
Abstract
Rationale:
Direct oral anticoagulants (DOAC) are highly effective in preventing ischaemic strokes in people with atrial fibrillation (AF). However, it is unclear how soon they should be started after acute ischaemic stroke (AIS). Early initiation may reduce early risk of recurrence but might increase the risk of haemorrhagic complications.
Aim:
To estimate the safety and efficacy of early initiation of DOACs compared to late guideline-based initiation in people with AIS related to AF.
Methods and design:
An international, multicentre, randomised (1:1) controlled, two-arm, open, assessor-blinded trial is being conducted. Early treatment is defined as DOAC initiation within 48 h of a minor or moderate stroke, or at day 6–7 following major stroke. Late treatment is defined as DOAC initiation after day 3–4 following minor stroke, after day 6–7 following moderate stroke and after day 12–14 following major stroke. Severity of stroke is defined according to imaging assessment of infarct size.
Sample size:
ELAN will randomise 2000 participants 1:1 to early versus late initiation of DOACs. This assumes a risk difference of 0.5% favouring the early arm, allowing an upper limit of the 95% confidence interval up to 1.5% based on the Miettinen & Nurminen formula.
Outcomes:
The primary outcome is a composite of symptomatic intracranial haemorrhage, major extracranial bleeding, recurrent ischaemic stroke, systemic embolism or vascular death at 30 ± 3 days after randomisation. Secondary outcomes include the individual components of the primary outcome at 30 ± 3 and 90 ± 7 days and functional status at 90 ± 7 days.
Discussion:
ELAN will estimate whether there is a clinically important difference in safety and efficacy outcomes following early anticoagulation with a DOAC compared to late guideline-based treatment in neuroimaging-selected people with an AIS due to AF.
Introduction and rationale
Anticoagulation therapy with direct oral anticoagulants (DOACs) is highly effective in preventing recurrent ischaemic events in people with strokes related to atrial fibrillation (AF). 1 However, it is unclear how soon DOACs should be started after acute ischaemic stroke (AIS). Randomised controlled trials comparing DOACs with vitamin K antagonists for prevention of AF-related ischaemic strokes excluded people with a recent AIS. 2 Early anticoagulation with DOACs may reduce the risk of early recurrent ischaemic events but might also increase the risk of haemorrhagic complications (particularly intracranial bleeding), thus outweighing any beneficial effect.
In the absence of randomised-trial evidence, the European Heart Rhythm Association (EHRA) and the European Society of Cardiology suggest following the ‘1–3–6–12 day rule’ for initiation of anticoagulation following AF-related transient ischaemic attack (TIA), minor, moderate and severe AIS. 3 Thus, anticoagulation can be initiated on day 1 in people with TIA, on day 3 in those with mild stroke, on day 5–7 in those with moderate stroke and on day 12–14 in those with major stroke. 3 This is based on the observation that larger infarcts are more likely than smaller ones to undergo haemorrhagic transformation.4,5 Many countries, societies and expert opinions have now adopted this recommendation.2,6,7 However, real-world data suggest that DOACs might be safely used earlier than recommended following AIS, although this evidence is limited by selection bias or small randomised controlled trials.2,8,9 Furthermore, the risk of both recurrent ischaemic and haemorrhagic stroke is highest in the first 2 days following stroke onset, meaning that randomised trials are needed to establish whether it is safe and beneficial to start DOACs early after AIS. 10 Neuroimaging selection may help minimise the risk of intracranial haemorrhage.4,5
The Early versus Late initiation of direct oral Anticoagulants in post-ischaemic stroke patients with atrial fibrillatioN (ELAN) aims to estimate the safety and efficacy of early initiation of DOACs compared to late guideline-based initiation in imaging-selected participants with AIS and AF.
Methods
Study design
ELAN is an international, multicentre, randomised (1:1) controlled, two-arm, open, assessor-blinded trial comparing early versus late initiation of DOACs in people with AIS and AF. The trial is being conducted in more than 90 stroke units in Europe, India, the Middle East and Japan. The first participant was enrolled in November 2017.
Participant population
ELAN will randomise 2000 people with an AIS and AF. Inclusion and exclusion criteria are listed in Table 1. ELAN has a gender policy aiming at an equal representation of the sexes.
Inclusion and exclusion criteria.

