
Abstract
Background
The optimal timing of anticoagulation following acute ischaemic stroke or TIA in patients with atrial fibrillation (AF) is a frequent challenge. Early initiation of anticoagulation can reduce the risk for recurrent ischaemic events, but may lead to an increased risk for intracerebral haemorrhage.
Aim
The Prospective Record of the Use of Dabigatran in Patients with Acute Stroke or TIA (PRODAST) study was initiated to investigate outcome events under antithrombotic therapy after ischaemic stroke or TIA in patients with AF. The main objective is to compare the three-month rates of major haemorrhagic events between early (≤ 7 days) versus late (> 7 days) administration of dabigatran or treatment with vitamin-K antagonists started at any time. Occurrences of ischaemic and major haemorrhagic events will be evaluated to determine the optimal time point for initiation or resumption of anticoagulation.
Design and Methods
PRODAST is a prospective, multicenter, observational, non-interventional post-authorization safety study. 10,000 patients with recent (≤ 1 week from index event) ischaemic stroke or TIA and non-valvular AF were recruited at 86 German sites starting in July 2015. The observational plan includes a baseline visit, documentation of data during hospitalization and a telephone-based, central follow-up at three months after the index event. The primary endpoint is the major bleeding rate within three months. Secondary endpoints include rates of recurrent ischaemic or haemorrhagic stroke, TIA, systemic embolism, myocardial infarction and death.
Summary
PRODAST will provide important real-world data on safety and efficacy of antithrombotic therapy after acute stroke and TIA in patients with AF.
Keywords
Introduction
Atrial fibrillation (AF) is the most common cardiac arrhythmia and the cause of ischaemic stroke (IS) or transient ischaemic attack (TIA) in up to 25–30% of patients.1,2 With a relative risk reduction (RRR) of 60–70%, oral anticoagulation is highly effective in the prevention of AF-related IS. 3 The benefit of vitamin-K antagonists (VKA) and non-vitamin-K–dependent oral anticoagulants (NOAC) for secondary stroke prevention in AF patients is clearly established. According to meta-analyses of randomized controlled trials (RCT) and observational studies, NOAC are at least as effective as VKA to prevent IS and are associated with a reduced risk for haemorrhagic complications compared to VKA.4-7 However, it is still uncertain at which time after a TIA or an acute IS anticoagulant therapy should ideally be started or resumed. In this context, the secondary preventive benefit has to be weighed against the potential risk of bleeding and, in particular, the risk of intracerebral haemorrhage (ICH) due to haemorrhagic transformation (HT) of the infarct. Importantly, both the risks for recurrent IS and for HT are high in the first few weeks after an acute IS: according to observational studies, the risk of IS recurrence in AF patients without anticoagulant therapy is estimated to be 0.5–1.3% per day (d) in the first two weeks after IS. 8 Patients with recent IS (< 7–14 d) were excluded in the RCTs RE-LY, 9 ROCKET-AF, 10 ARISTOTLE 11 and ENGAGE 12 that led to the approval of dabigatran, rivaroxaban, apixaban and edoxaban, respectively. Data on early use of NOAC after acute IS derived from RCTs are currently limited to only few and small studies.13,14
According to observational studies, a starting interval between 4 and 14 d after acute IS is recommended. 15 However, this finding could not be confirmed in a more recent study. 16 Previous studies are limited by sample size, confounding and especially confounding by indication. Therefore, the recommendations for the ideal timing of anticoagulation after acute IS are still conflicting. Given the lack of evidence, guidelines are based merely on expert opinions.17-20 Recent surveys on this topic among stroke specialists revealed an absence of consensus.21,22 In conclusion, there is a clear need for closing the evidence gap regarding the timing of anticoagulation in patients with acute IS and AF. Thus, real-world data on the efficacy and safety of anticoagulant therapy regimens in the acute and early post-acute phase following IS are highly warranted. In order to address these requirements, the ‘Prospective Record of the Use of Dabigatran in Patients with Acute Stroke or TIA’ (PRODAST; ClinicalTrials.gov Identifier: NCT02507856) study has been initiated in 2015. The study’s recruitment target was reached in November 2020 and the assessment of the vital status is currently ongoing. Analyses regarding the primary endpoints are scheduled to be completed by the mid of 2022.
