
Abstract
The bioluminescence resonance energy transfer (BRET) technology is a widely used live cell-based method for monitoring protein-protein interactions as well as conformational changes within proteins or molecular complexes. Considering the emergence of protein-protein interactions as a new promising class of therapeutic targets, we have adapted the BRET method in budding yeast. In this technical note, we describe the advantages of using this simple eukaryotic model rather than mammalian cells to perform high-throughput screening of chemical compound collections: genetic tractability, tolerance to solvent, rapidity, and no need of expensive robotic systems. Here, the HDM2/p53 interaction, related to cancer, is used to highlight the interest of this technology in yeast. Sharing the protocol of this BRET-based assay with the scientific community will extend its application to other protein-protein interactions, even though it is toxic for mammalian cells, in order to discover promising therapeutic candidates.
Keywords
Introduction
Protein-protein interactions (PPIs) govern virtually all cellular processes and thus offer a tremendous panel of opportunities for therapeutic intervention. Targeting PPIs instead of single proteins provides a means to increase drug specificity and efficacy. Because the interface of a particular PPI is formed by the combination of interacting domains of two particular proteins, it will display a higher level of uniqueness in comparison to, for instance, the catalytic pocket of an enzyme, which is often well conserved throughout a whole enzyme class. As examples, all of the active human protein kinases use adenosine triphosphate (ATP) to phosphorylate their substrates. On the market since 2001, imatinib mesylate (Gleevec, Novartis, Basel, Switzerland), which targets the ATP-binding site of the tyrosine kinase Bcr-Abl, was the first targeted therapy developed for chronic myelogenous leukemia (CML). During cancer treatment, point mutations may arise on a single cavity to escape drugs targeting the catalytic pocket while keeping the enzymatic activity. Concerning CML treatment with imatinib mesylate, the main cause of therapy failure is related to mutations affecting principally the ATP-binding cleft and notably a key residue, termed the gatekeeper, located at the back of this pocket. 1 To circumvent such resistance phenomenon, one potential therapeutic strategy can be the inhibition of PPIs required for activation of signaling pathways downstream of Bcr-Abl (strategy explored in ref. 2). Indeed, eluding a PPI inhibitor (P2I2) while preserving the interaction would require a second compensatory mutation in the binding partner, which would be much less probable. The challenge is thus to discover small molecules that disrupt protein-protein complexes. To this end, robust, facile high-throughput methods, preferentially in a living cellular context, are needed (screening assays are reviewed in ref. 3).
The yeast-based bioluminescence resonance energy transfer (BRET) assay described herein offers a powerful method to discover small-molecule inhibitors of PPIs. In this technical note, we use the HDM2/p53 interaction (of particular interest for cancer therapy 4 ) and its small-molecule inhibitor Nutlin-3 to exemplify this scalable method.
BRET Technology in Molecular Screening
BRET is a naturally occurring phenomenon that can be observed in the sea pansy Renilla reniformis and is similar to an existing method for assessing PPI, the Förster or more commonly called fluorescence resonance energy transfer (FRET). In FRET, one fluorophore (the “donor”) transfers its excited-state energy to another fluorophore (the “acceptor”), which emits fluorescence at a longer wavelength. In both methods, the donor and acceptor are genetically or chemically fused to candidate proteins or compounds. In BRET, a luciferase is used as the energy donor to avoid the consequences of donor excitation in FRET (for instance, the damage of tissues by the excitatory light, photobleaching, and simultaneous excitation of the acceptor by the donor excitatory light). In the presence of its substrate, bioluminescence from the luciferase occurs, and the transfer of energy leads to the excitation of the acceptor fluorophore, if the donor and acceptor are close enough (<10 nm), which can occur upon a molecular interaction between the fused proteins. BRET is a highly versatile technique that can be used to measure protein interactions in vitro (using purified proteins, crude cell membranes, or other cell fractions) in cultured cells and in vivo. 5 Based on the BRET method, different screening assays have been designed (reviewed in ref. 6).
Why Is Budding Yeast a Suitable Alternative Model to Set up a Cellular Screening Assay and to Maximize Hit Rates?