AF: atrial fibrillation; aPTT: activated partial thromboplastin time; CT: computed tomography; INR: International Normalised Ratio; MRI: magnetic resonance imaging; DAPT: dual antiplatelet therapy; DOACs: direct oral anticoagulants; HI: haemorrhagic infarction; PH: parenchymatous haematoma.
Randomisation and blinding
Based on the infarct size on CT and/or MRI prior to randomisation, participants are classified as having experienced minor, moderate or major ischaemic stroke (Figure 1 and Table 2). 11 Infarct size is classified by the treating team and in case of rapid clinical improvement after admission, especially after intravenous thrombolysis and thrombectomy, study teams are strongly encouraged to repeat imaging before randomisation. The qualifying imaging for stroke classification is the last imaging performed prior to randomisation. Participants are assigned in a 1:1 ratio to one of the two treatment arms using deterministic minimisation implemented via a web-based data management system (secuTrial) to ensure concealment of allocation. Randomisation is performed within 48 h after symptom onset in participants with minor and moderate stroke and at day 6–7 in participants with major stroke (Figure 2). Allocation is stratified by trial site, age (<70 vs ⩾70 years), stroke severity (minor, moderate or major stroke) and NIHSS score (<10 vs ⩾10).
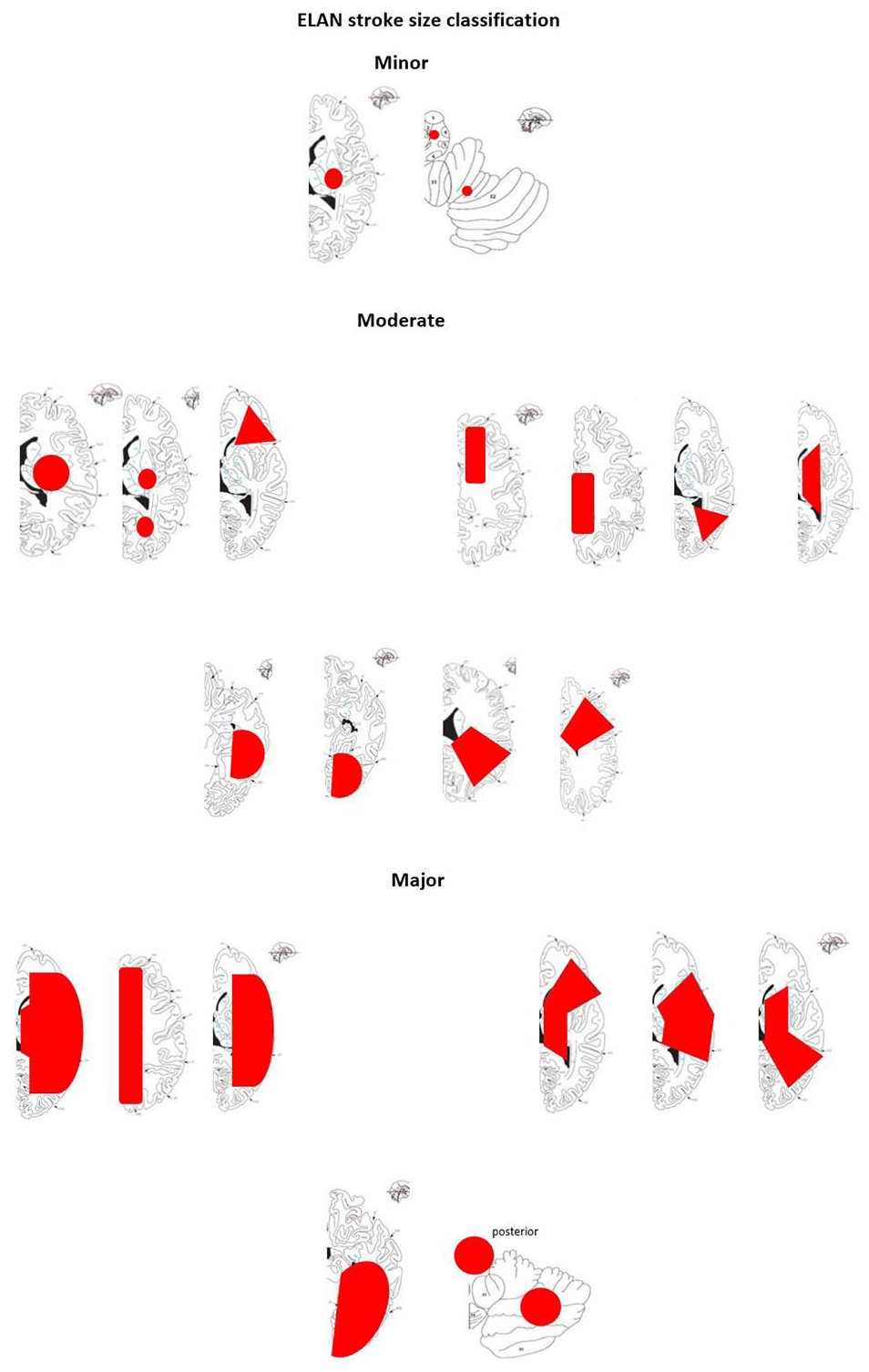
Stroke size classification.