Design and Methods
PRODAST is a prospective, multi-centre, observational, non-interventional post-authorization safety study (PASS) and recruited 10,000 patients with acute IS or TIA and non-valvular AF at 86 German sites starting in July 2015. The main objective of PRODAST is to collect real-world data on important outcome events after initiation of antithrombotic treatments for the secondary prevention of IS in AF patients. The following antithrombotic treatments will be considered as categories: Dabigatran, other NOAC, VKA, antiplatelet therapy, parenteral anticoagulant therapy and no antithrombotic treatment.
Inclusion and exclusion criteria. For patients who were not able to provide informed consent by themselves, this was given by a legal representative. In the case that it was not possible to obtain informed consent before the start of data collection because of the medical condition of a patient without a legal representative, the investigator was authorized to decide on inclusion in the study and the appropriate consent was provided as soon as possible. AF needs to be documented by 12 lead electrocardiogram (ECG), ECG rhythm strip, monitor print-out, pacemaker/ICD electrocardiogram, Holter ECG (duration of AF episode of at least 30 seconds) or written physician’s diagnosis prior to index event.

Procedures
PRODAST is an observational study, and thus, all therapeutic procedures are conducted at the discretion of the treating physicians according to the Summary of Product Characteristics (SmPC). The study collects information on demographics, lifestyle factors, clinical characteristics of the index event including stroke severity, outcome and infarct size, AF characteristics, medical history and concomitant diseases, vital and laboratory parameters, antithrombotic treatment, treatment compliance and concomitant therapies.
The study time points are depicted in Figure 1. In addition to a detailed observation of in-hospital treatment, all patients except those discharged with NOAC other than dabigatran undergo a central follow-up at three months by standardized telephone interviews with patients and general practitioners. Furthermore, in these patients vital status is assessed one year after index event. For a detailed description of the collected data, see the online supplement. Schedule of study time points. In the case that the central follow-up could not be performed, the vital status was assessed and, if necessary, the cause of death was obtained.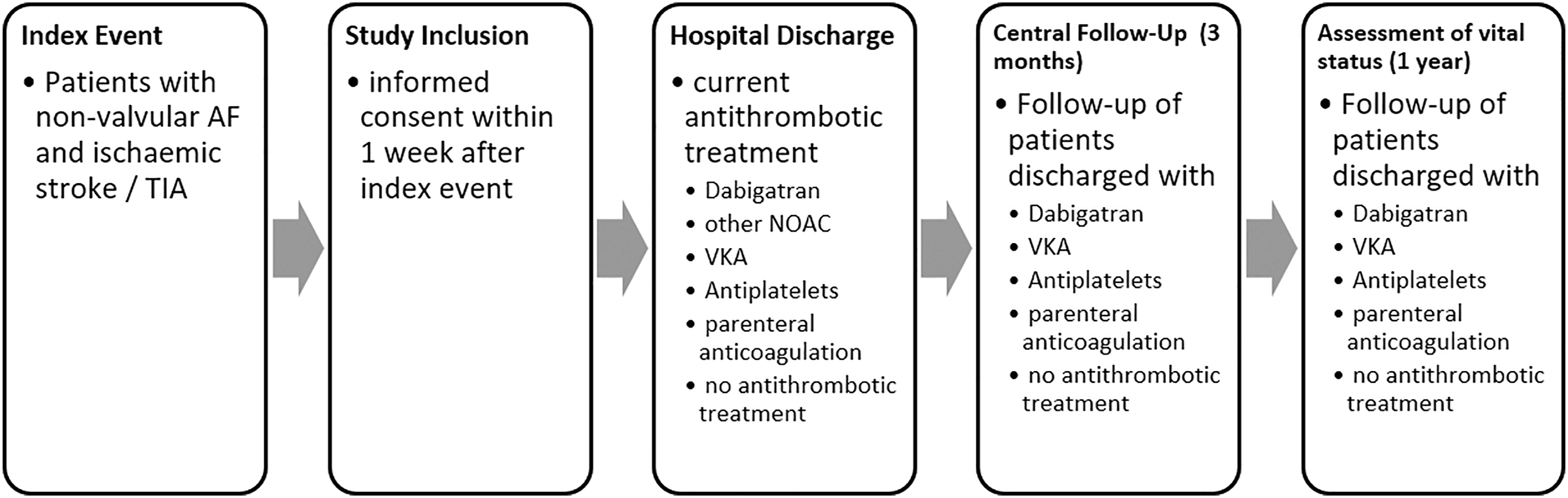
Outcomes
The primary endpoint is defined as a major bleeding event within three months following the index event. A major bleeding event is defined as a fatal bleeding, intracranial, intraocular, intraspinal, retroperitoneal, intraarticular or intramuscular bleeding causing a compartment syndrome, or as a clinically overt bleeding associated with a decrease in the haemoglobin concentration of > 2g/dL or with indication for transfusion of two or more units of whole blood or packed cells or indication for surgical intervention. Symptomatic intracranial haemorrhages are defined as an intracranial bleed as diagnosed with CT or MRI with accompanying