
Abstract
Protein–protein interactions (PPIs) are attractive but challenging targets for drug discovery. To overcome numerous limitations of the currently available cell-based PPI assays, we have recently established a fully reversible microscopy-assisted fluorescent two-hybrid (F2H) assay. The F2H assay offers a fast and straightforward readout: an interaction-dependent co-localization of two distinguishable fluorescent signals at a defined spot in the nucleus of mammalian cells.
We developed two reversible F2H assays for the interactions between the tumor suppressor p53 and its negative regulators, Mdm2 and Mdm4. We then performed a pilot F2H screen with a subset of compounds, including small molecules (such as Nutlin-3) and stapled peptides. We identified five cell-penetrating compounds as potent p53–Mdm2 inhibitors. However, none exhibited intracellular activity on p53–Mdm4. Live cell data generated by the F2H assays enable the characterization of stapled peptides based on their ability to penetrate cells and disrupt p53–Mdm2 interaction as well as p53–Mdm4 interaction.
Here, we show that the F2H assays enable side-by-side analysis of substances’ dual Mdm2–Mdm4 activity. In addition, they are suitable for testing various types of compounds (e.g., small molecules and peptidic inhibitors) and concurrently provide initial data on cellular toxicity. Furthermore, F2H assays readily allow real-time visualization of PPI dynamics in living cells.
Introduction
Protein–Protein Interaction Assays in Drug Discovery
Protein–protein interactions (PPIs) mediate virtually every cellular function. Essential processes are guided by a complex spatial and temporal interplay of proteins. Because PPIs are affected in various diseases, including cancer, diabetes, and viral infections, therapeutically important PPIs are attractive targets for drug discovery.1, 2
To analyze PPIs, a variety of test systems has been established (reviewed in Colas 3 ). Numerous biochemical assay technologies are available to study PPIs in vitro. These assays are relatively simple and reliable, and some of them can be scaled up for high-throughput screening (HTS). Their drawback, however, is that they evaluate isolated and concentrated proteins lacking their native context. In contrast, cell-based assays offer superior information on a compound’s activity in a physiologically relevant cellular environment.4,5 Because cellular assays are characterized by higher complexity and variability, they often demand advanced technical expertise and frequently suffer from lower throughput.
Probably the most widely used technique to analyze PPIs intracellularly is the yeast two-hybrid (Y2H) system. 6 Y2H is easily scalable; however, it is known also to generate numerous false positives. 7 Furthermore, a mammalian system would be more favorable for drug discovery (e.g., to preserve the human posttranslational modification patterns). One elegant approach to detect PPIs in mammalian cells is based on protein complementation that reconstitutes a functional enzymatic or fluorescent reporter from the interaction of two proteins or protein fragments. 8 Because the reconstitution is often irreversible, this method is not well suited to screen for compounds disrupting an interaction once it is formed. An alternative approach applies Förster resonance energy transfer (FRET) and bioluminescence resonance energy transfer (BRET) assays. 9 For FRET, a robust and affordable solution for HTS is not available yet, whereas BRET has been repeatedly reported to be applied in HTS. 10 Small changes in the positioning of the donor and acceptor, however, result in signal decline, which does not necessarily reflect a splitting of the PPI under investigation.9 –11 Finally, microscopy-based methods have been introduced that take advantage of subcellular protein redistribution. The use of such assays is restricted to proteins that undergo subcellular translocation on interaction or are engineered to do so. 12 In summary, no ideal system to monitor reversible PPIs in mammalian cells is available yet. Typically, several methods have to be combined to reliably screen for inhibitors of PPIs and confirm the mechanism of action in independent systems (e.g., Chen et al. 13 and Zhai et al. 14 ).
F2H Assays to Evaluate PPI Inhibitors
As an alternative to currently available cell-based PPI assays, we recently developed a microscopy-assisted fluorescent two-hybrid (F2H) assay. 15 The F2H principle uses the tethering strategy, taking advantage of an array of lac-operator sequence repeats stably integrated into the genome of specifically modified mammalian cells.16,17 The lac-operator repeats from an inert PPI platform. A green fluorescently tagged bait protein is immobilized at the platform via its fusion to the lac-repressor (LacI) protein. Thus, the LacI region anchors the fluorescent bait on the DNA region containing the lac operator, resulting in the formation of a clearly detectable “green spot” in the cellular nucleus.
To assess the degree of PPI, co-localization of the red fluorescent prey protein at the same spot is evaluated. This optical readout of the F2H assay is straightforward and reliable, which makes it suitable for automated image acquisition, segmentation, and analysis. The nuclear binding platform in the F2H assay provides an increased signal window because the two co-localizing PPI partners become visible at a precise single spot. In contrast, other comparable assays exploit the translocation or co-localization of proteins within the blurry space of the whole nucleus.12,18 Moreover, the F2H assay enables examination of not only PPIs in static endpoint assays but also their dynamics in living cells at real time.
To date, the F2H principle has been validated on more than 50 different interactions, including cell cycle–dependent PPIs.15,19,20 Proteins from different subcellular compartments, such as the nucleus, cytoplasm, and mitochondria, have been successfully subjected to F2H analysis.
p53–Mdm2 as a Model Interaction to Test in F2H
For our screening approach, we have chosen the widely studied interactions between the tumor suppressor p53 and its regulatory binding partners, Mdm2 and Mdm4. These important interconnected interactions are implicated in the pathogenesis of various cancers. They have been the focus of many pharmaceutical companies and academic institutions in recent decades. Until now, however, there has been a lack of reversible cell-based assays for concurrent analysis of both PPIs: p53–Mdm2 and p53–Mdm4. Our aim was to develop two comparative assays, based on the F2H principle, that enable side-by-side analysis of antagonistic compounds.