Ischaemic stroke size classification.

Ischaemic stroke size classification is based on: Paciaroni et al. 11
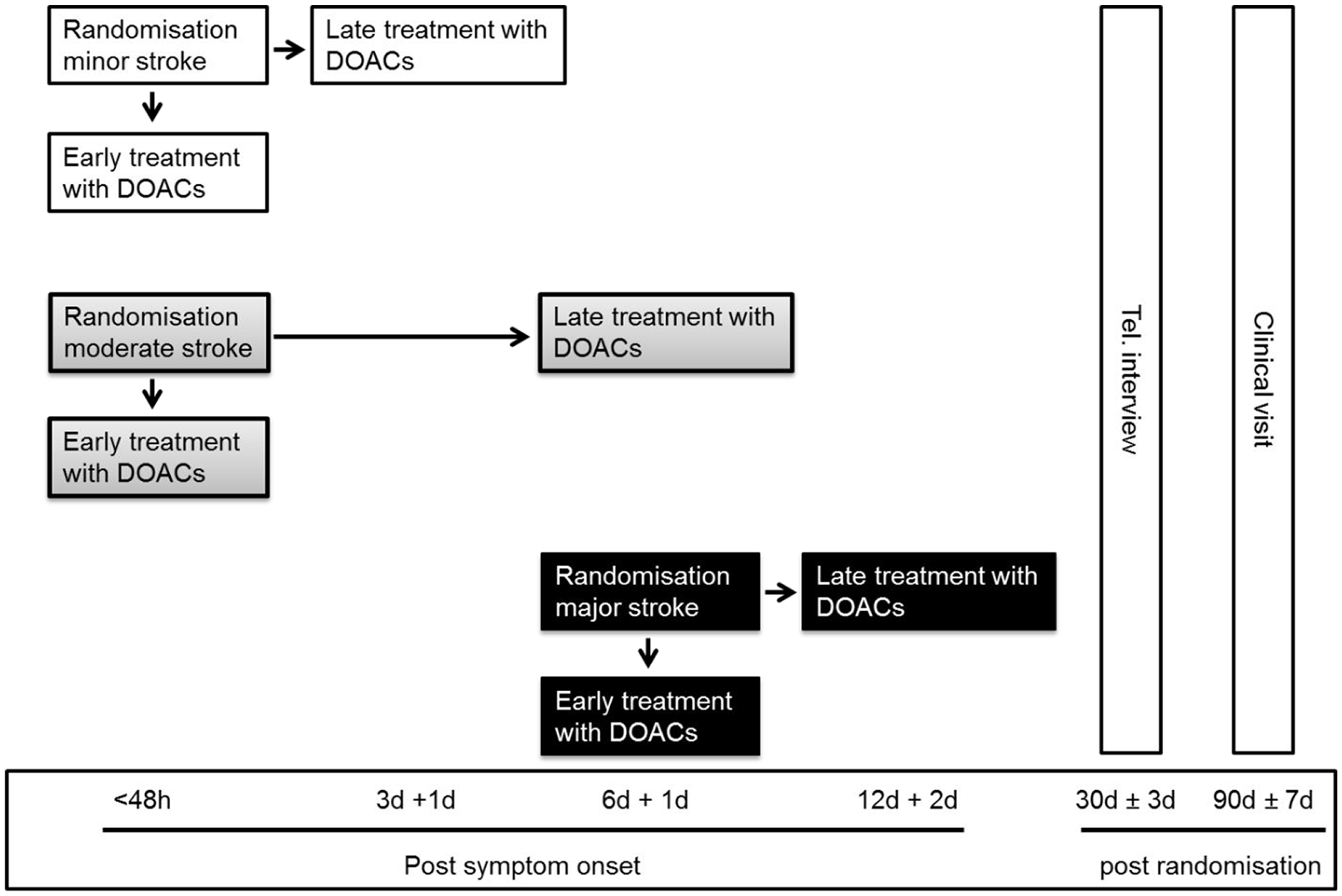
Trial schedule.
