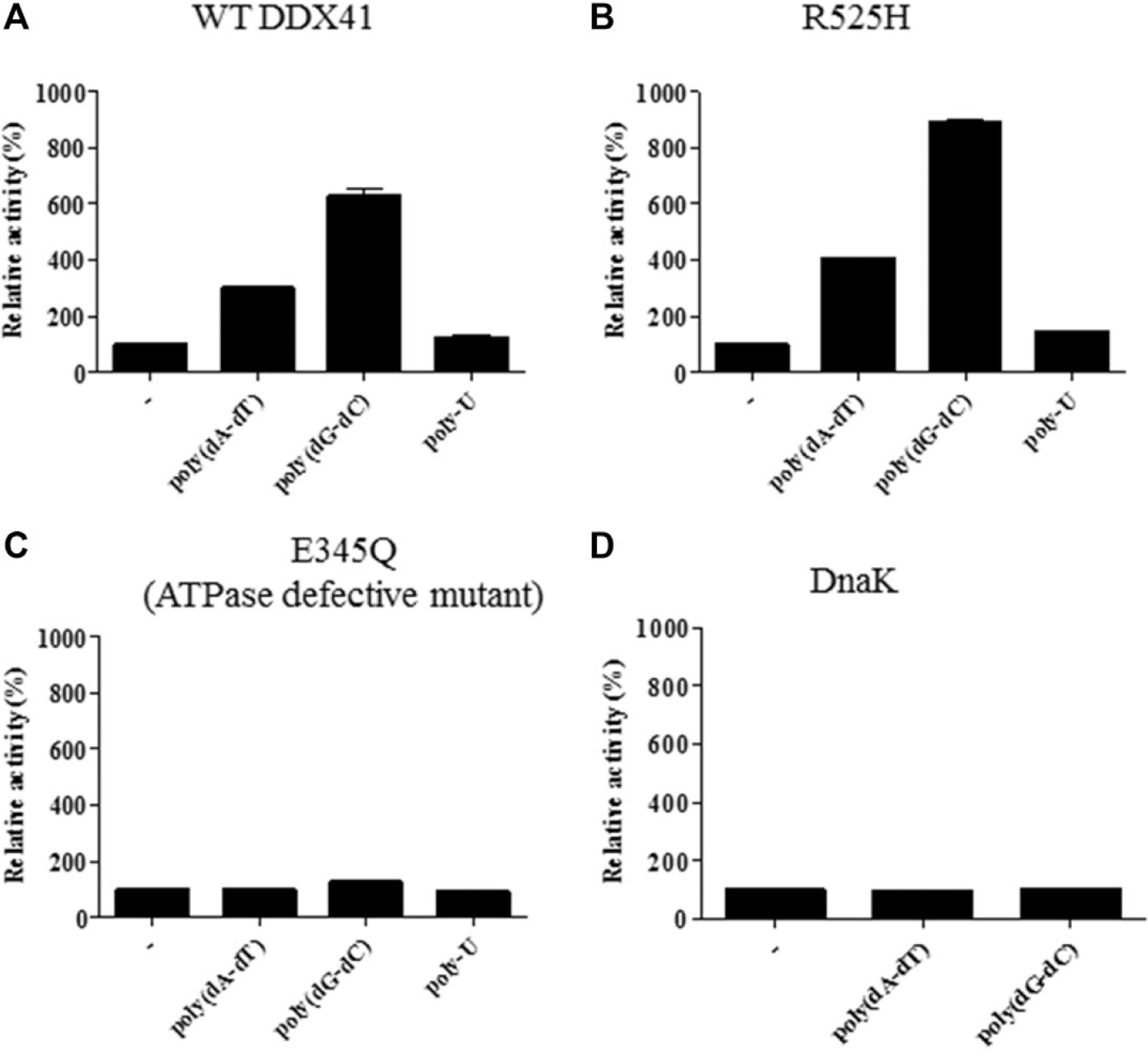
Substrate specificities of DDX41, R525H, E345Q, and DnaK. The substrate specificities were confirmed with 5 µg/mL poly(dA–dT), poly(dG–dC), and poly-U in the presence of 100 µM ATP and 500 nM enzyme. DDX41 (
The apparent Km values for dsDNA and ATP were determined according to the Michaelis–Menten equation, and their values are shown in Figure 3 and Supplementary Tables S3 and S4 , respectively. DDX41 and R525H showed the same affinities for both substrates. The substrate concentrations under the screening conditions were 70 µM ATP and 0.2 µg/mL poly(dG–dC). The concentrations of the substrates were close to the apparent Km values to maximize the diversity of screening hits 17 and to maintain the assay quality throughout the screening process.