p53 plays a major role in cell cycle control, apoptosis, and senescence. In cancer, up to 50% of tumors exhibit p53 mutations, mainly in the DNA-binding domain, whereas the remaining half of tumors display various other mechanisms to inactivate p53. 21 p53 transactivates the expression of Mdm2, which is a negative feedback loop and the major mechanism to control p53. Mdm2, also known as Hdm2, not only can bind directly to p53 and inhibit its transcriptional activity, but also targets p53 for degradation by ubiquitinylation. Furthermore, the structural homolog Mdm4, also known as MdmX or HdmX, can bind to Mdm2, increasing the efficiency of p53 ubiquitinylation. At the same time, Mdm4 can also bind directly to p53, inhibiting its transactivational property. Both Mdm2 and Mdm4 are oncogenes, and they are frequently overexpressed in tumors with nonmutated p53. 22 To reactivate wild-type p53 in tumors, peptides and small molecules, such as Nutlins, have been developed that inhibit the interaction of p53 and Mdm2.22,23 So far, however, most identified compounds (e.g., Nutlin-3) show poor antiproliferative activities when Mdm4 is overexpressed. Therefore, to identify dual Mdm2–Mdm4 antagonists is a valid route for drug discovery. 22
Outline of the p53–Mdm2 and p53–Mdm4 F2H Screens
In this work, we first developed the p53–Mdm2 assay and the complementary p53–Mdm4 PPI assay based on the F2H principle. Then, we used these F2H assays to evaluate a collection of 19 “blinded” small-molecule compounds selected from an ELISA-screened library and provided by Janssen Pharmaceutica. Nutlin-3 served here as the positive control to inhibit the p53–Mdm2 interaction. 23 Because no small-molecule compound is available yet to efficiently inhibit the p53–Mdm4 interaction (in cells), we validated the reversibility of this interaction with a peptide-based approach. 24
In this article, we describe and validate how the two assays enable comparative cell-based screening for dual-action inhibitors. Furthermore, we demonstrate that these assays evaluate the activity of the test compounds in the intracellular environment in combination with their cell permeability and cellular toxicity.
Materials and Methods
DNA Constructs
The F2H-bait and F2H-prey vectors and the generic sequences for the cloning primers are described in detail in
15
and .
25
These vectors served for generating the following constructs: LacI–GFP–p53, an in-frame fusion of LacI, NLS–TagGFP (Evrogen, Moscow, Russia), and the 1–81 amino acid (aa) protein region of the human TP53 (GenBank: BAC16799.1); RFP–Mdm2, a fusion of mCherry and a 7–134 aa protein region of the human MDM2 (GenBank: AAM78554.1); and RFP–Mdm4, a fusion of mCherry with a truncated version (1–129 aa) of MDM4 (GenBank: AAB62928.1). More details can be extracted from the
Cell Culture and Transfections
Transgenic baby hamster kidney (BHK) cells containing multiple lac-operator repeats 16 were cultured in DMEM (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS) and 50 μg/ml gentamycin (PAA Laboratories, Piscataway, NJ) in humidified 37 °C, 5% CO2 incubators. For microscopy, cells were split to 50% confluence into μClear 96-multiwell plates (No. 655090, Greiner, Frickenhausen, Germany) and simultaneously co-transfected with the expression constructs using Lipofectamine® 2000 (Life Technologies, Carlsbad, CA) according to the manufacturer’s instructions. Typically, the transfection efficiency for double-transfected cells ranged between 5% and 10%. Experiments with less than 2% double transfectants were discarded.
Incubations with Compounds and Peptides
For the endpoint F2H assays, the chemical compounds were added into cell growth media in increasing concentrations 20 h after transfection, followed by incubation for 3 h under normal cell-growth conditions. Then, the cells were fixed with 3.7% formaldehyde in phosphate-buffered saline (PBS) for 10 min at room temperature and counterstained with DAPI (4′,6-diamidino-2-phenylindole) to highlight the nuclei. For the live-imaging experiments, the compounds were applied in concentration as specified in the text, and imaging was carried out during the treatments.
Compounds 1–19 were provided by Janssen Pharmaceutica. Nutlin-3 23 was taken as a reference inhibitor of p53–Mdm2 (Cayman Chemical, Ann Arbor, MI). For the p53–Mdm4 interaction, the stapled peptides sMTide-02 and sMTide-02A 24 were used to validate the assay. The incubations with the peptides were carried out for 8 h in a serum-free medium. All stock solutions were prepared at 5 or 10 mM in dimethyl sulfoxide (DMSO).
Microscopy and Analysis
Image acquisition was performed automatically at 30 frames per well using an IN Cell Analyzer 1000 (GE Healthcare, Little Chalfont, UK), and settings are specified in the
Enzyme-Linked Immunosorbent Assay (ELISA)
A detailed description of the materials and the ELISA protocol is included in the
Passive Permeability and Transepithelial Transport Assay
LLC-PK1 pig kidney epithelial cells were stably transfected with the human multidrug-resistance gene (MDR-1) to express the ABC transporter P-glycoprotein (P-gp). 26 LLC-PK1–MDR1 cells were seeded onto 24-well cell-culture inserts (Millicell®-PCF, 0.4 µm, 13 mm ∅, 0.7 cm2; EMD Millipore, Billerica, MA) at 400,000 cells/cm2 and cultured for 5 days in Medium 199 supplemented with 10% FBS and 100 U/ml penicillin–streptomycin. On the day of the experiment, test compounds were diluted in OPTI-MEM (Life Technologies, Carlsbad, CA) with 1 w/v % bovine serum albumin to final stock concentrations of 1 µM. These compound stock solutions were then applied to the apical side of the cellular monolayers, and transport in the apical-to-basolateral (A-to-B) direction was assessed following a 120 min incubation period (37°C, 5% CO2) in triplicate. Samples from the acceptor and donor compartments were collected, and absolute test-compound concentrations were measured using liquid chromatography–tandem mass spectrometry, and quantified via a calibration curve. Apparent permeability (Papp × 10−6 cm/sec) was measured for each test compound in the presence and absence of GF120918 (5 µM). 27 Monolayer integrity was assessed by including fluorescein (1 µM). Reference compounds carbamazepine and loperamide (substrate for P-gp 27 ) were included as positive controls for high permeability and P-gp functionality.