Treatment
Any DOAC with marketing authorisation for the prevention of stroke and systemic embolism in the respective countries can be used. Early treatment means initiation of DOAC within 48 h in participants with minor and moderate stroke, or on day 6–7 in those with major stroke. Late treatment means initiation of treatment in participants with minor stroke on day 3–4, those with moderate stroke on day 6–7 and with major stroke on day 12–14. The late treatment times were chosen to be consistent with the ‘1–3–6–12 day rule’. 3
Clinical and imaging evaluation
All trial procedures are summarised in Supplemental Table 1. The primary outcome is assessed at 30 ± 3 days after randomisation by a structured telephone interview conducted by trained medical personnel unaware of the treatment allocation. If the person is unable to participate in the interview, the next of kin or treating physician is asked. For every reported outcome event (bleeding, stroke, embolism and/or death), corresponding source documents are collected. An independent clinical event committee (CEC) reviews these documents and adjudicates all outcome events. The CEC also reviews serious adverse events and unclassified events to identify potential unreported outcome events. A central imaging core lab evaluates all clinical imaging data prior to randomisation as well as imaging performed up to 90 ± 7 days after randomisation.
Primary outcome
The primary outcome is a composite binary endpoint. The occurrence of at least one of the following up to 30 ± 3 days after randomisation is considered as an outcome event: symptomatic intracranial haemorrhage, major extracranial bleeding, recurrent ischaemic stroke, systemic embolism or vascular death.
Symptomatic intracranial haemorrhage, including subdural, epidural, subarachnoid and intracerebral haemorrhage, is defined as a haemorrhage that leads to a clinical worsening and hospitalisation or prolongation of hospitalisation, and is assessed by the treating physician to be the likely cause of the new neurological symptom or the death. Major extracranial bleeding (major bleeds are those that result in death or are life-threatening) is defined as clinically overt bleeding accompanied by one or more of the following: decrease in haemoglobin of ⩾2 g/dl over a 24-h period; transfusion of ⩾2 units of packed red blood cells; or bleeding occurring in a critical part of the body (intraspinal, intraocular, pericardial, intraarticular, intramuscular with compartment syndrome, retroperitoneal). For bleeding in a critical area (e.g. gastrointestinal) or organ to be classified as major extracranial bleeding it must be associated with a symptomatic clinical presentation. 12
Secondary outcomes
Secondary outcomes are the individual components of the primary endpoint at 30 ± 3 and 90 ± 7 days after randomisation, favourable outcome at 90 ± 7 days defined as mRS ⩽ 2, mRS shift analysis at 30 ± 3 and 90 ± 7 days, individual components of major extracranial bleeding at 30 ± 3 and 90 ± 7 days, all-cause mortality at 30 ± 3 and 90 ± 7 days, drug compliance measured after 30 ± 3 days and the difference between treatment randomised and treatment received. The main safety endpoints are symptomatic intracranial haemorrhage, major extracranial bleeding and vascular death. The main efficacy endpoints are prevention of recurrent ischaemic stroke and systemic embolism, as well as favourable outcome at 90 ± 7 days.
Other outcomes of interest
Further relevant endpoints are myocardial infarction at 90 ± 7 days, TIA and undetermined stroke at 30 ± 3 and 90 ± 7 days, major cardiovascular events at 90 ± 7 days as a composite of stroke, myocardial infarct, heart failure or cardiovascular death, NIHSS at 90 ± 7 days and silent brain lesions at 90 ± 7 days.
Data safety monitoring board (DSMB)
An independent DSMB is monitoring the trial. The DSMB met after the first 250 patients reached data maturity and again after the first 500 patients. Thereafter it meets at least once a year.
Hypothesis and statistical analysis
The main aim of ELAN is to estimate the effect of early versus late initiation of DOACs in AIS patients. Therefore, no specific statistical hypothesis will be tested. The analysis plan will focus on estimating the treatment effect and its uncertainty by calculating 95% confidence intervals.