worsening of neurological symptoms. The three-month major bleeding event rate will be compared in patients who received dabigatran early (≤ 7 days) with those who received dabigatran after 7 days or vitamin-K antagonists at any time. Secondary endpoints include the three-month rate of recurrent IS or haemorrhagic stroke, TIA, systemic embolism, myocardial infarction, death from all causes and a composite endpoint consisting of stroke, systemic embolism, life-threatening bleeding as well as death from any cause at three months and at one year after the index event. The initial assessment of endpoints occurring during hospitalization is performed by the study sites. Following central and on-site data verification, unclear outcome events are confirmed by an independent clinical adjudication committee (CAC), which will also review outcome events occurring during the follow-up period, taking into account all available information. In unclear cases, medical records will be retrieved from the general practitioner in addition.
Statistical analysis
This observational post-authorization study was not designed to test a formal primary null hypothesis. However, with recruiting 10,000 patients the follow-up was projected to include approximately 6,000 participants. For example, under the assumption that the risk of major bleeding is doubled (1% versus 0.5%) in patients with early versus late administration of dabigatran with a group size of 1,800 or 600, respectively, the power to detect this difference is projected as 84%. Accordingly, the power to detect a doubling of bleeding risk in patients with VKA treatment versus late administration of dabigatran (2% versus 1%) with a group size of 2,400 or 1,800, respectively, is projected to be over 99%.
Starting from the day of index event (or from the day of treatment change), each patient’s observed time under treatment will be included until the occurrence of the target event or be censored at the end of follow-up, or at death, respectively. If the treatment regimen under investigation is changed, the respective observed person times under the particular treatment regimens will be considered for the analysis. The hazard rate ratio will be analysed using Cox-proportional hazards regression including time-dependent coefficients in order to investigate a potential difference in risk of developing a major bleeding event under early or late initiation of anticoagulation. The analysis will be adjusted for potential confounding factors, as discussed below. Minimal sufficient adjustment sets will be identified using directed acyclic graphs (DAGs) under the use of DAGitty V3.0 (see online Supplementary Figure 1). 23 Delayed antithrombotic effects of treatment regimens as described in Supplementary Table 1 will be considered in sensitivity analyses. All models will be stratified by prevalent use of antithrombotic treatment at index and adjusted for age.
Time-dependent treatment effects will be analysed in all eligible patients. Data will be completed with multiple imputations. Sensitivity analyses will be performed on all complete cases. The estimated hazard rates will be presented for the different medication regimens.
