
Abstract
Introduction:
Drug-induced liver injury (DILI) is a significant cause of drug attrition and market withdrawal, underscoring the importance of the early assessment of hepatotoxicity during drug development. Disruption of bile acid (BA) homeostasis can precipitate liver injury, making the regulation of BA by the liver essential to prevent DILI. Conventional bile salt export pump (BSEP) inhibition assays have poor predictive value, as they do not consider the BA feedback mechanism that mitigates hepatotoxicity. In this study, we present an innovative approach for the preclinical evaluation of the BA-induced hepatotoxic potential of drug candidates.
Methods:
The C-DILI™ Assay employs two distinct media conditions to differentiate between BA-dependent and BA-independent cytotoxicity by measuring LDH release and ATP depletion in sandwich-cultured human hepatocytes. Seventy-one drugs were evaluated, including 49 unblinded and 22 blinded compounds with varied BSEP inhibition profiles and DILI risk.
Results and Discussion:
The assay successfully identified 14 drugs with BA-dependent and 7 with BA-independent hepatotoxicity. For instance, troglitazone (Trog) demonstrated BA-dependent cytotoxicity, whereas cyclosporine A exhibited BA-independent cytotoxicity. Notably, antagonism of farnesoid X receptor (FXR) emerged as a common mechanism underlying BA-dependent toxicity, consistent with FXR’s critical role in BA homeostasis. In the blinded assessment, the assay detected nine drugs with BA-dependent cytotoxicity, affirming its utility in elucidating this mechanism of liver toxicity. Of these, six drugs had documented preclinical or clinical hepatotoxicity findings, thus corroborating the strategy’s value in preclinical safety evaluation. This approach provides a comprehensive and clinically relevant framework for the preclinical prediction of BA-dependent liver toxicity, thereby strengthening preclinical DILI safety assessments. Given the varied clinical presentations and mechanisms of DILI, this strategy should be integrated into a multifaceted preclinical DILI assessment paradigm.
Introduction
Drug-induced liver injury (DILI) remains the most common reason for drug candidate clinical failure or withdrawal from the market. 1 The liver plays an important role in protecting and maintaining whole-body homeostasis in addition to its role as a primary drug clearance organ via uptake, metabolism, and basolateral and biliary excretion mechanisms. In these roles, the liver is susceptible to DILI. Therefore, it is important to illuminate the mechanisms of DILI and develop methodologies to evaluate a drug candidate’s potential to cause liver injury early in drug development to mitigate clinical liver injury risk.
Drug-induced disruption of bile acid (BA) homeostasis has been established as a common DILI mechanism. The intracellular concentration of BA must be stringently regulated to maintain the health of hepatocytes and prevent the initiation of programmed cell death pathways via endoplasmic reticulum stress.2–4
Historically, inhibition of the biliary efflux of BA via bile salt export pump (BSEP) has been a major focus of DILI screening paradigms in early drug development to reduce the liability risk.5–7 While BSEP plays an important role in the biliary clearance of BA, the effectiveness of such inhibition screens is belied by the BA feedback mechanism triggered by BSEP inhibition.8,9 The accumulation of BA via BSEP inhibition activates the BA feedback mechanism via farnesoid X receptor (FXR), resulting in the increased basolateral efflux of BA, thereby preventing a BA-induced hepatotoxic event.8–11 Therefore, assessing BSEP inhibition to triage drug candidates is likely an ineffective DILI screening strategy and may in fact be counterproductive in removing pharmacologically effective nonhepatotoxic molecules from drug development.
Herein, we demonstrate the novel employment of two different hepatocyte culture media, standard and BA sensitization [presence of 250 µM total BA pool + free fatty acids (FFA)], with sandwich-cultured human hepatocytes (SCHH) to discriminate BA-dependent from BA-independent DILI using LDH leakage and ATP depletion as standard biochemical endpoints. We evaluated 71 different drugs, 49 unblinded and 22 blinded with various BSEP inhibition profiles and DILI incidence, using this approach, namely the C-DILI™ Assay.