The unicellular baker’s yeast Saccharomyces cerevisiae is a proven model for fundamental and applied research. For example, basic cellular processes occurring in human cells are well conserved in yeast (e.g., the control of the cell division cycle). This safe organism is also genetically well defined, because its entire genome was sequenced in 1996. Moreover, yeast growth and division can be precisely controlled, and different strategies can be adopted to maximize hit rates in yeast-based assays: (1) Enhancing the limited cell permeability to small molecules by alteration of yeast membrane composition. Classically, erg6 gene mutation (involved in the ergosterol biosynthesis, one of the components of fungal membranes) was found to increase permeability to a growing list of chemical compounds, in particular to small lipophilic drugs. 7 (2) Screening molecules against nonpreformed complexes.3,8 It is now considered essential that small molecules have to be delivered to the cellular system before the synthesis of one of the two interacting proteins. The use of inducible promoters, such as the GAL1 promoter, enables the small molecules to first interact with one of the two partners, prior to protein complex formation. Indeed, in a BRET study conducted in mammalian cell lines, in which cells were transiently transfected with fusion genes to monitor the HDM2/p53 interaction, the inhibitor Nutlin-3 was not able to completely disrupt a preformed complex. 9 In our experimental setup, the use of inducible protein expression allowed complete inhibition of the HDM2/p53 interaction (see section “Monitoring the Effect of a P2I2 Using a BRET Based Assay in Yeast: The Inhibition of the HDM2/p53 Interaction by Nutlin-3 as Proof of Concept”). (3) The development and selection of stable strains in which the interacting proteins can be expressed, which can be achieved in 1 to 2 wk. This is in sharp contrast to mammalian cells, in which obtaining stable lines is classically time-consuming and sometimes arduous. (4) The possibility of expressing mammalian proteins involved in toxic or death response in mammalian cells. (5) The rapid expansion of yeast compared with mammalian cells, allowing for the acquisition of the proper amount of cells needed to perform high-throughput screening (HTS) experiments in less time. Besides all technical benefits described above, this is one of the main advantages of this model. Indeed, proteins implicated in the cell cycle or apoptosis control may impair the development of such screening assays in mammalian cells. Nevertheless, some limitations exist when using yeast, such as (1) the reduced sensitivity to some classes of compounds due to efficient drug efflux pumps; (2) when overexpressed in yeast, some human proteins can be toxic (e.g., some tyrosine kinases); (3) some protein interactions depend on posttranslational modifications that do not exist in yeast and cannot be screened (e.g., absence of tyrosine phosphorylations in yeast); and (4) some protein interactions might involve folding/conformations that depend on chaperones that may not exist or may not function in yeast. However, easier genetic manipulations of yeast allowed the development of strains expressing human genes counteracting cell death. These models may thus represent a major advantage of yeast for such screening purpose. The study of cyclin-dependent kinase 5 (CDK5)/p25 interaction highlights this key advantage of yeast. 8 Indeed, p25 is a 25 kDa pathological proteolytic fragment of p35, one of the physiological regulatory subunits of CDK5 kinase. A previous study has shown that even low basal level expression of p25 is toxic for mammalian cells, and thus, stable cell lines were obtained only if the tau protein is constitutively coexpressed. 10 In yeast, we were able to establish a stable inducible p25 yeast strain that does not require tau coexpression as a suitable model to study the CDK5/p25 interaction. 8
Here, we detail a robust and scalable BRET-based cellular screening assay developed in the budding yeast, which supports the discovery of inhibitors of PPIs.
Materials and Methods
Chemicals
D(+)-galactose and D(+)-raffinose were purchased from ACROS Organics (Geel, Belgium). Yeast nitrogen base without amino acids and Bacto peptone were purchased from BD Biosciences (Franklin lakes, NJ). D(+)-glucose, Bacto yeast extract, dimethylsulfoxyde (vehicle control), 3,3′5-triiodo-L-thyronine sodium salt (T3), Nutlin-3, Nutlin-3a, FK-506 monohydrate, and the different amino acids complements were purchased from Sigma (St. Louis, MO).
Reagents
Coelenterazine h was purchased from Interchim (Montluçon, France) and Nano-Glo Luciferase Assay Substrate from Promega (Madison, WI). erg6 yeast mutant strain was obtained from Euroscarf No.Y00568 BY4741; MATa; his3Δ 1; leu2Δ 0; met15Δ 0; ura3Δ 0; YML008c::kanMX4 (www.euroscarf.de). The antibodies used are anti-GFP (#ab290; Abcam, Cambridge, UK) and anti-RLuc (#MAB4400; Millipore, Billerica, MA).
Equipment
A Mithras LB940 fluorometer/luminometer (Berthold Technologies, Bad Wildbad, Germany) microplate reader was used with the following emission filters:
RLuc/NLuc: counting time, 2.00 s; emission filter (No. 39450), 480 nm (±10 nm)
eYFP: counting time, 2.00 s; emission filter (No. 39451), 530 nm (±12.5 nm)
Measurement operation is performed by well, and each plate is read three times in a cycling manner.