Results
In Vitro Selection of the Test Compounds
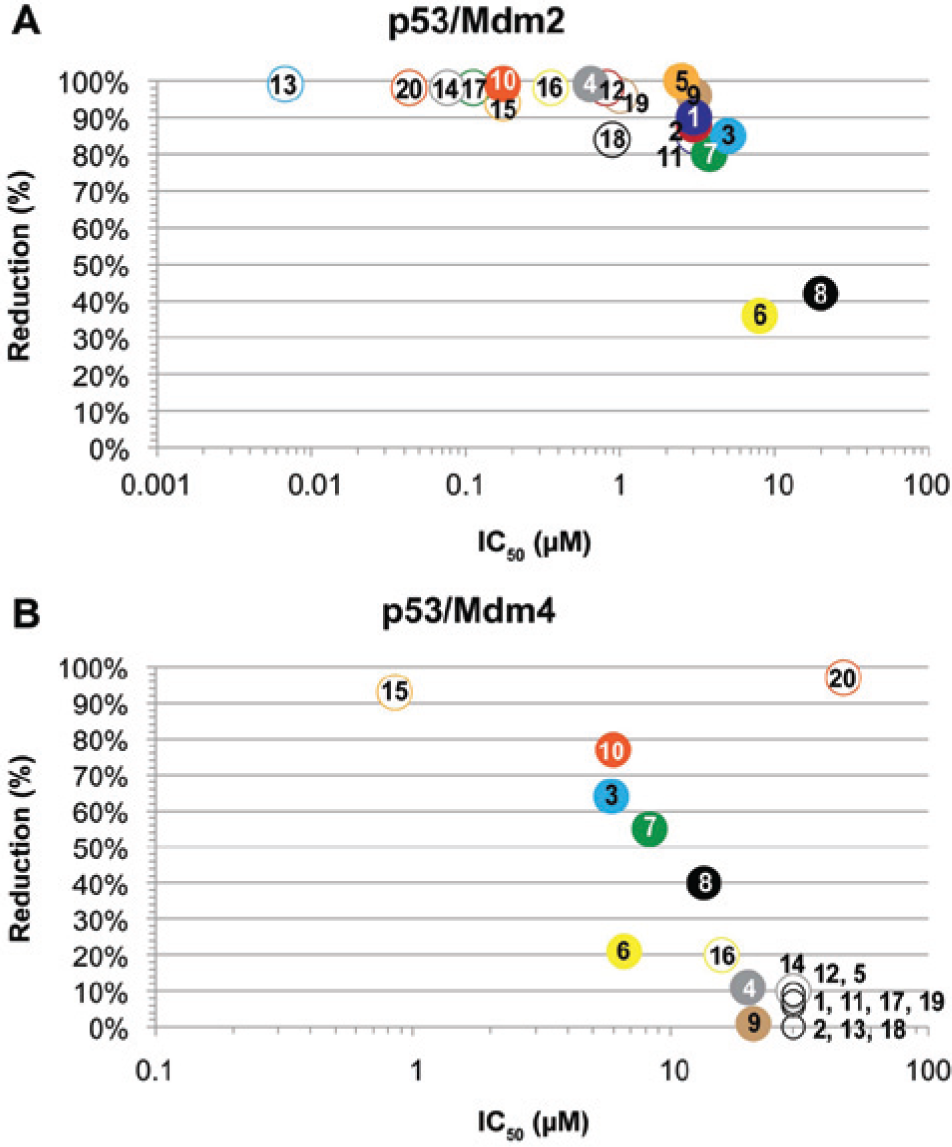
Compounds have been derived from sandwich ELISA experiments in which p53 was coated onto the surface of wells of 96-well microtiter plates. To allow blocking of the p53-binding site and/or inducing of structural changes before complex formation, Mdm2 or Mdm4 was pre-incubated with the compounds prior to addition to the p53. PPIs were detected and quantified by applying antibodies against Mdm2 or Mdm4. Nineteen compounds of various chemical classes identified during previous work at Janssen Pharmaceutica were selected for this project and tested side-by-side in duplicates for a second time in Mdm2 and Mdm4 ELISAs. The results of this second experiment are displayed in
Figure 1
; exact values are provided in
(
The p53–Mdm2 ELISA reveals that all selected compounds show a significant dose-dependent reduction of the p53–Mdm2 interaction. Twelve out of 19 compounds reached ≥90% reduction of the p53–Mdm2 complexes. Fifty percent of the compounds demonstrated submicromolar half-maximal inhibitory concentrations (IC50). Two compounds showed an IC50 lower than 100 nM, which is in the range of the well-known reference compound Nutlin-3 23 , which showed an IC50 of 43 nM in this experiment.
For the p53–Mdm4 interaction, we observed a completely different landscape for the selected compounds. Only 4 out of 19 reduce the binding of Mdm4 to p53 more than 50%, and only one has the potency to reduce the complex formation more than 90%. Also, the correlating IC50s are significantly increased compared to the p53–Mdm2 inhibition. Only one compound could be identified that exhibits submicromolar inhibitory activity at a maximal (93%) reduction rate. Three tested compounds show an IC50 in the sub-10 μM range achieving a 50–80% reduction of PPI complex formation. Consistently, Nutlin-3 shows 1000-fold decreased activity for inhibiting the p53–Mdm4 formation compared to its potency against the p53–Mdm2 formation. It still, however, achieves a 97% reduction of the p53–Mdm4 complex formation with an IC50 of 47 μM.
Establishment and Validation of the F2H Cellular Assays to Monitor p53–Mdm2 and p53–Mdm4 Interactions
Then, we applied the F2H principle to establish a cell-based assay for direct, comparative analysis between the PPIs of p53–Mdm2 and p53–Mdm4. We designed mammalian expression constructs containing the interacting regions of the proteins, each fused to a respective fluorescent tag. In addition to the fluorescent tag, the bait construct carried a fusion to the LacI protein enabling targeting to the lac-operator array on the genomic DNA of the genetically modified BHK cells.
Initially, multiple combinations of constructs had to be cloned and tested for functionality. Critical parameters in this regard were the length of the individual protein domains, the composition of the triple-fusion bait construct, the orientation of the fluorescent protein in the prey construct, and the assignment as either bait or prey (
The combinations as displayed in Figure 2 were finally identified to work best. LacI fused N-terminally to GFP was linked at its C-terminus to the first 81 amino acids of p53. For the prey construct, RFP was fused N-terminally to a fragment of Mdm2 consisting of amino acids 7–134 or to the first 129 amino acids of Mdm4.