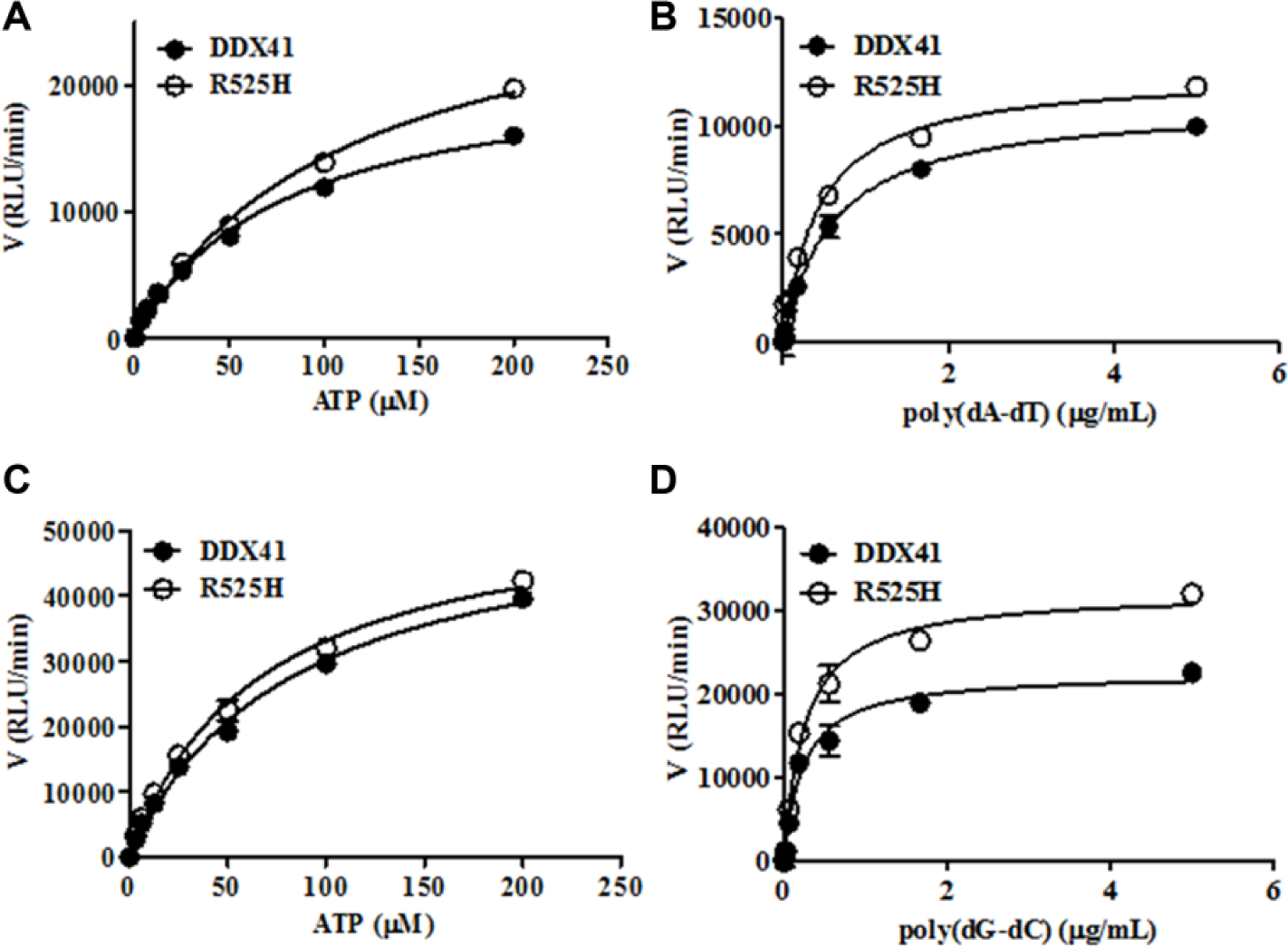
Determination of the kinetic constants for adenosine triphosphate (ATP) and dsDNA (poly[dA–dT] and poly[dG–dC]) in ATPase reactions with recombinant DDX41 or R525H protein. The assay was performed with 5 µg/mL dsDNA and increasing concentrations of ATP (
Identification of Novel R525H Inhibitors with HTS
A diverse library of more than 500,000 small-molecule compounds from the Takeda Pharmaceutical Company Limited was screened at a concentration of 3 µM against R525H, as shown in the workflow ( Fig. 4A ). The assay quality was deemed acceptable because the Z′ factor was 0.78 and the signal-to-noise ratio was 6.1. The histogram of the primary screening in Figure 4B shows a normal distribution curve with a median value of –0.69 and a standard deviation (SD) of 7.8. The hit frequency was 0.5% when the threshold was set at ≥31% of the median + 3SD inhibitory activity. These primary hit compounds through a process of deconvolution were picked, and their inhibitory potency for EIF4A3 was tested with the ADP-Glo assay to eliminate the nonselective compounds. In this step, 12% of the initial hits were eliminated and 180 compounds with no activity against eIF4A3 were selected ( Fig. 5A ). All compounds showed the same inhibitory activities for DDX41 and R525H ( Fig. 5B ), and none of the compounds showed inhibitory activity for DnaK ( Fig. 