Discussion
PRODAST is the largest prospective study to date addressing the timing of anticoagulation after acute AF-related IS or TIA. Although meanwhile a couple of observational studies have been conducted on this topic, results and interpretation are inconsistent. The ‘Early Recurrence and Cerebral Bleeding in Patients With Acute Ischemic Stroke and Atrial Fibrillation’ (RAF) study (n = 1,029) found that initiation of anticoagulation within a window of 4 to 14 days after IS might be associated with a lower incidence of recurrent IS as well as HT 15 which has led to a corresponding recommendation in the guidelines of the American Heart Association (AHA). According to more recent results from the ‘Initiation of Anticoagulation after Cardioembolic stroke’ (IAC) study (n = 1,289), however, no relevant differences in occurrence of ischaemic or haemorrhagic events were found when comparing the starting interval of 4–14 days with earlier or later administration of anticoagulation. 16 In accordance, neither the ‘Clinical relevance of Microbleeds in Stroke’ (CROMIS-2) study 24 (n = 1,355) showed a difference between an early (≤ 4d) or later (> 4d) start of anticoagulation in regard to outcome measures, nor did the ‘Stroke Acute Management with Urgent Risk-factor Assessment and Improvement’ (SAMURAI-NVAF) study (n = 499; ≤ 3d vs. > 3d). 25 Other smaller studies were conducted yielding heterogeneous findings.
As a result of the low level of evidence, there are heterogeneous concepts in starting regimens of anticoagulant therapy in clinical practice. Recent surveys among stroke specialists in the United States and the United Kingdom revealed a lack of consensus in decision making.21,22
Observational studies are prone to confounding, and regarding this research question, confounding by indication is a special matter of concern. Factors may influence the risk for recurrent IS and haemorrhagic complications after acute IS, and these predictors are likely to be considered by treating physicians in deciding for or against an early administration of anticoagulants after IS. The most important predictor for an HT is infarct size. Numerous studies provided evidence that the bleeding risk increases with larger infarct volumes. 26 Vice versa, patients with larger infarcts are also at higher risk for a recurrent IS. 27 Age and traditional vascular risk factors are also associated with both the risks for haemorrhagic and ischaemic events, 28 while measures of atrial cardiopathy, cardiac thrombi, previous recurrent TIA or IS are more predisposed to another cerebral ischaemic infarction. 29 On the contrary, patients with renal insufficiency may be at higher risk for HT. 26 Acute stroke therapies, that is, thrombolysis and mechanical thrombectomy, are further factors influencing the balance between risks for HT or re-ischaemia. 30 The multitude of confounders that are to be considered in the timing of anticoagulation underscores the need for large sample sizes in order to obtain reliable estimates of real-world safety and efficacy measures. Accordingly, we will assess the prevalence of risk factors for recurrent IS and HT at hospital admission and discharge and will perform accordingly adjusted analyses. It is furthermore possible that therapies will be changed or discontinued in a relevant number of patients. To account for time-varying risks and the possibility of treatment changes, the treatment will be modelled as a time-dependent exposure in the proportional hazards regression. Strengths of this study include the large sample size, the multi-centre design, the detailed characterization of demographic, clinical factors and adverse events as well as the standardized central follow-up. Limitations arise from the non-randomized study design, which requires the above-mentioned statistical methods. In addition, the restriction of patients who undergo follow-up due to the scope of the PASS design may bias the results of the long-term observation. However, PRODAST will also contribute to a better understanding of which anticoagulant treatment strategies are selected for particular patient characteristics, thus providing further insight into this potential bias.
Other prospective studies are currently underway to address the issues regarding anticoagulation after stroke. The ‘Registry of Acute Stroke Under Novel Oral Anticoagulants-prime’ (RASUNOA-prime) study prospectively recruits 4,000 patients with acute ischaemic stroke and 1,000 patients with ICH and AF. Participants need to be hospitalized within 24 hours after index event and undergo a 90-day follow-up. NOAC and VKA-treated patients are recruited in a balanced fashion. Primary endpoint is the rate of major bleeding in IS patients and the rate of secondary haematoma expansion in patients with ICH. 31
Currently, three RCTs (Early Versus Late Initiation of Direct Oral Anticoagulants in Post-ischaemic Stroke Patients With Atrial fibrillation (ELAN), Optimal Delay Time to Initiate Anticoagulation After Ischemic Stroke in Atrial Fibrillation (START) 32 and Optimal Timing of Anticoagulation After Acute Ischaemic Stroke: a Randomised Controlled Trial (OPTIMAS)) are recruiting stroke patients with different designs to compare NOAC starting regimens. Results from the ‘Timing of oral anticoagulant therapy in acute ischemic stroke with atrial fibrillation’ (TIMING) study 33 have not been published so far but recently been presented at the European Stroke Organisation Conference 2021. 34 In TIMING, 888 IS patients with AF were randomized in a 1:1 fashion to an early (≤ 4 d) or delayed (5–10 d) start of NOAC therapy.