Outline of the p53–Mdm2 and p53–Mdm4 F2H assays. Schematic drawings of the assay fit bait and prey constructs. Exemplary images of baby hamster kidney (BHK) cells co-transfected with the respective expression vectors, showing the interactions. Scale bar, 5 µm.
Thus, BHK cells were transiently co-transfected with expression vectors encoding LacI–GFP–p53 and RFP–Mdm2 or RFP–Mdm4, respectively. As expected, LacI–GFP–p53 forms a distinct “green spot” in the nucleus of transfected cells. The interactions with Mdm2 or Mdm4 become visible by co-localization of the red fluorescent signal at the same nuclear green spot (
Fig. 2
). Unaltered RFP expressed as a negative control shows a diffuse distribution (
This co-localization of the fluorescent signals at a defined nuclear structure is well suited for a high-content profiling and screening approach. Subsequently to the automated image acquisition, a routine software-assisted analysis was developed based on the multitarget-analysis package of the IN Cell Workstation. Initially, nuclei, cells, and the interaction “spot” were segmented. Then, a decision tree was set up, and interactions (in %) were determined as the ratio of the cells showing co-localization of the fluorescent signals to the total number of double-transfected cells with a “green spot.” A minimum of 150 co-transfected cells was usually evaluated per well.
Screening of 19 Compounds for Inhibition of the p53–Mdm2 Interaction in an Endpoint F2H Assay
In a next step, we validated the reversibility of the p53–Mdm2 F2H assay using Nutlin-3, a potent small-molecule inhibitor of this interaction. 23 BHK cells expressing p53–LacI–GFP and Mdm2–RFP were incubated for 3 h with increasing concentrations of Nutlin-3. We observed that, compared to DMSO-treated cells, Nutlin-3 disrupts the p53–Mdm2 interactions in a dose-dependent manner: We detected a reduction of complexes from 91 ± 6% in untreated cells to 53 ± 12% upon treatment with 10 µM Nutlin-3 (mean ± SD; five independent experiments). Incubation with 40 µM Nutlin-3 reduces the interactions further down to 23 ± 12 %.
Then, we determined the intracellular activity of the small molecules earlier characterized in ELISA. The 19 compounds were administered at 0.1–10 µM concentration to the medium of BHK cells and incubated for 3 h.
In total, four independent experiments were performed in duplicate. The results are summarized in
Figure 3A
and
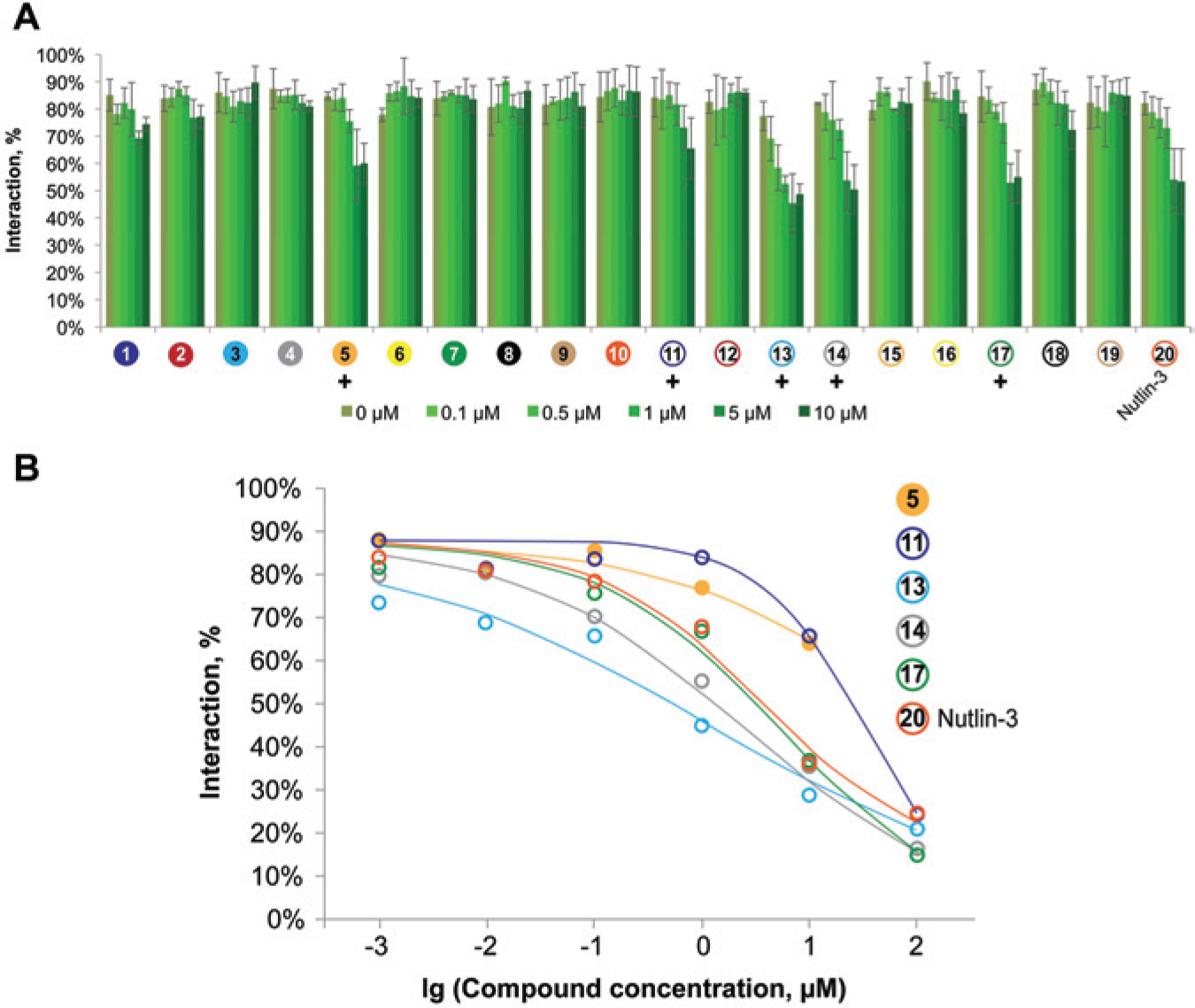
Comparison of the abilities of 19 compounds to disrupt p53–Mdm2 interactions in an endpoint fluorescent two-hybrid (F2H) assay. Baby hamster kidney (BHK) cells were co-transfected with LacI–GFP–p53 and RFP–Mdm2, and incubated with the compounds and the positive control Nutlin-3 in duplicates for 3 h. Cells were fixed, followed by automated imaging and analysis of interactions. (
To analyze the activity of those compounds in more detail, we extended the concentration range and conducted the duplicate 6-point dose–response F2H assays starting at a maximum of 100 µM ( Fig. 3B ). These experiments verified previous results. In addition, the concentration–response curves permit direct comparison of the compound activities.