5C ). Moreover, an in-house database was searched to obtain the compounds, which showed no activity against other target molecules such as kinases, G protein–coupled receptors, and other enzymes, and 43 compounds were selected. The presence of false-positive compounds that interacted as intercalators with poly(dG–dC) was a concern, so an intercalator displacement assay using TO-PRO-3 was performed to remove such compounds. The threshold was set at 29% of the mean of the low control + 3SD of the low control, and a total of 13 of the 43 compounds showed displacement activity ( Fig. 5D ). The assay to remove the compounds quenching of TO-PRO-3 fluorescence was also performed under the condition without poly(dG-dC), and four compounds were eliminated. Ultimately, we identified 26 hit candidate compounds, such as compound 1 [(4E)-N-hydroxy-1-phenyl-2,3-dihydroquinolin-4(1H)-imine, CAS Registry No. 10258-02-3] and compound 2 (4-amino-6-((2-amino-1,6-dimethylpyrimidin-4(1H)-ylidene)amino)-1-methyl-2-phenylquinolinium chloride hydrochloride or the tautomer, CAS Registry No. 115230-55-2), which interacted selectively with DDX41 and R525H from the results described above ( Fig. 5E ). The structures were confirmed by single crystal X-ray analysis ( Suppl. Fig. S1, S2 ), and the purities of the compounds were shown to be 95% or more by liquid chromatography–mass spectrometry.