Materials and Methods
Chemicals, hepatocytes, and reagents
Transporter Certified™ primary human hepatocytes (Table 1) were obtained from Thermo Fisher Scientific (Waltham, MA) and BioIVT (Westbury, NY). Primary human hepatocytes were cultured with proprietary cell culture media formulations developed by Qualyst Transporter Solutions (QTS), a division of BioIVT, including QualGro™ Seeding Medium, QualGro™ Culture Induction Medium, QualGro™ C-DILI Culture Medium (5 mM glucose), and QualGro™ C-DILI Sensitization Medium (5 mM glucose, 250 µM BA pool, 1 mM FFA).
Primary Hepatocytes Utilized Across Study

BA, bile acid; FXR, farnesoid X receptor.
The base medium (DMEM) for all media formulations used by QTS and additional supplements, including fetal bovine serum, used for cell culture were from Gibco (Carlsbad, CA) and Corning (Tewksbury, MA), respectively. Forty-nine commercial compounds listed in Table 2 were purchased from Cayman Chemicals (Ann Arbor, MI). Dimethyl sulfoxide (DMSO) and chenodeoxycholic acid (CDCA) were purchased from Sigma Aldrich (St. Louis, MO); deoxycholic acid (DCA) was purchased from Santa Cruz Biotechnology (Dallas, TX). All other BAs, including glycine-DCA, glycine-CDCA, and glycine-cholic acid, were purchased from Steraloids (Newport, RI). Oleate and palmitate were purchased from Sigma Aldrich. DY268 was purchased from Tocris (Bristol, UK). Deuterated-taurocholate (d8-TCA) was purchased from Martex (Minnetonka, MN).
Clinical Evidence of Marketed or Withdrawn Drugs Associated with Drug-Induced Liver Injury
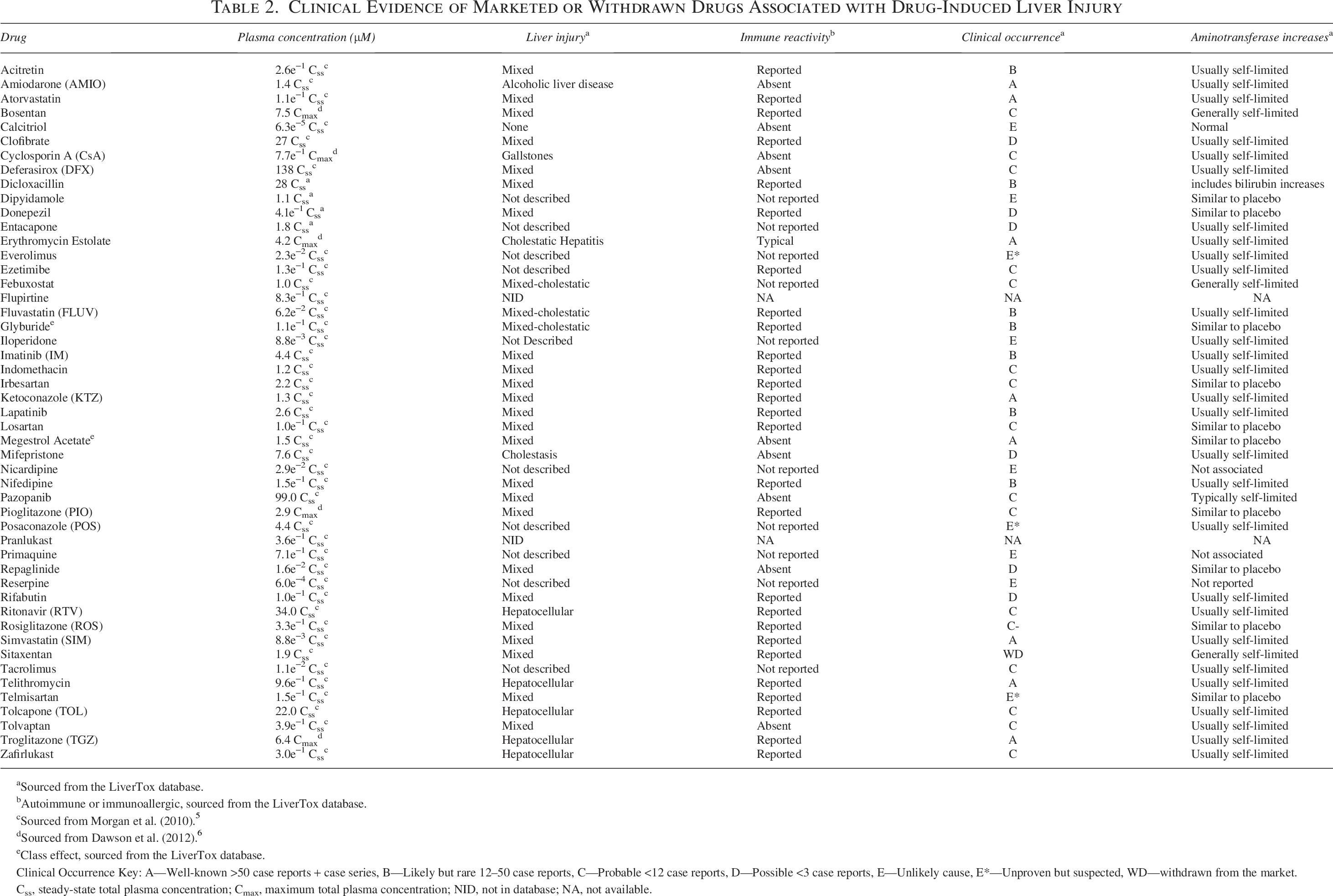
Sourced from the LiverTox database.