F2H Screen of 19 Compounds for Inhibition of the p53–Mdm4 Interaction
Next, we screened the compounds for disrupting the p53–Mdm4 interaction. Because there is no potent small-molecule inhibitor of this interaction available to date showing convincing activity at the cellular level, we validated the reversibility of the p53–Mdm4 F2H assay with a peptide approach. Previously, we have shown that the stapled peptides sMTide-02 and -02A efficiently disrupt the p53–Mdm4 interaction. 24 In these experiments, double-transfected BHK cells were incubated for 8 h with increasing concentrations of the stapled peptides. The most efficient disruption was observed with sMTide-02, which reduced the interactions from 67 ± 4% (untreated cells) to 36 ± 5% (50 µM treated cells). 24
Thereafter, BHK cells expressing LacI–GFP–p53 and RFP–Mdm4 were incubated with 0.1–10 µM of the selected 19 compounds for 3 h, fixed, and analyzed. None of the compounds, however, showed significant inhibition of this interaction. Finally, we tried to increase compound concentrations but could not observe any inhibitory effect up to 50 µM concentration. Those dose-escalation experiments were run in parallel on both interactions to confirm compound integrity.
Real-Time Visualization of the PPIs Perturbed by Selected Compounds
The F2H assay allows detection of PPI dynamics in living cells, and we traced the disruption of p53–Mdm2 or p53–Mdm4 interactions by treatment with Nutlin-3 as well as with the respective stapled peptide sMTide-02 in real time. Antagonizing effects of the substances are visualized as disappearance of the “red spot” (i.e., the preys RFP–Mdm2 or RFP–Mdm4), whereas the bait (LacI–GFP–p53) is anchored at the interaction platform. The rates at which the PPIs were disrupted reveal the different efficacies of the test substances. The results show that Nutlin-3 begins to disrupt the p53–Mdm2 interaction during the first hour, whereas it has no effect on p53–Mdm4. The stapled peptide sMTide-02 does interfere with both PPIs. It is, however, more potent on Mdm2 than on Mdm4 (disruption after 2 h vs. 5 h) (
Fig. 4
). This direct side-by-side analysis between the individual peptides and Nutlin-3 on the two PPIs in real time underlines the major differences in their pharmacokinetics. Correlating movies are available online as
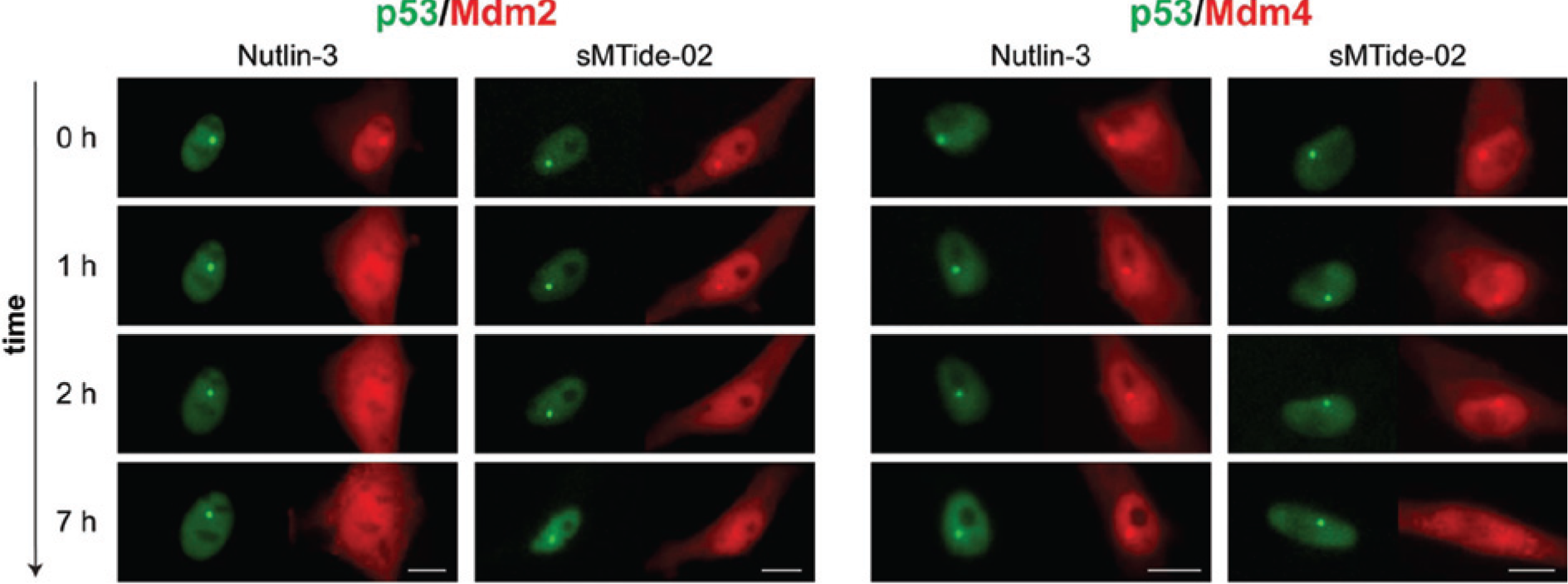
Real-time visualization of a compound’s ability to disrupt protein interactions. For the p53–Mdm2 interaction (left), selected time frames for Nutlin-3 (50 µM) activity show fading of red fluorescence from the “sport” but not from the rest of the nucleus after only 1 h, indicating disruption of the interaction. Stapled peptide sMTide-02 (50 µM) required 2 h to disrupt the p53–Mdm2 interaction. For the p53–Mdm4 interaction (right), Nutlin-3 is not able to disrupt the interaction within 7 h. Stapled peptide sMTide-02 could inhibit the interaction after 5 h of incubation (a 7 h time frame is shown). The complete time series can be found in
Cell-Viability Determination within the F2H Assay
Image-based cellular assays allow multiparametric data extraction. Therefore, data on the cytotoxicity of the applied compounds were derived in parallel. Analyzing the same sets of acquired images, we determined cell numbers by counting the morphologically appropriate nuclei in the images and normalized this to the DMSO control. Apoptotic or mitotic cells were discarded. No significant changes in cell count after 3 h of incubation with 0.1–10 µM compounds were detectable ( Fig. 5A ).