BRET Calculation
The BRET ratio was calculated by dividing the signal measured at 530 nm by the signal measured at 480 nm. Then, the BRET signal was calculated as the BRET ratio subtracted by the BRET background ratio (obtained when the donor protein was expressed alone) and multiplied by 1000 to express results in milliBRET (mBRET):

Statistical Analysis
Data were expressed as mean ± SD or as percentage ± %RSD (percentage relative standard deviation: SD × 100/mean). Statistical analyses were done by Student t test, and significance levels used are *p < 0.05, **p < 0.01, ***p < 0.001, and n.s. = not significant.
Vector Constructions
The centromeric vectors p415 (LEU2, GAL1 inducible promoter) and p416 (URA3, GPD constitutive promoter) were used to express the chimeric proteins. TRP1 selectable marker gene cannot be employed with erg6 mutant strain, because trp1-erg6 double mutant is synthetic lethal. Donor (HDM2-RLuc or HDM2-NLuc) proteins were cloned into p415, and acceptor proteins (p53-YFP and p53m[F19A]-YFP) were cloned into p416 (see Table 1 ; Suppl. Fig. S1 ) for more details. Both C- or N-terminal fusion vectors are available upon request.
List of the Yeast Plasmids Used in This Study.

Results and Discussion
PPIs control many cellular processes, including metabolic cycles, DNA transcription and replication, enzyme activity, different signaling cascades, and other processes. The importance of PPIs justifies the development of new powerful methods to understand the role of such interactions and to discover inhibitors. The protocol described below is based on the BRET technology, a nonradiative energy transfer, which can be applied to monitor proteins interactions and to identify potential inhibitors. The innovation of this HTS assay lies in the use of the budding yeast S. cerevisiae rather than mammalian cells, which facilitates, expedites, and reduces the cost of assay development.
Feasibility Study and Assay Optimization
A schematic representation of the BRET-based assay is provided in Figure 1A . As depicted, energy transfer occurs when the donor and the acceptor, respectively fused to the interacting proteins, are 1 to 10 nm apart. To monitor the BRET signal, two yeast strains are needed: one strain expressing the energy donor alone (for background detection, here HDM2-Luc) and the other expressing both the donor and the acceptor fusion proteins (here, HDM2-Luc and p53-YFP). An advantage of working with yeast is the possibility of modifying membrane permeability to favor the penetration of small druglike molecules inside the cell. An erg6 strain is thus chosen for the development of the BRET-based screening assay in order to increase the hit rate. Moreover, to screen against a nonpreformed complex, the protein expression of the energy donor is placed under the control of an inducible GAL1 (galactose-regulated) promoter. Once the yeast is transformed, expression of both chimeric proteins should be verified by sodium dodecyl sulfate polyacrylamide gel electrophoresis followed by immunoblotting ( Fig. 1B ). Induction using galactose is an effective way to control gene expression, as shown for HDM2-RLuc.