Autoimmune or immunoallergic, sourced from the LiverTox database.
Sourced from Morgan et al. (2010). 5
Sourced from Dawson et al. (2012). 6
Class effect, sourced from the LiverTox database.
Clinical Occurrence Key: A—Well-known >50 case reports + case series, B—Likely but rare 12–50 case reports, C—Probable <12 case reports, D—Possible <3 case reports, E—Unlikely cause, E*—Unproven but suspected, WD—withdrawn from the market.
Css, steady-state total plasma concentration; Cmax, maximum total plasma concentration; NID, not in database; NA, not available.
Pfizer provided 22 compounds to BioIVT in a blinded manner using a unique identifier with molecular weight information. Ten of these compounds, previously published or not Pfizer proprietary, were unblinded following the conclusion of the study, including PF-06273340, PF-04895162, TAK875 (fasiglifam), PF-01428777, 25-desacetyl-3-formyl RIF (rifampicin metabolite), montelukast, zafirlukast, CP-724714, panadiplon, and ADX10059; however, 12 proprietary molecules remained blinded.
Preparation of sandwich-cultured hepatocytes
SCHH were established by thawing Transporter Certified cryopreserved human hepatocytes (Table 1) according to the manufacturer’s instructions. Once thawed, the hepatocytes were seeded (∼0.05 million cells/well) into 96-well collagen coated plates purchased from Corning. Cells were allowed to attach for 2–4 hours, then rinsed, and fed with QualGro Seeding Medium. Following 18–24 hours, the seeding medium was removed, and the cells were fed and overlaid with QualGro Culture Induction Medium supplemented with 0.25 mg/mL Matrigel (Corning). Cells were then maintained in QualGro Culture Induction Medium until day 4 of culture.
Compound solubility assessments
Prior to beginning the experiment, stock solutions of all test compounds were prepared. The stock solutions were prepared at 1000X the desired treatment concentration using DMSO as the common solvent. All test compounds were visually assessed for solubility in QualGro C-DILI Culture Medium (standard medium) and QualGro C-DILI BA Sensitization Medium, containing a fixed concentration of total BA (250 µM BA pool) and FFA (1 mM), as previously described. 8 Briefly, previously prepared stock DMSO solutions (1 µL) were spiked into 1 mL of warm media and maintained at 37°C overnight. Any cloudiness or precipitation indicated incomplete solubility, and a lower concentration of the test compound was evaluated.
Measuring BA biliary efflux
The effects of cyclosporine A (CsA), troglitazone, pioglitazone, and rosiglitazone on the hepatobiliary disposition of BA were determined using SCHH as described above in the 96-well format and treated as follows: Stock solutions of CsA and Trog (1000X final concentration) were prepared in DMSO and then diluted directly into QualGro Culture Induction Medium on the day of assay to yield the desired final concentrations. Hepatocytes were treated with CsA, troglitazone, pioglitazone, or rosiglitazone at 1, 5, 10, 25, 50, and 100 µM for 2 hours on day 5 of culture. Following the 2-hour exposure period, the cultures were rinsed, and a disposition study with d8-TCA was performed using B-CLEAR® technology, as previously described.8,12,13 The plates were frozen at −80°C until processed for bioanalysis to determine protein content and disposition of d8-TCA. The biliary excretion index (BEI), biliary clearance (ClBiliary), and total accumulation were determined. Protein content was determined using the Pierce® BCA Protein Assay Kit (Thermo Fisher Scientific) following the manufacturer’s instructions.