The ‘Apixaban for Early Prevention of Recurrent Embolic Stroke and Hemorrhagic Transformation’ (AREST) trial randomly assigned patients with acute TIA, small- or medium-sized stroke to an early administration of Apixaban at day 0 to 3 for TIA, day 3 to 5 for small-sized IS and day 7 to 9 for medium-sized IS or to warfarin at 1 week after TIA, or 2 weeks after IS. 14 After the guideline recommendations regarding anticoagulation after stroke due to AF were changed in favour of administration of DOAC, the AREST trial was prematurely stopped after enrolment of only 91 patients. No symptomatic haemorrhages were observed in apixaban-treated patients, while one occurred in the warfarin study arm. Five asymptomatic haemorrhagic transformations were found in each group. 14 However, due to the very small sample size, no meaningful conclusion regarding the risk of an early administration of anticoagulants after stroke can be drawn from this investigation.
Thus, results from the RCTs ELAN, START and OPTIMAS are eagerly awaited. However, it remains to be shown whether patients in the real-world setting have comparable benefits and harms of anticoagulation compared to those treated in RCTs. In this context, restrictions due to the respective inclusion and exclusion criteria, comorbidities or patient compliance play a decisive role. Observational studies like this are therefore of utmost relevance as a complementary accompaniment to data derived from RCTs. Given all these considerations, PRODAST will significantly contribute to our understanding on how the timing of anticoagulation affects the outcomes of stroke patients in daily practice.
Conclusion
PRODAST will provide important and highly relevant data on safety and efficacy of anticoagulation for secondary prevention after acute IS and TIA. To date, this is the largest prospective study dedicated to the issue of anticoagulant therapy in the early phase after acute IS or TIA due to AF.
Supplemental Material
sj-pdf-1-eso-10.1177_23969873211060219 – Supplemental Material for Rationale, Design and Methods of the Prospective Record of the Use of Dabigatran in Patients with Acute Stroke or TIA (PRODAST) Study
Supplemental Material, sj-pdf-1-eso-10.1177_23969873211060219 for Rationale, Design and Methods of the Prospective Record of the Use of Dabigatran in Patients with Acute Stroke or TIA (PRODAST) Study by Gerrit M. Grosse, Christian Weimar, Nils Kuklik, Anika Hüsing, Andreas Stang, Marcus Brinkmann, Christoph C. Eschenfelder, Hans-Christoph Diener and on behalf of the PRODAST Investigators in European Stroke Journal
Footnotes
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Christoph Eschenfelder is an employee of Boehringer Ingelheim. The other authors do not declare potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The PRODAST study is funded by an unrestricted grant from Boehringer Ingelheim.
Ethical approval
The PRODAST study is conducted in accordance with national law and the 1964 Helsinki Declaration and its later amendments, and according to the recommendations of the guidelines on Good Clinical Practice and Good Epidemiological Practice. Ethical approval was given by the institutional review board of the University Duisburg-Essen.
Contributorship
CW, AS, MB, CCE and HCD conceived the study. CW, NK, AH, AS, MB, CCE and HCD were involved in the protocol development, gaining ethics and regulatory approvals, data management and conceiving the statistical analysis plan. GMG wrote the first draft of the manuscript. All authors reviewed and edited the manuscript and approved the final version of the manuscript.
Guarantor
HCD
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.