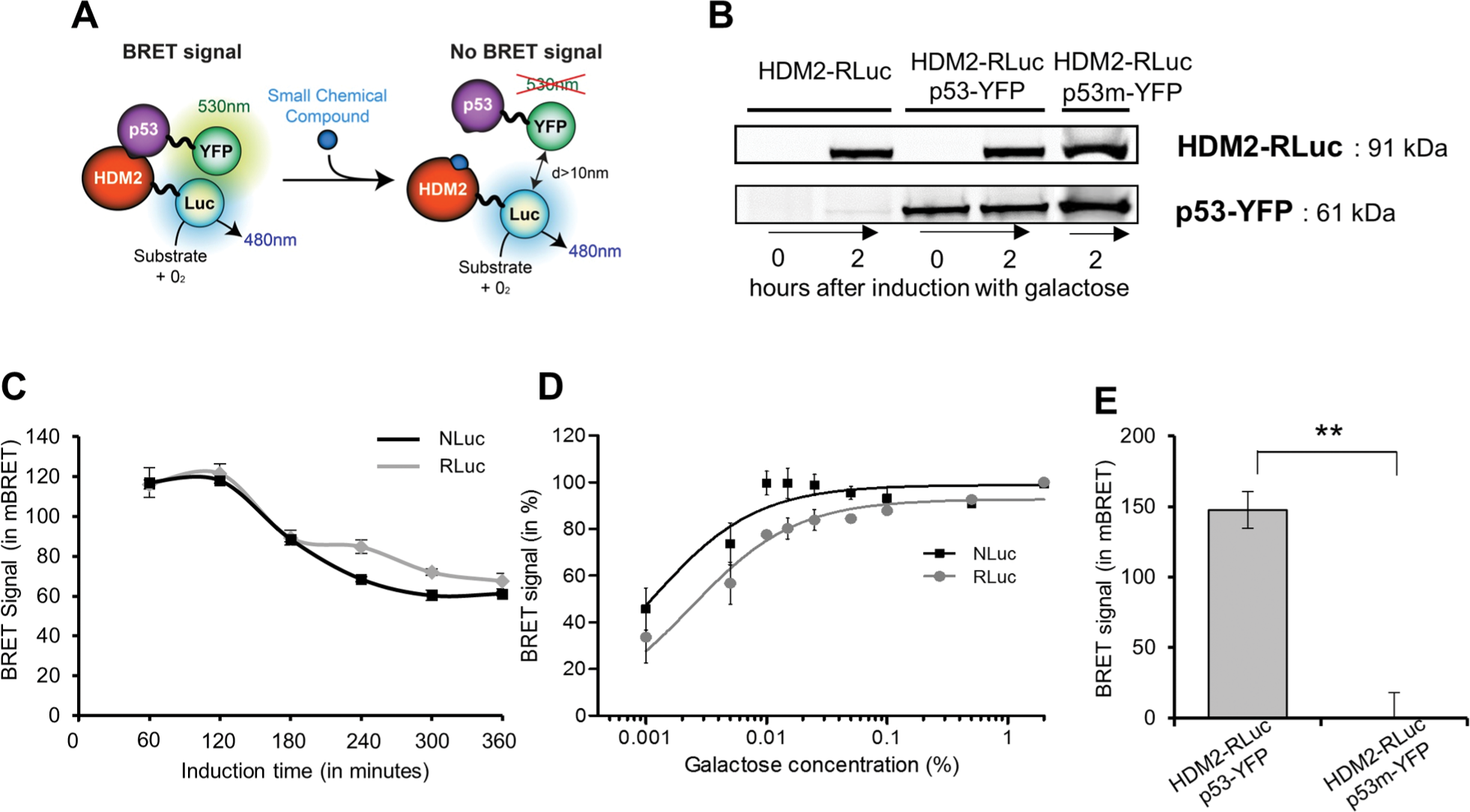
Yeast bioluminescence resonance energy transfer (BRET) technique validation and optimization. (
To obtain a significant BRET signal, donor and acceptor groups should be in closed proximity, but an absence of the BRET signal does not necessarily mean that there is no interaction; it could be due to a nonoptimal orientation of the BRET partners. To circumvent this problem, it is crucial to test different combinations in which the proteins of interest are fused to donor and acceptor at the C- or N-terminal extremities and using several linker peptides with flexibility (for example, GS or GGS repetitions). Indeed, one of the orientations would be favorable over the others and lead to maximal BRET signal. In our case, donor and acceptor groups fused at the C-terminal extremities are considered for screening.
The optimal expression level of donor fusion protein should be evaluated by measuring the BRET signal in response to increasing galactose concentration (from 0.001% to 2%) and time of induction (up to 6 h). This evolution of the BRET signal is protein interaction dependent and must be monitored for each new PPI. The conditions to choose for the screening are those that produce the highest measured BRET signal. The use of other donor proteins can be envisaged. NLuc (NanoLuc, Promega) is a smaller and brighter luciferase (19.1 kDa) than RLuc. It presents a high thermal stability and a strong activity over a broad pH range, and its emission peak (465 nm) is suitable for BRET assay. Moreover, NLuc signal is stable for long time (up to 1 h). Therefore, NLuc was tested in the same way as RLuc. In the case of HDM2/p53, the highest BRET signal was measured after 2 h, in the presence of 2% galactose ( Fig. 1C , D ). As expected, the NLuc signal intensity is higher compared with RLuc (almost 20-fold in our hands) at all inducer concentrations assayed (not shown). The BRET signal was 20 to 50 mBRET higher with NLuc versus RLuc at low inducer concentration (≤0.05%), whereas it was comparable when the donor saturated the acceptor (galactose concentration >0.1%; Fig. 1D ). NLuc can thus be employed usefully to overcome problems of low expression levels of some heterologous proteins.
Another requirement to validate a screening assay is to demonstrate the specificity of the BRET signal, which can be done either by using a mutation that destroys the interaction or by performing a donor saturation assay. For this purpose, the F19A mutation, involving a key residue for the interaction with HDM2, 11 was introduced in p53. It caused a complete loss of the BRET signal, thus confirming the specificity of the signal obtained using wild-type proteins ( Fig. 1E ). Otherwise, when information about a point mutation abolishing PPI is lacking, nonspecific BRET signal, due to random collisions between donor and acceptor, can be measured by cotransforming yeast with YFP alone (cloned in the same vector of the acceptor fusion protein) and donor fusion protein.