Bioanalysis of d8-TCA
Following hepatobiliary disposition studies, the quantitation of d8-TCA was conducted as previously described. 8
Measuring cytotoxicity
On day 4 of culture, SCHH were utilized in the C-DILI Assay (patent pending). Briefly, SCHH hepatocytes were exposed to test articles for ≤24 hours beginning on day 4 of culture. Compound DMSO stocks (1000X) were diluted directly into QualGro C-DILI Culture Medium and QualGro C-DILI BA Sensitization Medium. Following the exposure period, LDH leakage in media samples was determined using the CytoTox-ONE™ homogeneous membrane integrity fluorescence assay from Promega (Madison, WI). Subsequently, ATP was determined using the CellTiter-Glo™ luminescent cell viability assay from Promega. Both assays were performed on the same wells, namely LDH measurements using media and ATP measurements using cell lysates, in parallel according to the manufacturer’s instructions. Luminescence and fluorescence were measured with a BioTek Synergy 4 Plate Reader (Winooski, VT). Each treatment group was conducted in triplicate wells.
Evaluation of FXR antagonism
Four independent experiments were conducted in SCHH to evaluate treatment effects on FXR activation. To assess FXR activation status, the mRNA content of well-established FXR target genes, including OSTβ and/or FGF19, was assessed following exposure.8–11
In the first three independent experiments, SCHH were prepared and treated on day 4 of culture with CsA (10 µM), CDCA (30 µM) alone, CsA (10 µM) + CDCA (30 µM), DY268 (5 µM) + CsA (10 µM) + CDCA (30 µM), deferasirox (400 µM) + CsA (10 µM) + CDCA (30 µM), ketoconazole (26 µM) + CsA (10 µM) + CDCA (30 µM), troglitazone (50, 75, 100 µM) + CsA (10 µM) + CDCA (30 µM), pioglitazone (100 µM) + CsA (10 µM) + CDCA (30 µM), or rosiglitazone (100 µM) + CsA (10 µM) + CDCA (30 µM) for 24 hours.
In the first FXR antagonism study, we included a cytotoxicity assessment to rule out these effects on gene expression results. Cytotoxicity was measured in parallel using ATP as previously described above. SCHH utilized for the parallel cytotoxicity assessment were treated under the same conditions as previously described above.
We examined the disruption of FXR activation in SCHH by treatment in the following order: (1) troglitazone dose, (2) pioglitazone and rosiglitazone, and (3) deferasirox and ketoconazole. DY268, a potent FXR antagonist, was utilized as a positive control in these experiments.8,14–16 Following exposure, hepatocytes were washed with HBSS 3X and lysed by the addition of 0.1 mL of Qiagen RLT Buffer (Germantown, MD) supplemented with an equal volume of Qiagen Qiazol. Lysates were frozen at −80°C until processed.
In the fourth independent experiment, FXR antagonism was measured differently. On day 4, hepatocytes were treated under QualGro C-DILI Culture Medium and QualGro C-DILI BA Sensitization Medium containing the solvent control DMSO (0.1%), negative control (CsA), positive control (troglitazone), or test articles. Following 12 hours of exposure, hepatocytes were washed with HBSS 3X and lysed by the addition of 0.1 mL of Qiagen RLT Buffer supplemented with an equal volume of Qiagen Qiazol. Lysates were frozen at −80°C until processed.