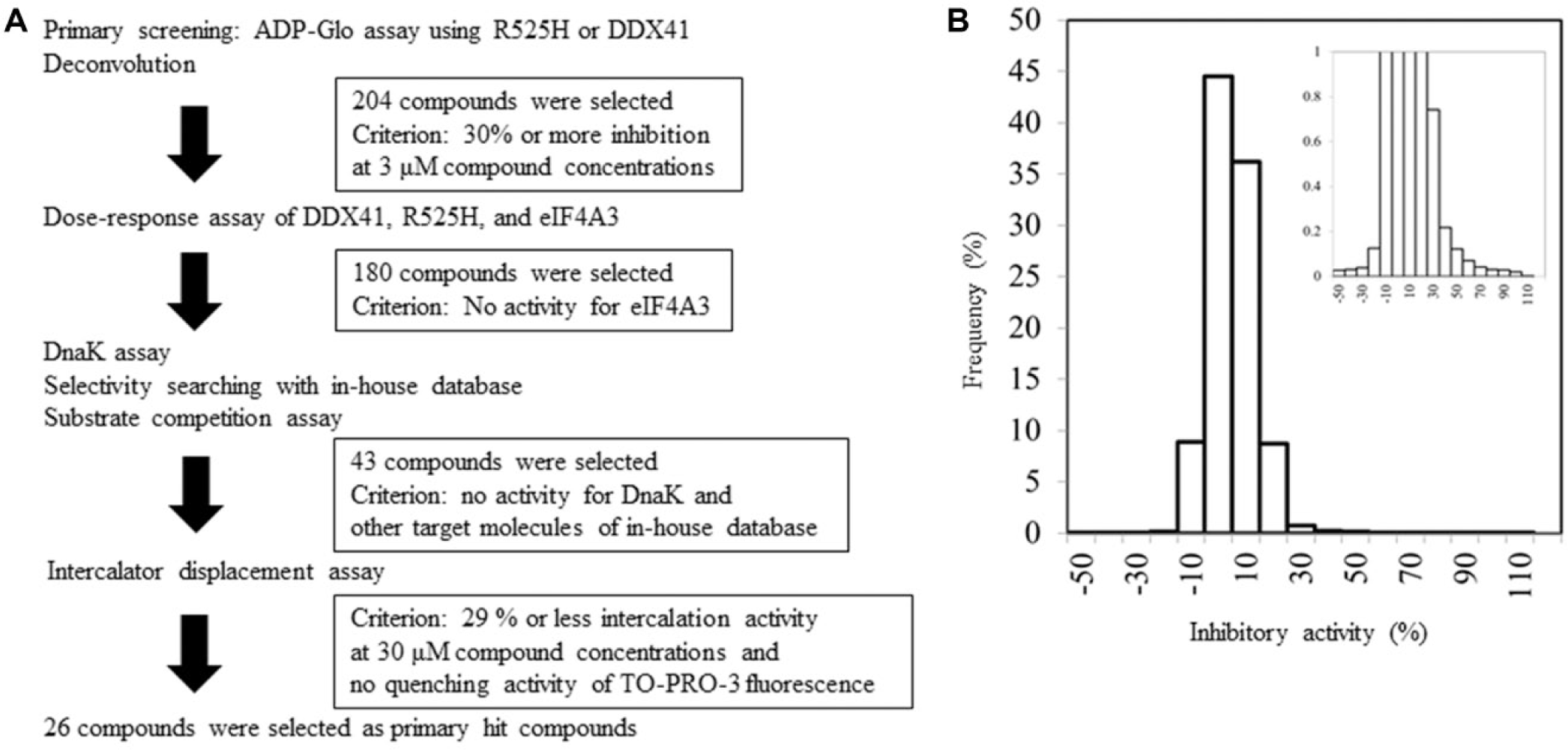
Screening cascade and results of primary screening. The high-throughput screening cascade consisted of the primary screening, profiling assay, hit validation, and characterization (
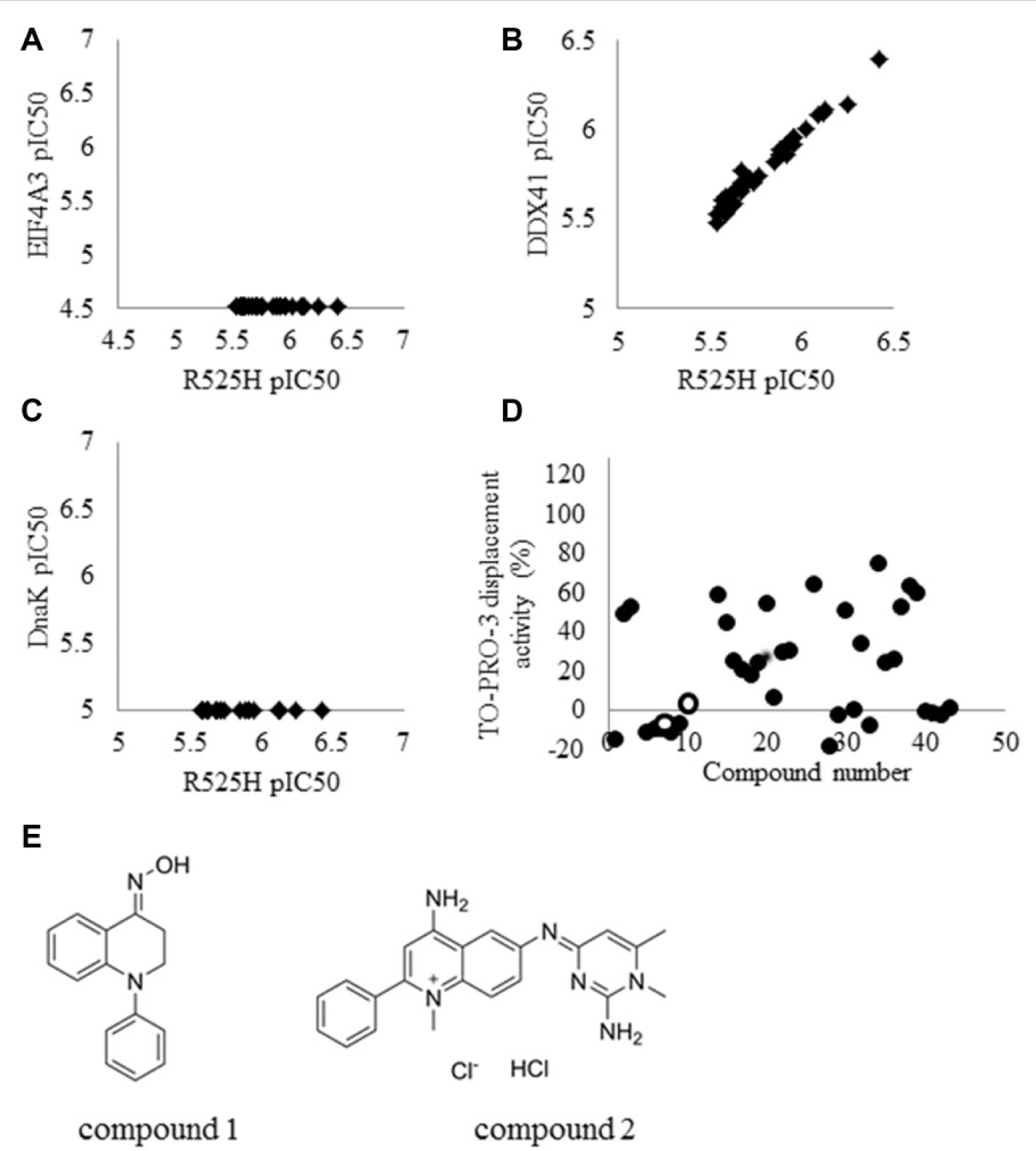
Results of profiling assays. The primary hit compounds were analyzed with a selectivity assay using eIF4A3 (
Characterization of the Hit Compounds
To understand the mechanism of inhibition, substrate competition assays were performed. The rates at which the compounds inhibited R525H were examined under the four conditions described in the Materials and Methods section. Representative hit compounds, exemplified by compound 1 and compound 2, showed the theoretical shift in their IC50 values when the poly(dG–dC) concentration was increased ( Fig. 6A , B ). However, the IC50 values for both compounds shifted only slightly when the ATP concentration was increased. These results indicate that these hit compounds may be competitive inhibitors of DDX41 with respect to the poly(dG-dC) substrate and noncompetitive inhibitors with respect to the ATP substrate ( Fig. 6C , D ). Although compounds such as DNA intercalators were ruled out by the TO-PRO-3 competition assay, there may be other modes of DNA binding besides intercalation, such as minor groove binding, major groove binding, and electrostatic binding to the backbone. Further analyses would be necessary to draw definitive conclusions on the inhibitory mechanism. When the preincubation time (before the reaction was initiated by the addition of the substrates) for the enzyme and the test compound was set at 0 min or 120 min, the inhibitory activities of the compounds remained the same, and it was confirmed that the compounds are not time-dependent inhibitors ( Fig. 6E , F ).