In the classical donor saturation assay, the BRET signal is measured in response to increasing amounts of acceptor (achieved by increasing inducer concentration), whereas the donor is kept constant. The expected result for a specific BRET signal is a hyperbolic curve. Nevertheless, inducible donor fusion protein is preferred for HTS, as besides the possibility of directly verifying that the expression of the protein is well induced, another advantage of inducing the donor instead of the acceptor is the ability to reach the maximal BRET signal upon induction. Indeed, donor saturation by the acceptor occurs quite instantly as a huge amount of the acceptor is already present because of its constitutive expression.
Monitoring the Effect of a P2I2 Using a BRET-Based Assay in Yeast: The Inhibition of the HDM2/p53 Interaction by Nutlin-3 as Proof of Concept
p53, also called the guardian of the genome, is negatively modulated by HDM2. Disrupting the HDM2/p53 interaction may thus offer a new strategy for cancer therapy. To this end, in 2004, the chemical compound Nutlin-3 was discovered using surface plasmon resonance. 12
We have examined the effect of Nutlin-3 on the HDM2-p53 BRET signal in our assay. The BRET signal decreased in a dose-dependent manner ( Fig. 2A ). As shown in Figure 2B , the inhibitory effect of Nutlin-3 and its enantiomer Nutlin-3a 12 has also been verified using NLuc as donor. As expected, Nutlin-3a, known to be more potent on the HDM2/p53 inhibition than Nutlin-3, showed a higher inhibitory effect (Nutlin-3 IC50 = 28.6 µM, Nutlin-3a IC50 = 8.8 µM) on the HDM2/p53 interaction.
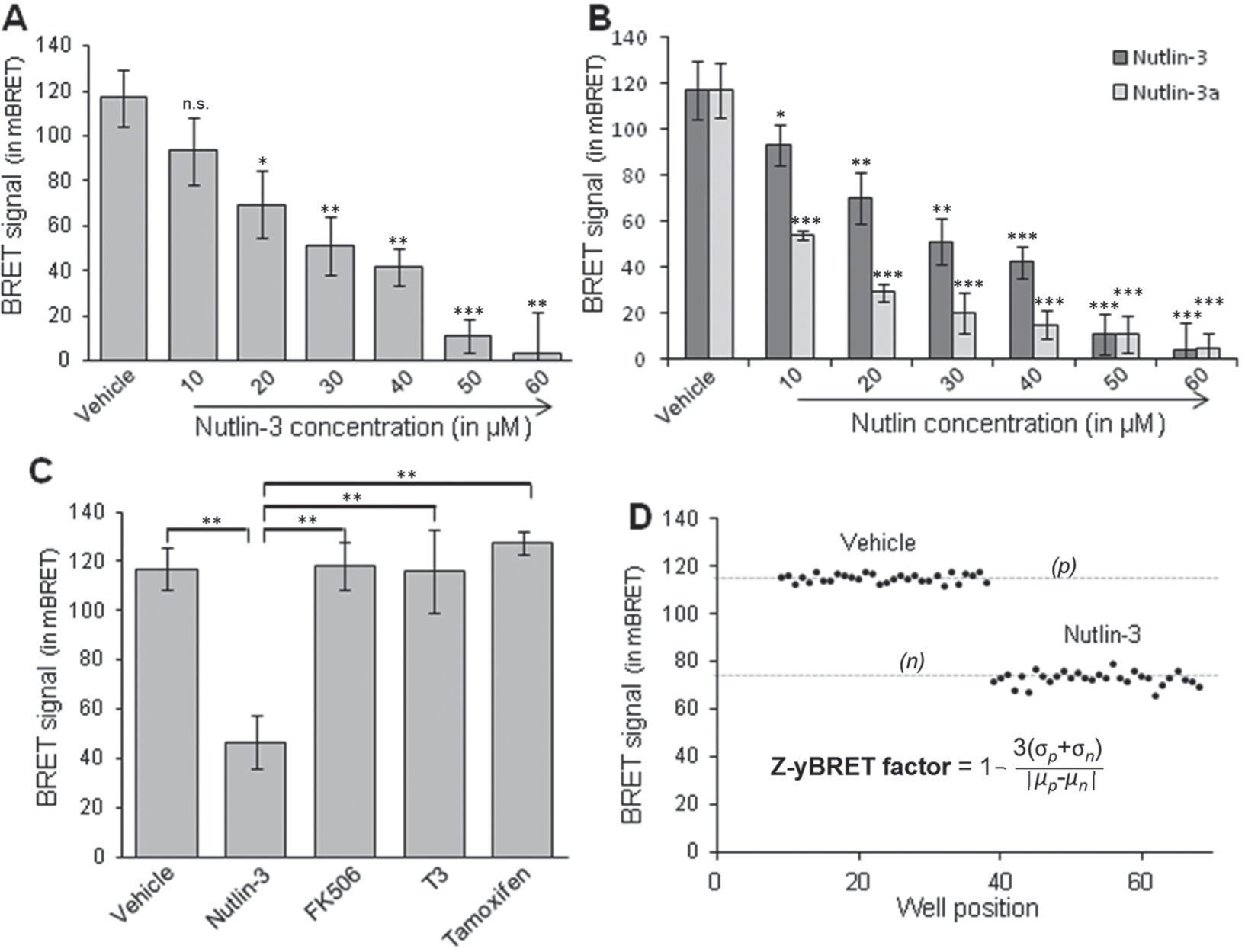
Monitoring the effect of a P2I2 using bioluminescence resonance energy transfer (BRET) in yeast. (
The effect of other known PPI inhibitors was also evaluated on the HDM2/p53 interaction using this BRET-based assay. As shown in Figure 2C , no effect was observed on the BRET signal using FK506, T3, or tamoxifen, which inhibit the FKBP12/TGFβR, NCoR/TRα, and CDK5/p25 13 interactions, respectively.