Total RNA isolation and qRT-PCR
Lysates were thawed, and then, total RNA was isolated from each treatment group from pooled triplicate wells using the Qiagen RNeasy kit following the manufacturer’s instructions. RNA was stored at −80°C until processed. Isolated RNA was quantified using the Quant-iT™ RiboGreen® RNA Assay Kit (Thermo Fisher Scientific) following the manufacturer’s instructions. The pooled total RNA (250 ng) was converted into cDNA following the manufacturer’s procedure for the High-Capacity cDNA Archive Kit (Thermo Fisher Scientific). cDNA from SCHH was analyzed from each reverse transcription (RT) reaction using gene-specific TaqMan® assays (Thermo Fisher Scientific) for OSTβ (Assay# Hs01057182_m1) and/or FGF19 (Assay# Hs00192780_m1). Glyceraldehyde 3-phosphate dehydrogenase (Assay# Hs99999905_m1) was used as the endogenous house-keeping gene to normalize each sample. Amplifications were performed on a ViiA™ 7 Real-Time PCR System (Thermo Fisher Scientific) in the relative quantification mode for 45 amplification cycles using standard conditions for TaqMan-based assays. Threshold cycle (CT) determinations were performed by the ViiA™ 7 system software program for all target and endogenous control genes. Relative-fold mRNA content was determined for each treatment group relative to the endogenous control gene expression and the calibrator, namely 0.1% DMSO vehicle control. Confidence intervals of 95% were calculated for each target gene relative quantitation (RQ) mean by the ViiA™ 7 system software program.
BA biliary efflux
To quantitate BA biliary efflux, the BEI and biliary clearance were determined as previously described.8,12,13
Data analysis
All calculations were performed using Microsoft Excel for Microsoft 365 MSO Version 2412 (Redmond, WA), unless otherwise stated. Statistical analyses were performed using GraphPad Prism Software Version 9.5.1 (La Jolla, CA), and p-values were defined in figure legends, where applicable.
Hepatobiliary disposition results, including BEI and ClBiliary of d8-TCA, were analyzed using two-way analysis of variance (ANOVA), followed by Tukey’s multiple comparison test. The statistical significance of ATP content and LDH leakage results was evaluated using two-way ANOVA, followed by Dunnett’s multiple comparison test.
Curve fit analysis of SCHH ATP content was analyzed using the nonlinear regression analysis tool of GraphPad Prism software. Using the comparison-of-fit tool, three-parameter and four-parameter (variable slope) nonlinear regression models were analyzed for the best fit of ATP depletion under both culture conditions. Neither of the curve fits was applicable to ATP depletion in SCHH treated with ritonavir under standard conditions. However, four-parameter (variable slope) curve fits were the best (p < 0.0001) to describe ATP depletion in SCHH treated with ritonavir under BA sensitization conditions (R2 = 0.992) and in SCHH treated with simvastatin under standard (R2 = 0.985) or BA sensitization conditions (R2 = 0.987).
Results
Disruption of BA disposition by thiazolidinediones
Following exposure to CsA or thiazolidinediones, the biliary clearance of d8-TCA was significantly (p < 0.05) reduced by >90% of the solvent control in a dose-dependent manner in SCHH treated with CsA or troglitazone at the highest concentrations assessed (Fig. 1A). Although the biliary clearance of d8-TCA was reduced in dose-dependent manner in SCHH treated with rosiglitazone, the reduction in clearance was not reduced by 50% at the concentrations examined. In contrast, no significant decreases (p > 0.05) in d8-TCA biliary clearance were observed in SCHH following pioglitazone treatment. These results demonstrated that CsA, troglitazone, and rosiglitazone, but not pioglitazone, disrupt hepatic BA clearance.