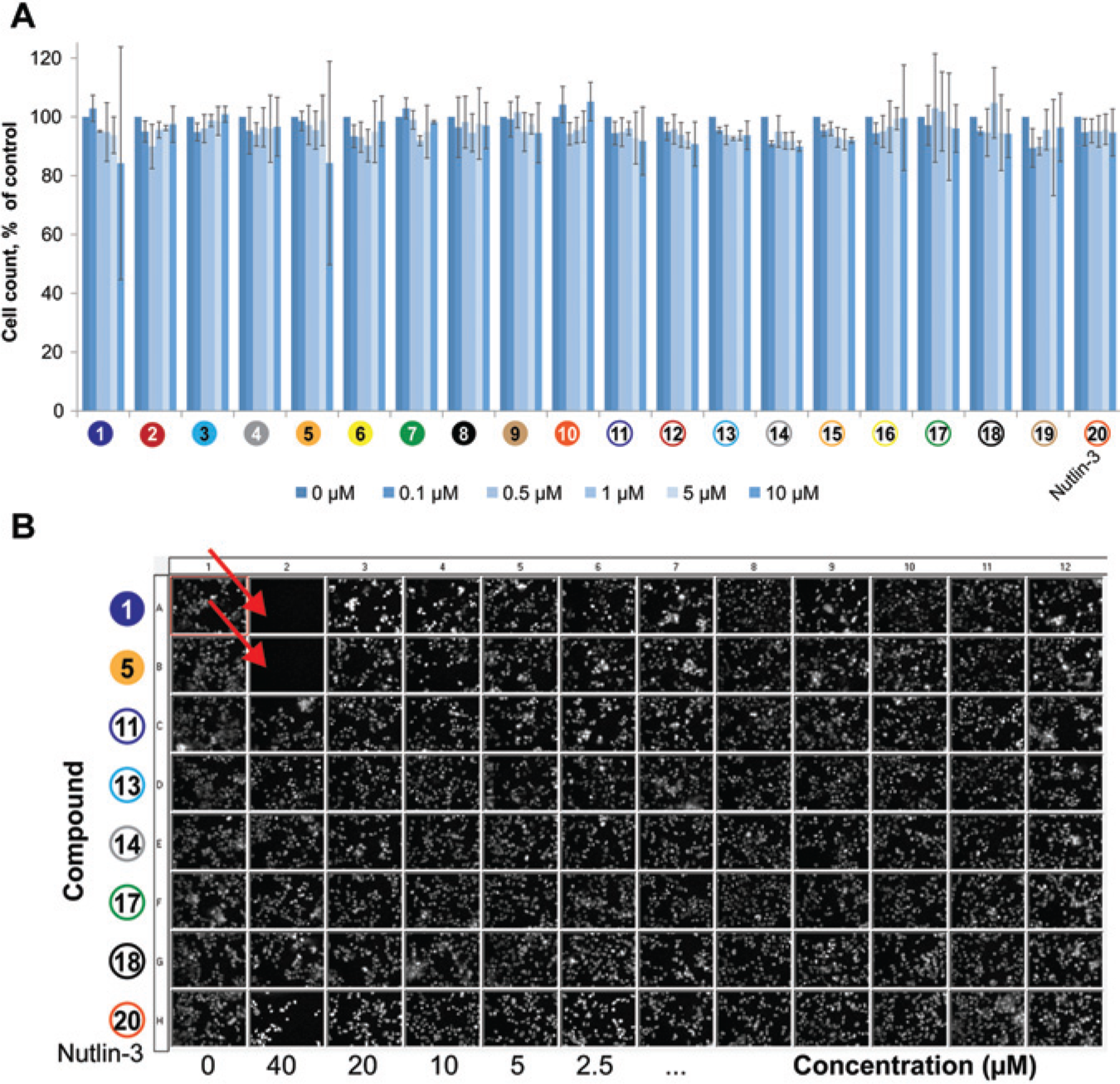
Estimation of cell viability from the fluorescent two-hybrid (F2H) data sets. (A) Morphologically appropriate nuclei were counted in the images acquired during the F2H screen. No consistent changes in cell numbers could be observed when compounds were applied in the concentration range of 0.1–10 µM (3–5 experiments; data are means ± SD). (
Incubations with higher concentrations of some of the compounds in combination with longer incubation times resulted in a reduced cell number, however. We observed a dramatic decrease in the total cell number after incubation with compounds 1 and 5 at 40 µM for 3 h. This can be visualized on thumbnail images of the DAPI staining ( Fig. 5B ). Taken together, these results show that with the F2H assay, additional data on cell proliferation and compound cytotoxicity can be gathered in a simple and reliable manner.
Cell Permeability Assessment
For some compounds, marked differences were observed between in vitro activity in ELISA and activity determined in the cell-based F2H assay. Compounds 10 and 15 had very similar potency against Mdm2 in vitro compared to compounds 5, 14, or 17. In the cell-based F2H assay, however, compounds 10 and 15 were totally inactive throughout the tested concentration range, while 5, 14, and 17 efficiently disrupted the cellular p53–Mdm2 complex.