Prior to starting a large screening campaign, pilot screenings are used to predict if it would be feasible in a high-throughput setting. For this purpose, the calculation of a coefficient called the Z′-factor is recommended (see ref. 14 for details). This statistical dimensionless parameter is essential to evaluate and validate the quality of HTS assays. The Z′-factor is defined in terms of four parameters: the means and standard deviations of both the positive (p) and negative (n) controls (µp, σp and µn, σn). Usually, the negative control is determined without the tested enzyme or using noninduced conditions. Considering this yeast-based screening assay, it is experimentally inconceivable to use the same yeast strain to obtain a “noninduced” negative control. Indeed, in the noninduced condition (yeast growing in raffinose-based media without galactose), no reliable BRET signals can be achieved. We therefore decided to adapt the calculation using a known inhibitor, Nutlin-3. To characterize this estimated coefficient (named Z-yBRET factor in Fig. 2D ), we prepared a 96-well plate and measured the BRET signal obtained with the compound vehicle (DMSO; p, positive controls) or 20 µM of Nutlin-3 (n, negative controls) on the HDM2/p53 interaction. The formula used for the calculation is reported in Figure 2D and is similar to the one of the Z′-factor. 14 According to the scatter plot obtained, we observed a significant difference between the two sets of samples (separation band), confirming that this method is adapted for screening, with a calculated factor coefficient of 0.67 ( Fig. 2D ). This value indicates a suitable difference between maximal signal and inhibited values (or background) together with low variability. Moreover, when the more efficient inhibitor Nutlin-3a was used, the calculated factor coefficients were 0.70 and 0.85 using RLuc or NLuc as donor, respectively (not shown).
Nevertheless, if any P2I2s are already described for the targeted PPI, values obtained for strains transformed with Luc-fusion protein and YFP alone can be also used as negative control. For our model interaction, HDM2/p53, we obtained a factor coefficient of 0.80 (using RLuc as donor protein) and 0.94 (when using NLuc; not shown). These results indicate that this yeast-based screening method is suitable for use in a full-scale HTS.
Screening
On day 1, 10 to 20 yeast colonies were picked from selection plates (SD-Ura-Leu media, SD: synthetic dextrose medium) and inoculated in liquid media (see Suppl. Data for a step-by-step detailed protocol). The two yeast strains, transformed by the donor and acceptor expression vectors or the donor alone and the empty acceptor plasmid p416GPD, were grown overnight (12 h) at 29 °C in 5 mL of liquid SR-Ura-Leu (SR: synthetic raffinose medium). Raffinose 2% (final concentration) is a carbon source that neither represses nor induces the Gal promoter and that allows prompt donor expression upon galactose (inducer) addition. On day 2, the cultures were diluted to an optimal optical density of 1 (A600nm = 1) suitable for screening in fresh SR-Ura-Leu. Then, the 96-well plates were filled with 36 µL of yeast culture (according to the plate map, see Fig. 3 ). As depicted, the first