Sample size calculation
The sample size is estimated based on the precision of the estimation as reflected by the width of the confidence interval around the treatment effect estimate, that is the risk difference for the primary outcome. With 1802 patients, an assumed event rate in the late treatment group of 5% at the trial end, and an assumed risk difference of −0.5% (i.e. an assumed event rate of 4.5% in the early treatment group), the upper limit of the 95% confidence interval will be up to 1.5% (based on Miettinen and Nurminen’s formula) favouring the control group. This means that the resulting 95% CI will exclude values suggesting that early treatment increases the rate of the composite primary outcome by more than 1.5%. To account for possible missing outcome data, we plan to randomise 2000 participants.
Statistical analysis
For the primary analysis, to avoid bias due to a small number of events, we will compare the event rate between late treatment and early treatment using a logistic regression model corrected for the bias via a penalised likelihood method. 13 The effect measure will be the odds ratio. Unadjusted analysis using Mantel-Haenszel risk difference will also be calculated along with the Miettinen and Nurminen confidence interval as sensitivity analyses. Details are provided in the statistical analysis plan. The variable mRS (scale with seven levels) will be analysed using mixed-effects ordered logistic regression. Continuous outcome data will be analysed using linear regression. Time-to-event outcomes will be described using Kaplan-Meier curves and analysed using penalised survival methods. 14 The use of three stratification factors combined with over 90 recruiting sites may lead to imbalances in the randomisation process. However, to overcome this problem the deterministic minimisation method has been implemented for allocation to reduce the impact of imbalances.
Interim analysis
Regular monitoring of outcome data, especially haemorrhage and ischaemic events, will be performed by the DSMB. The DSMB charter sets out thresholds for treatment effects and criteria based upon which it can recommend an early stopping of the trial or additional analysis. The thresholds are indicative of potential unacceptable harmful effects but are not binding.
Study organisation and funding
ELAN is an investigator-initiated clinical trial. The sponsor of the trial is the University Hospital Bern (Inselspital) and the trial is supported by grants from the Swiss National Science Foundation (32003B_197009; 32003B_169975), the Swiss Heart Foundation, the UK Stroke Association (2017/02) and the Intramural Research Fund (20-4-5) for Cardiovascular Diseases of the National Cerebral and Cardiovascular Centre, Japan. The clinical trial is managed by the Neuro Clinical Trial Unit at the Department of Neurology, University Hospital Bern, Switzerland. The database, central data monitoring and statistical analyses are performed by the CTU Bern at the University of Bern, Switzerland.
Ethical approval
Ethical approval for the study was granted by the Cantonal Ethics Commission (KEK) in Bern, Switzerland and subsequently by all local authorities and/or, if applicable, by national lead ethics committees and competent regulatory authorities at all participating sites.
Trial status
On 30 March 2022, 1649 patients had been randomised into the ELAN trial. Information on baseline characteristics of the first 1000 patients randomised is provided in Supplemental Table 2.
Discussion
ELAN is a global pragmatic randomised controlled trial addressing an important unanswered clinical dilemma, whether it is safe and beneficial to start anticoagulation therapy with DOACs early on after an AF-related AIS. Observational studies suggest that the risk of recurrent stroke is seven times higher than the risk of haemorrhagic transformation early on after recent stroke, yet the fear of harming the patient by starting anticoagulation too early prevents many physicians from doing so. 15 In the absence of evidence, many physicians worldwide have adopted the ‘1–3–6–12 day rule’. 3 This approach is supported by an expert opinion statement by the European Stroke Organisation. 4 However, it may be beneficial to start DOAC therapy earlier. The ELAN trial therefore compares an earlier treatment start (i.e. within 48 h of a minor or moderate stroke and at day 6–7 after a major stroke) with this current standard of care.
Given that haemorrhagic transformation is dependent on lesion size, ELAN uses an imaging-based approach to exclude patients with early parenchymal haemorrhage, which can be easily detected on a CT scan prior to randomisation. Infarct size on imaging prior to randomisation is also used to classify participants according to whether they have minor, moderate or major stroke. This is in contrast to the EHRA guideline, which classifies stroke severity based on the NIHSS score. 3 We chose our approach because the NIHSS score is strongly influenced by infarct location as well as lesion size.4,5 For example, patients with a deep or brainstem stroke can have a high NIHSS score but a low infarct volume, and patients with large cerebellar or non-dominant hemispheric infarction may have a relatively low NIHSS.