One reason for this observation could be differences in cell permeability. Therefore, we tested a small subset of compounds covering a range of potencies in the F2H assay in a cell-based permeability model ( Table 1 ). Due to a depletion of stock availability, compounds 5 and 11 were not included in this subset. The transepithelial transport of test compounds through a monolayer of LLC-PK1 cells stably transfected with (human) MDR1 was assessed. Measurement of the apparent permeability in the A-to-B direction in the presence of the P-gp inhibitor GF120918 reflects the passive permeability [Papp A-to-B + GF120918 (×10−6 cm/sec)], while in the absence of GF120918 [Papp A-to-B − GF120918 (×10−6 cm/sec)] it indicates the ability of the test compound to be actively transported by P-gp.
Comparison of the Activities of the Selected Compounds in Vitro, Their Cell Permeability, and Their Inhibitory Effect in an F2H Assay.
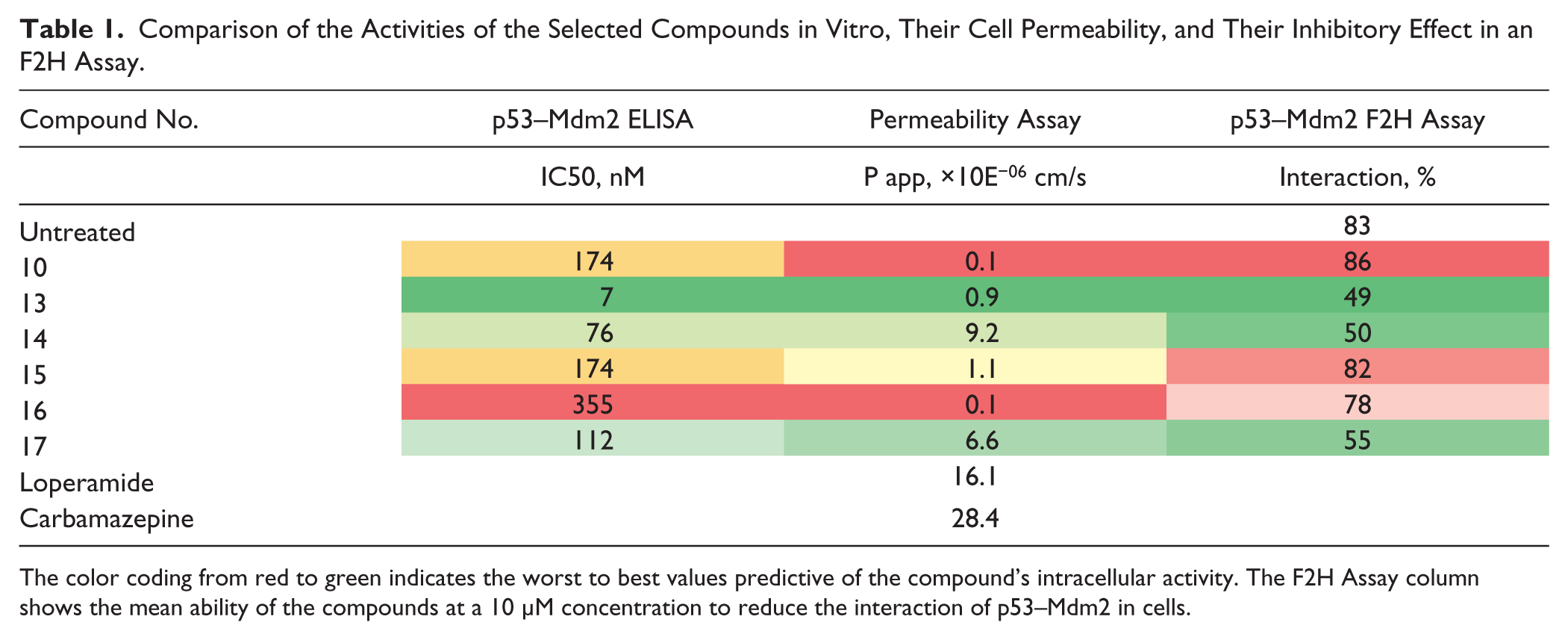
The color coding from red to green indicates the worst to best values predictive of the compound’s intracellular activity. The F2H Assay column shows the mean ability of the compounds at a 10 µM concentration to reduce the interaction of p53–Mdm2 in cells.
The integrity of the cellular monolayer was assessed in all wells through the inclusion of the low-permeability marker compound fluorescein. The positive control for active transport by P-gp was loperamide, 27 and maximum permeability of this compound is observed in the presence of the P-gp inhibitor GF120918. Carbamazepine was used as a P-gp-independent, highly permeable control molecule.
In Table 1 , only the cell-permeability values (Papp × 10−6 cm/sec) at the presence of the P-gp inhibitor are displayed. The results reveal that compounds 14 and 17, which exhibit good cellular activity splitting the p53–Mdm2 interaction, demonstrated high permeability in the cellular-permeability assay. In contrast, compounds 10 and 16, shown to be inactive in living cells, also demonstrated very low cell permeability. Compound 13 showed low permeability; however, its extraordinary in vitro potency at the single-digit nanomolar level likely reduced the impact of poor permeability, rendering this compound active in the cell-based F2H assay. In contrast, the similarly low-permeable compound 15 failed, presumably due to its 25-fold lower potency compared to compound 13.
Discussion
In this report, we present two simple and fully reversible cellular high-content assays to analyze p53–Mdm2 and p53–Mdm4 PPIs in living mammalian cells using the F2H principle. With these F2H assays, we successfully monitored and compared the ability of 19 selected chemical compounds as well as cell-penetrating stapled peptides to inhibit the interactions of p53 with Mdm2 or Mdm4.
The rationale for the F2H principle is based on the observation that proteins are freely moving in the cell, unless they are transiently immobilized on interactions with certain cellular components. 28 Here, we anchor the green-fluorescent bait protein (p53) at a defined spot in the nucleus, formed by the lac-operator array on the genomic DNA.16,17 Then, we assay for co-localization of the red-fluorescent prey proteins (either Mdm2 or Mdm4) at that nuclear spot. On incubation with test compounds, automated image acquisition and software-assisted image analyses were carried out, either as an endpoint assay or in real time.