and 12th columns of the 96-well plate were dedicated to controls, and eight wells were filled by the control strain expressing the BRET donor only (used for the background BRET ratio calculation). Then, 0.4 µL of the tested molecules, at the appropriate concentration, was added. Alternatively, and depending on liquid-handling systems available in the lab, yeasts can be added to the 96-well plate already containing the tested compounds. We experimentally observed that yeast tolerates up to 3% of DMSO (solvent for chemical compounds). Eighty different compounds can be analyzed per 96-well plate. Finally, to induce the GAL1 promoter, 3.6 µL of galactose was added from a 10× stock solution (20% or otherwise optimized), and the plate was placed in a shaking platform for 2 h at 29 °C. We used here the induction time optimized as described in section “Feasibility Study and Assay Optimization,” and reported on Figure 1C . A few minutes before the end of the incubation, a fresh dilution of luciferase substrate was prepared. For RLuc donor substrate, coelenterazine h was solubilized in ethanol and diluted in phosphate-buffered saline (PBS) to 5 µM final concentration in the wells. The Nano-Glo Luciferase Assay Substrate was first diluted in PBS and used as 5000× final dilution in the wells. The multiwell plate was then loaded in the BRET reader, and BRET values were determined as described in section “BRET Calculation.” The analysis of a 96-well plate is represented in Figure 3 . A positive hit was defined as a chemical compound producing a significant inhibition of the BRET signal. To avoid false-positives, standard deviation obtained on 96 wells can be used to set a threshold; for example, in our experiments, a decrease of 25% of the BRET value was considered as an inhibited signal: this value was willingly higher than 3× the standard deviation of control samples. Special attention should be paid to those molecules that might interfere with the absorption properties of the BRET-based assay (fluorescent, colored compounds). To distinguish between false-positive hits or bona fide specific inhibitors, these compounds should be tested against a BRET signal produced from an unrelated PPI. In case a hit molecule is identified, it should be confirmed by a new BRET measurement performed in triplicate and also tested on the control strain expressing only the energy donor, to confirm that it does not interfere with the luciferases or the luciferins (substrates; see details in Suppl. Data ). This homogeneous assay (no wash steps are required) can be easily adapted to perform HTS of large libraries of chemical compounds. With this protocol, up to 800 compounds can be screened manually per day.
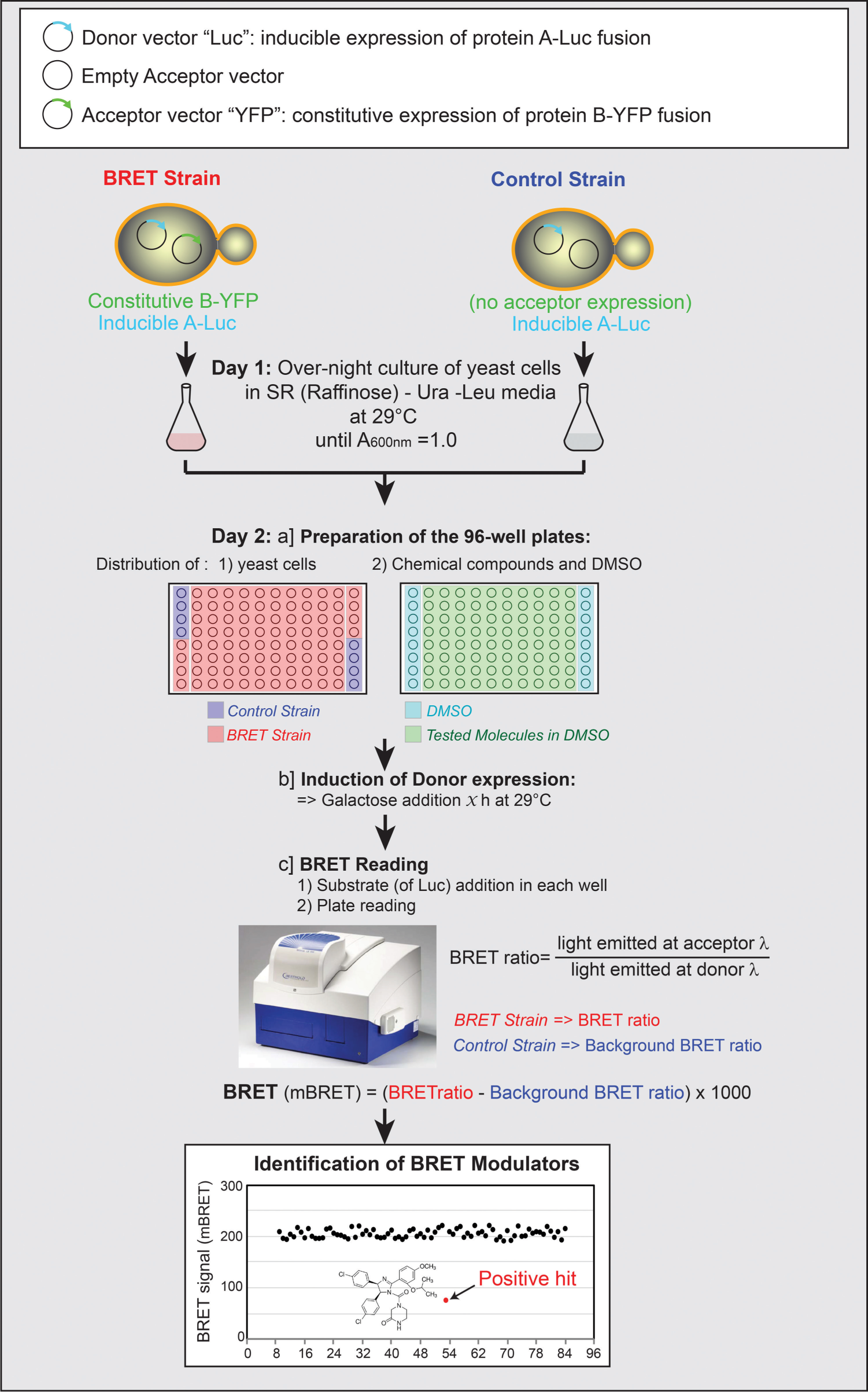
Workflow of the proposed bioluminescence resonance energy transfer (BRET)–based screening assay in yeast. In the first (A1 to H1) and in the 12th column (A12 to H12) of each 96-well plate, strains expressing the protein A-Luc donor fusion protein alone (control strain, A1:D1, and E12:H12) and both donor and acceptor fusion proteins (BRET strain, E1:H1, and A12:D12) are treated with DMSO as controls. The other 80 wells are dedicated to the screening of 80 compounds. Luc = RLuc or NLuc.