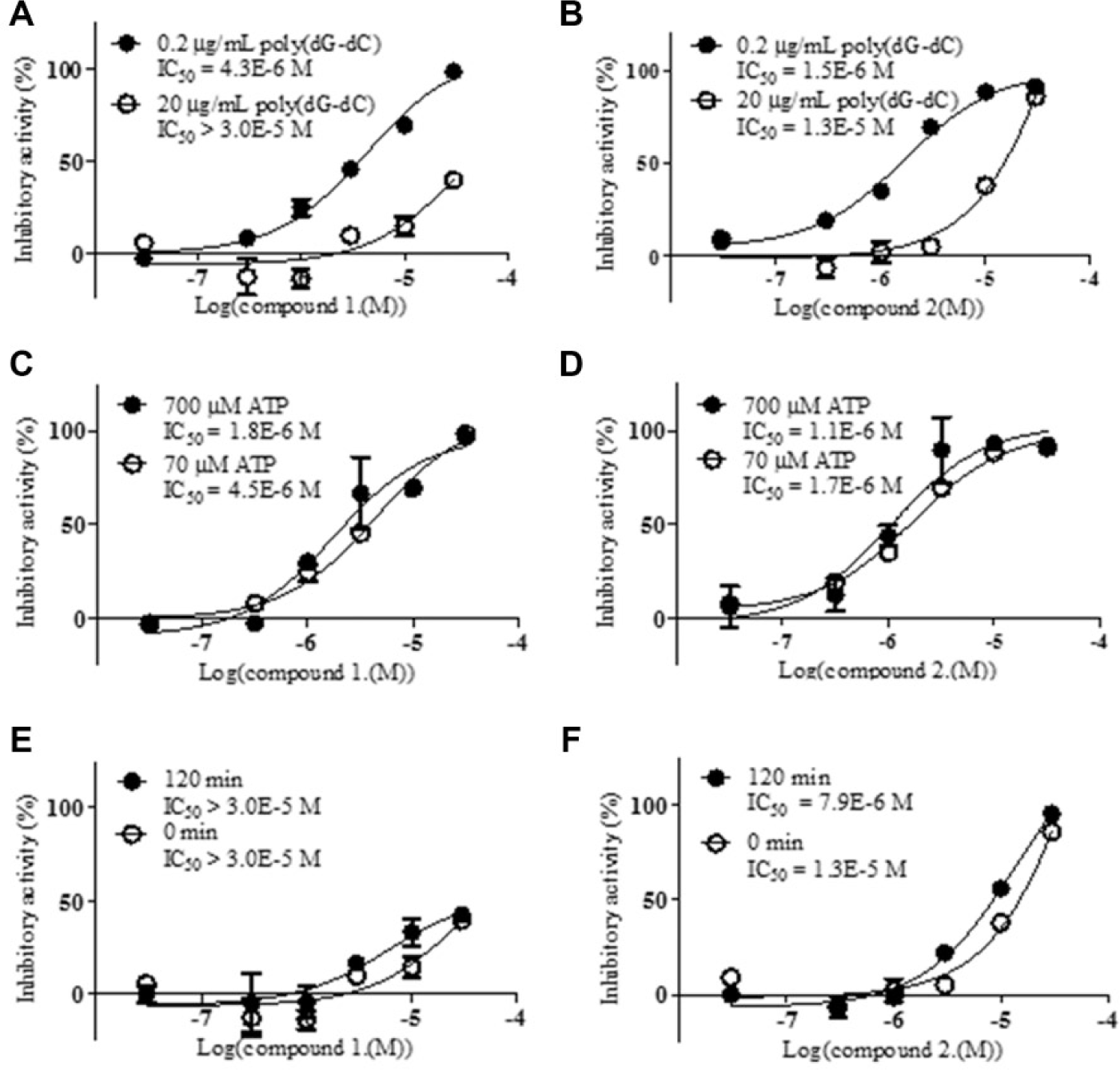
Profiling assays of compound 1 and compound 2 with R525H. The results for compound 1 are shown in (
Discussion
Our results show that the ATPase activities of human DDX41 and R525H are dependent on ATP and dsDNA, either poly(dG–dC) or poly(dA–dT). Novel inhibitors of DDX41 and R525H were identified with HTS. Two hit compounds identified with several assays showed selective inhibitory activities.