In the ELAN trial we will estimate the treatment effect and the degree of precision by calculating the odds ratio of the predefined outcomes and the corresponding 95% CI. The trial has not been designed to statistically test a specific statistical hypothesis, nor is it a non-inferiority trial. The rationale for this decision is twofold. First, when we designed the trial, there was a lack of high-quality data on event rates in this setting, making it difficult to identify an appropriate non-inferiority margin. Second, the assumed low event rate would require a very large trial to assess either superiority or non-inferiority and this would not necessarily provide greater clarity concerning patient management. ELAN is already one of the largest trials in this population with many participating sites that have been enrolling patients over several years. Although we propose a different analytic approach to that often seen in clinical trials, this should not hinder interpretation of trial data or their clinical utility. We also believe that the complexity of managing patients with AF early on after AIS precludes simplified dichotomous decision-making and necessitates some leeway for individual decision-making. This is best supported by estimation rather than statistical hypothesis-testing, and, where there is insufficient clinical information, this is an accepted approach.16–18
ELAN is one of several contemporaneous randomised controlled trials comparing early versus late anticoagulation with DOACs in people with AIS and AF (Supplemental Table 3). ELAN differs from the Swedish TIMING (NCT02961348) and the British OPTIMAS (NCT03759938) trials by randomising people with minor and moderate strokes within 48 h, by its imaging-based approach and by comparing the ultra-early initiation with the 1–3–6–12 rule, which has become the standard of care for many physicians. In contrast to the American STAR (NCT03021928) trial, ELAN also includes patients with large infarct volumes. Furthermore, ELAN is a pragmatically designed global trial with sites in Europe, the Middle East and Asia, intending to provide easily applicable results for worldwide use. All four trials have their own strengths, and individual patient-data meta-analyses of all these trials are planned.
Summary and conclusions
ELAN will establish whether there is a clinically important difference in efficacy and safety outcomes of early treatment with a DOAC compared to late guideline-based treatment, in neuroimaging-selected people with an AIS related to AF. The ELAN trial has the potential to resolve a major clinical dilemma for many stroke physicians, to change future stroke guidelines and to benefit patients.
Footnotes
Appendix
Acknowledgements
Editorial assistance was provided by Susan Kaplan.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: This is an academic investigator-initiated trial.
Urs Fischer: Research support of the Swiss National Science Foundation (32003B_197009; 32003B_169975) and the Swiss Heart Foundation for the ELAN trial. Conflicting interests and disclosures outside the ELAN trial: research grants from Medtronic and Stryker; consultancy for Medtronic, CSL Behring, Alexion/Portola; Speakers honorarium from Abbott and Medtronic; DSMB member in the TITAN and IN EXTREMIS trials; Vice President of the Swiss Neurological Society
Sven Trelle: ST is affiliated with CTU Bern, University of Bern, which has a staff policy of not accepting honoraria or consultancy fees. However, CTU Bern is involved in design, conduct, or analysis of clinical studies funded by not-for-profit and for-profit organizations. In particular, pharmaceutical and medical device companies provide direct funding to some of these studies. For an up-to-date list of CTU Bern’s conflicts of interest see ![]()
Mattia Branca: MB is affiliated with CTU Bern, University of Bern, which has a staff policy of not accepting honoraria or consultancy fees. However, CTU Bern is involved in design, conduct, or analysis of clinical studies funded by not-for-profit and for-profit organizations. In particular, pharmaceutical and medical device companies provide direct funding to some of these studies. For an up-to-date list of CTU Bern’s conflicts of interest see ![]()
Georgia Salanti: None.
Maurizio Paciaroni: honoraria as a member of the speaker bureau of Sanofi-Aventis, Bristol-Myers Squibb, Daiiki Sankyo and Pfizer.
Cecilia Ferrari: None.
Stefanie Abend: None.
Seraina Beyeler: None.
Daniel Strbian: Unrestricted educational grant from Boehringer Ingelheim.
Goetz Thomalla: received fees as a consultant or lecturer from Acandis, Alexion, Amarin, Bayer, Boehringer Ingelheim, Bristol-Myers Squibb / Pfizer, Daiichi Sanyo, Portola, and Stryker, all outside the submitted work.
George Ntaios: reports speaker fees/advisory boards/research support from Abbott, Amgen, Bayer, Boehringer Ingelheim, BMS, and Pfizer; all paid to the University of Thessaly.