Correlation of ELISA and F2H Results
With the two F2H assays described here, we identified five small molecules out of a 19-compound collection that efficiently disrupt the intracellular p53–Mdm2 interaction, but not that of p53–Mdm4. The detailed comparison with ELISA data shows that all F2H-positive compounds also exhibited activity in vitro. More inhibitory candidates were identified in ELISA, however. Subsequent analyses disclosed that all of those few selected ELISA hits that did not show activity in F2H were characterized by no or poor cell permeability. This demonstrates that the F2H cellular assay may provide integral information about test substances, identifying those that can both penetrate cells and specifically abrogate interactions. For screening, the F2H offers a valuable filtering tool for hits from a primary in vitro HTS campaign and their confirmation by an independent test principle. For the hit-to-lead and lead-optimization processes, however, parallel ELISA and F2H assaying might be most helpful to explore the full potential for rational drug design as non-cell-permeable and/or low-active compounds can be valuable assets to determine structure–activity relationships (SARs).
That even a very potent molecule such as our positive control Nutlin-3 did not completely inhibit the complexes has been observed in a very similar cellular assay applying BRET, 11 although we did not observe saturation beyond 10 µM as reported there. A possible reason might be that Nutlin-3 does not split existing complexes. 11
Estimation of Compound’s Toxicity with F2H
In addition, F2H offers simultaneous estimation of the cytotoxicity of applied treatments. Here, we used counting nuclei to determine concentration ranges at which no severe cytotoxicity was observed. Cell-viability analysis can be easily extended to evaluate nuclear morphology and fluorescent intensities to acquire more detailed information. In addition, cells can be simultaneously stained for a desired necrosis or apoptosis marker exhibiting far-red fluorescence, which does not interfere with the F2H-interactions assessment.
Advantages, Limitations, and Validation
The most prominent feature of the F2H technology is the visualization of fully reversible PPI dynamics in real time in the context of a mammalian cell. To monitor the compound-induced disruption of a PPI, we traced the fluorescent signals of the F2H-interaction pairs using time-lapse imaging of the disappearance of the fluorescent “spot” of the prey protein. This reversibility feature is a distinct asset of the F2H approach in comparison to most other cellular PPI assays.
Another advantage of a cell-by-cell analysis is that interferences such as autofluorescence of compounds, modulation of the fluorescence of the fusion proteins, or decreasing cell viability can be easily extracted from the acquired images by analysis of multiple parameters. Overall, fluorescent intensities of the two fluorescent proteins should not change and, along with the number of cells, stay constant. Furthermore, as the F2H assesses the interaction in the nucleus using nuclear fluorescence as a reference, false positives from inhibition of the cytosol-to-nucleus translocation after compound addition, as exploited by similar assay types,12,18 will not score here. Finally, the F2H assay can be applied either as a fixed-endpoint assay for higher throughput or as a real-time (live) assay for detailed analysis of selected compounds.
Nevertheless, cell-based PPI approaches have some common limitations. A prerequisite of the F2H principle is the mobility of the bait and prey proteins, and hence their ability to encounter each other at the interaction spot. Turnover of the prey protein at the spot is also crucial for assay reversibility. We analyzed the bait (p53) and prey (Mmd2 and Mdm4) proteins and observed their full recovery after photo-bleaching at the spot region, confirming turnover of the unbound and bound fractions (data not shown). It is also important to consider that genetic fusion of the proteins of interest with fluorescent tags might alter their cellular properties. Overexpression of proteins as well as intentional targeting of the proteins of interest to “nonnative” cellular compartments, in our case the nucleus, might generate artifacts. Therefore, it is important to validate each assay with reference compounds that are described to be specific for a particular PPI. Such reference compounds are, however, often unknown or unavailable.
Frequently, a PPI has been characterized previously and reduced to domains or even single involved amino acids. As PPIs are often mediated by short alpha helical structures, the interaction site can be narrowed down to interacting peptides. Such interacting or even disrupting peptides can be used for the purpose of validating the assay, providing much higher flexibility. Here, we illustrate this approach by applying a Mdm4-binding cell-permeable stapled peptide 24 to inhibit the p53–Mdm4 interaction in the F2H assay, because a potent chemical compound is lacking. Herewith, we confirmed that disruption of the p53–Mdm4 interaction can also be clearly registered with F2H.
F2H versus Other p53–Mdm2 Cellular Assays
During the past decade, a variety of screening campaigns have been undertaken to identify small-molecule inhibitors of p53–Mdm2 interaction. The first potent small-molecule antagonists of this interaction were the cis-imidazoline compounds Nutlin-1, -2, and -3, reported in 2004. 23 By now, numerous small-molecule compounds typically incorporating a heterocyclic core, as well as peptidomimetic inhibitors, have been identified to disrupt the p53–Mdm2 binding.4,29 All of these compounds are, however, far less potent in reactivating the p53 pathway in vivo. One possible reason is the increase in expression of the other crucial negative regulator of p53, Mdm4. Despite its close homology to Mdm2, all currently available small-molecule antagonists of p53–Mdm2 interaction are several magnitudes less potent in disrupting p53–Mdm4 interaction. The current challenge is to obtain clinically appropriate molecules that inhibit both Mdm2 and Mdm4, are as potent in vivo as they are in vitro, and enable reactivation of the p53 pathway in a broad range of malignant cells.
Here, we introduced two novel, fully reversible PPI assays based on the F2H technology that have the potential to discover new inhibitors of p53–Mdm2 and p53–Mdm4 interactions. Furthermore, these assays enabled for the first time a direct side-by-side comparison of dual-action antagonists inside living cells.
Footnotes
Acknowledgements
We thank Jacqueline Gregor (ChromoTek) for her excellent technical assistance.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors U.R. and K.Z. declare competing financial interests as co-inventors of the F2H assay and as shareholders of ChromoTek. The F2H technology is covered by a patent application owned by the University of Munich and licensed to ChromoTek on an exclusive basis.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded in part by the GoBio grant of the German Federal Ministry of Education and Research (BMBF) to ChromoTek.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.