In this article, we used Nutlin-3 and Nutlin-3a, known inhibitors of the HDM2/p53 interaction, to validate the yeast BRET-based assay as a robust method to identify P2I2s. Indeed, as proof of concept, the cell-based screening assay described here has permitted the discovery, among more than 5000 compounds tested, of first-in-class chemical structures that can inhibit the CDK5/p25 interaction involved in various human diseases and notably in Alzheimer’s disease.8,13 Among these inhibitors, tamoxifen was discovered, and additional studies have demonstrated that treatment of neuronal cells with this compound affects Tau phosphorylation, a substrate of CDK5. 13 Contrary to these previous research articles, we comprehensively describe here all steps of the protocol (including putative troubleshootings), the statistical Z′-factor (adapted to BRET in yeast, the Z-yBRET factor), and finally the possibility of designing alternative versions of the screening assay by modifying the donors. In this study, we have improved this yeast BRET protocol by using the high-efficiency small donor luciferase NLuc rather than the RLuc used previously. This results in higher protein expression and signal intensity/stability and represents a significant improvement when dealing with poorly expressed heterologous proteins. New donor/acceptor couples are still being discovered and optimized, and their use may further enhance the BRET signals produced in our yeast PPI assay. Important advances have been made with the Nano lanterns, which are direct donor-acceptor fusion proteins optimized for BRET-based high bioluminescence. 15 New RLuc8 mutants fused to mutated acceptors such as mVenus, mTurquoise, and mKusabiraOrange2 lead to a BRET signal more than 8000 mBRET. 15
The protocol described herein associated with new advances in BRET couples should contribute to the discovery of new promising therapeutic candidates that inhibit a variety of PPIs.
Abbreviations
ATP Adenosine triphosphate
BRET Bioluminescence resonance energy transfer
CDK Cyclin-dependent kinase
CML Chronic myelogenous leukemia
FKBP12 12 kDa FK-506 binding protein
FRET Förster/fluorescence resonance energy transfer
HDM2 Human double minute 2
HTS High-throughput screening
mBRET milliBRET
NCoR Nuclear receptor co-repressor
NLuc Nano luciferase
P2I2 Protein-protein interaction inhibitor
PBS Phosphate-buffered saline
PPI Protein-protein interaction
RLuc Renilla luciferase
SD Synthetic dextrose medium
SR Synthetic raffinose medium
TGFβR Transforming growth factor β receptor
TR T3/Thyroid hormone receptor
YFP Yellow fluorescent protein
Footnotes
Acknowledgements
We would like to thank Marc Blondel for providing yeast vectors and Anne Mazars and Robin Fahraeus for materials related to p53 and HDM2. “USR3151 unit Roscoff” including the KISSf screening facility is supported by the following French networks: Biogenouest, IBiSA, and Cancéropôle Grand-Ouest (axis: natural sea products in cancer treatment).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by University of Parma FIL 2014 grant to Barbara Montanini and by a grant from the Interuniversity Consortium for Biotechnologies (CIB) for postdoctoral fellowship to Elisabetta Levati, Caroline Corbel is supported by the Région Bretagne (ALZACRIB project), and Stéphane Bach was supported by ARC (contract ARC3889), ANR/Investissements d’Avenir program by means of the OCEANOMICs project (grant No. ANR-11-BTBR-0008) and INCa (“NECROTRAIL” Program).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.