Leo H Bonati: has received an unrestricted research grant from AstraZeneca, and consultancy or advisory board fees or speaker’s honoraria from Amgen, AstraZeneca, Bayer, Bristol-Myers Squibb, Claret Medical, and InnovHeart, and travel grants from AstraZeneca and Bayer.
Patrik Michel: no disclosures/conflicts of interest.
Krassen Nedeltchev: Advisory boards: Bayer, Boehringer Ingelheim, Daichii Sankyo, BMS/Pfizer, unrestricted educational grants: Bayer, Daichii Sankyo, BMS/Pfizer, Alexion.
Thomas Gattringer: BMS Pfizer: speakers’ honoraria, travel support; Bayer: speakers’ honoraria, travel support; Boehringer Ingelheim: speakers’ honoraria, travel support, advisory board
Else C Sandset: Speaker Honoraria from Boston Scientific.
Peter Kelly: Advisory boards – Alexion, Novo Nordisk. Daichii Sankyo, BMS/Pfizer, Bayer have provided unrestricted grant funding for education and research to the Stroke Clinical Trials Network Ireland; Grant funding: Health Research Board Ireland.
Robin Lemmens: No personal disclosures but reports institutional fees for consultancy from BMS, Boehringer Ingelheim, Genentech, Ischemaview, Medpass, Medtronic and Pfizer.
Masatoshi Koga: honoraria from Daiichi-Sankyo, and research support from Daiichi-Sankyo and Nippon Boehringer Ingelheim.
Padmavathy N Sylaja: PN Sylaja – Funding for the RESTORE trial from Indian Council of Medical Research, Advisory board member of Medtronic PRAAN study, Steering committee member of the Angels initiative.
Diana Aguiar de Sousa: personal fees for AstraZeneca advisory board participation, travel support from Boehringer Ingelheim, DSMB participation for the SECRET trial (University of British Columbia), and speaking fees from Bayer outside the submitted work.
Natan M Bornstein: Pfizer Israel consultation fees and International speaker bureau; Bayer Israel –consultation fees; Boehringer Ingelheim – consultation fees and International speaker bureau.
Zuzana Gdovinova: received fees as a consultant or lecturer from Biogen, Boehringer-Ingelheim, MSD, Novartis, Pfizer, Sandoz, Schwabe, TEVA.
David Seiffge: reports travel support from Boehringer Ingelheim, advisory board fees from AstraZeneca, and DSMB participation for the SECRET trial (University of British Columbia). Speaker fees from Bayer.
Jan Gralla: Global PI of STAR (NCT01327989) and Swift Direct (NCT03192332) (Medtronic), Consultancy: CEC member of the Promise Study (Penumbra), Consultancy; Swiss National Foundation SNF grant for MRI in stroke.
Thomas Horvath: None.
Jesse Dawson: Speaker fees – Pfizer, BMS, Boeringher Inhgelheim, Daicchi Sankyo, Medtronic, Bayer Research funding – Pfizer, BMS.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: ELAN is an investigator-initiated clinical trial. The sponsor of the trial is the University Hospital Bern (Inselspital) and the trial is supported by grants from the Swiss National Science Foundation (32003B_197009; 32003B_169975), the Swiss Heart Foundation, the UK Stroke Association (2017/02) and the Intramural Research Fund (20-4-5) for Cardiovascular Diseases of the National Cerebral and Cardiovascular Centre, Japan. The clinical trial is managed by the Neuro Clinical Trial Unit at the Department of Neurology, University Hospital Bern, Switzerland. The database, central data monitoring and statistical analyses are performed by the CTU Bern at the University of Bern, Switzerland.
Informed consent
Written informed consent according to country-specific requirements is an inclusion criterion for the study.
Ethical approval
Ethical approval for the study was granted by the Cantonal Ethics Commission (KEK) in Bern, Switzerland and subsequently by all local authorities and/or, if applicable, by national lead ethics committees and competent regulatory authorities at all participating sites.
Guarantor
Professor Urs Fischer.
Contributorship
The following authors designed the ELAN trial protocol: UF, ST, MB, GS, MP, DS, GT, GN, LB, PM, KN and JD. UF, MB and JD drafted the publication of the protocol and all other authors have revised the manuscript and made critical comments.
Trial registration
ClinicalTrials.gov Identifier: NCT03148457
ORCID iDs
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.