DnaK was present in the purified DDX41 and R525H enzymes, detected with mass spectrometry and Western blotting. The DnaK protein is a molecular chaperone and folding catalyst, with ATPase activity in the presence of ATP. 18 We tried to prepare high-purity enzymes by removing the contaminated proteins, such as DnaK, with anion exchange column chromatography. However, this caused the aggregation of DDX41. It is possible that DnaK assists in the folding of DDX41 in E. coli. The activities of DDX41/R525H and DnaK could be differentiated based on their dsDNA-dependent ATPase activities because the human E345Q ATPase defective mutant and DnaK showed no dsDNA-dependent ATPase activity. Therefore, we set assay conditions in the presence or absence of dsDNA to represent the maximum and minimum controls to detect the activities of DDX41 and R525H.
DDX41 may have both ATPase and helicase activities, but no assay for helicase activity has yet been reported. The Km values for dsDNA against DDX41 and R525H were very similar. We speculate that these values would differ if the enzymes were analyzed with helicase activity assays. Pause et al. 16 reported that mutants R365Q and R365K of eIF4A3, whose position corresponds to the conserved residue R525 in DDX41, exhibited dramatic reductions in the helicase activity for dsRNA but did not show a significant difference in ATPase activity. 19 The poly(dG–dC) and poly(dA–dT) used in this experiment were purchased from Sigma, and the lengths of these dsDNAs were not accurately characterized. If the sequence of the dsDNA or dsRNA recognized by DDX41 is determined and a helicase assay is established, the hypothesis that DDX41 and R525H have different helicase activities can be tested. 20
Several compounds were identified with HTS. In parallel with HTS, we developed a selectivity assay to verify the specificity of the enzymatic inhibition. The hit compounds, compound 1 and compound 2, displayed inhibitory activities with IC50 values of 4.3 µM and 1.5 µM, respectively, against human R525H mutant under the HTS conditions used and no activity for eIF4A3 and DnaK. Although we initially selected EIF4A3 because of its ready availability, the further selectivity analysis is necessary using another helicase with more similar sequences with DDX41. In general, the DNA- or RNA-unwinding activities of helicases are dependent on their ATPase activity. Therefore, these hit compounds may also inhibit the helicase activity of DDX41 and R525H. Our HTS results suggest that DDX41 is a potential drug target because no inhibitors of DDX41 have yet been identified. There may be a prominent binding site for small-molecule inhibitors in DDX41 that overlaps the poly(dG–dC)–binding site. The crystal structures of the DDX41-bound inhibitors could clarify their mode of inhibition and facilitate the optimization of lead compounds.
It has been reported that DDX41 interacts directly with several proteins including STING and TBK1, which is a crucial adaptor molecule for cytoplasmic DNA receptors. 10 The silencing of DDX41 expression in murine myeloid dendritic cells led to a marked reduction in IFN and proinflammatory cytokine production in response to cytosolic DNA. 21 Besides its role in immunity, the human R525H mutation is associated with familial MDS and AML. Xenograft experiments with cell lines in which DDX41 was knocked down demonstrated DDX41-enhanced tumor growth, 1 but the mechanism behind this is still not understood. We have identified inhibitors of both DDX41 and R525H, with which it should be possible to conduct biochemical analyses. The clarification of their efficacy in tumor cells, the molecular mechanism of DDX41, and the crystal structure of DDX41 when bound to these compounds will have a significant impact on research into DDX41. If it is possible to create a DDX41- or R525H-selective inhibitor by lead generation, the significance of the mutation could be clarified.
In conclusion, our study provides evidence that DDX41 has dsDNA-dependent ATPase activity, dependent on poly(dG–dC) or poly(dA–dT), and presents new chemical tools for further investigation of the multiple functions of DDX41.
Footnotes
Acknowledgements
We thank Dr. Shoichi Okubo for performing cDNA cloning and Mr. Hironobu Maezaki for advice about compounds. We also thank Dr. Naoki Tarui and Mr. Junji Matsui for their supervision of the research and their valuable suggestions.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.