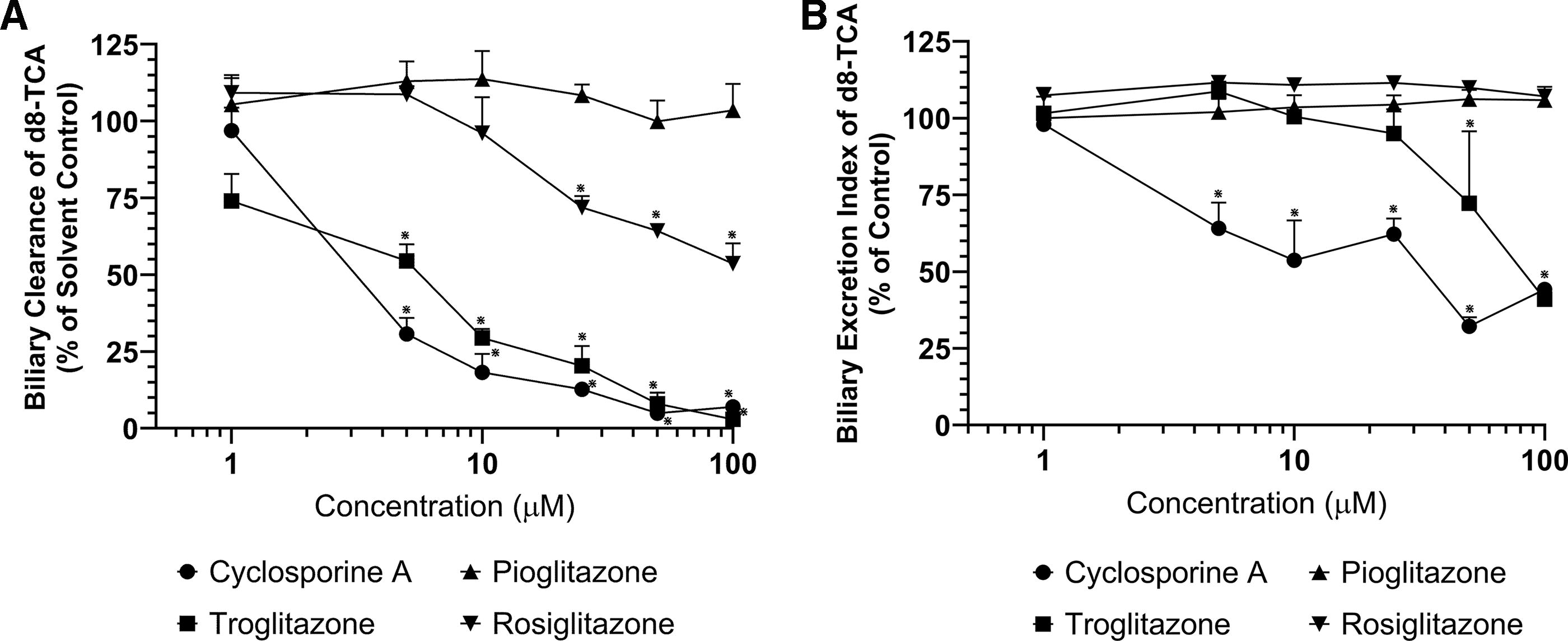
Following 2-hour exposure to CsA or thiazolidinediones, the biliary clearance and efflux of the model BA d8-TCA was evaluated in SCHH utilizing B–CLEAR® technology.
The BEI, a measure of biliary efflux, of d8-TCA was reduced significantly (p < 0.05) in a dose-dependent manner in SCHH treated with either CsA or troglitazone, suggesting that these compounds inhibit BSEP across the concentrations examined (1–100 μM, Fig. 1B). No reduction in d8-TCA biliary efflux was observed in SCHH treated with either pioglitazone or rosiglitazone, suggesting that neither drug inhibits the biliary efflux of BA via BSEP. Although rosiglitazone reduced the biliary clearance of d8-TCA, the lack of an effect on the biliary efflux of the model BA suggests that rosiglitazone only inhibits the uptake of BA.
FXR antagonism by thiazolidinediones
Following 24-hour treatment with the prototypical BSEP inhibitor CsA (10 μM), the prototypical FXR agonist CDCA (30 μM), 17 the FXR antagonist DY268 (5 μM), 14 troglitazone, pioglitazone, rosiglitazone, or a combination of these treatments, FXR activation was assessed in SCHH using the expression of FGF19, a sentinel marker of FXR activation.8,10,18 CsA (10 μM) increased FGF19 mRNA content 1.9-fold above the solvent control, whereas CDCA (30 μM) increased FGF19 mRNA content 23-fold above the solvent control (Fig. 2A). A synergistic increase in FGF19 mRNA content of 60-fold was observed in SCHH co-treated with CsA (10 μM) and CDCA (30 μM; Fig. 2A). As expected, DY268 treatment reduced the synergistic induction of FGF19 mRNA content by 90.3% when co-treated with CsA (10 μM) + CDCA (30 μM). A similar effect was observed in the co-treatment including troglitazone (50–100 µM), CsA (10 μM), and CDCA (30 μM; Fig. 2A). In this co-treatment, troglitazone reduced the synergistic induction of FGF19 mRNA content in a dose-related manner. These results suggested FXR activation was inhibited in the presence of troglitazone.
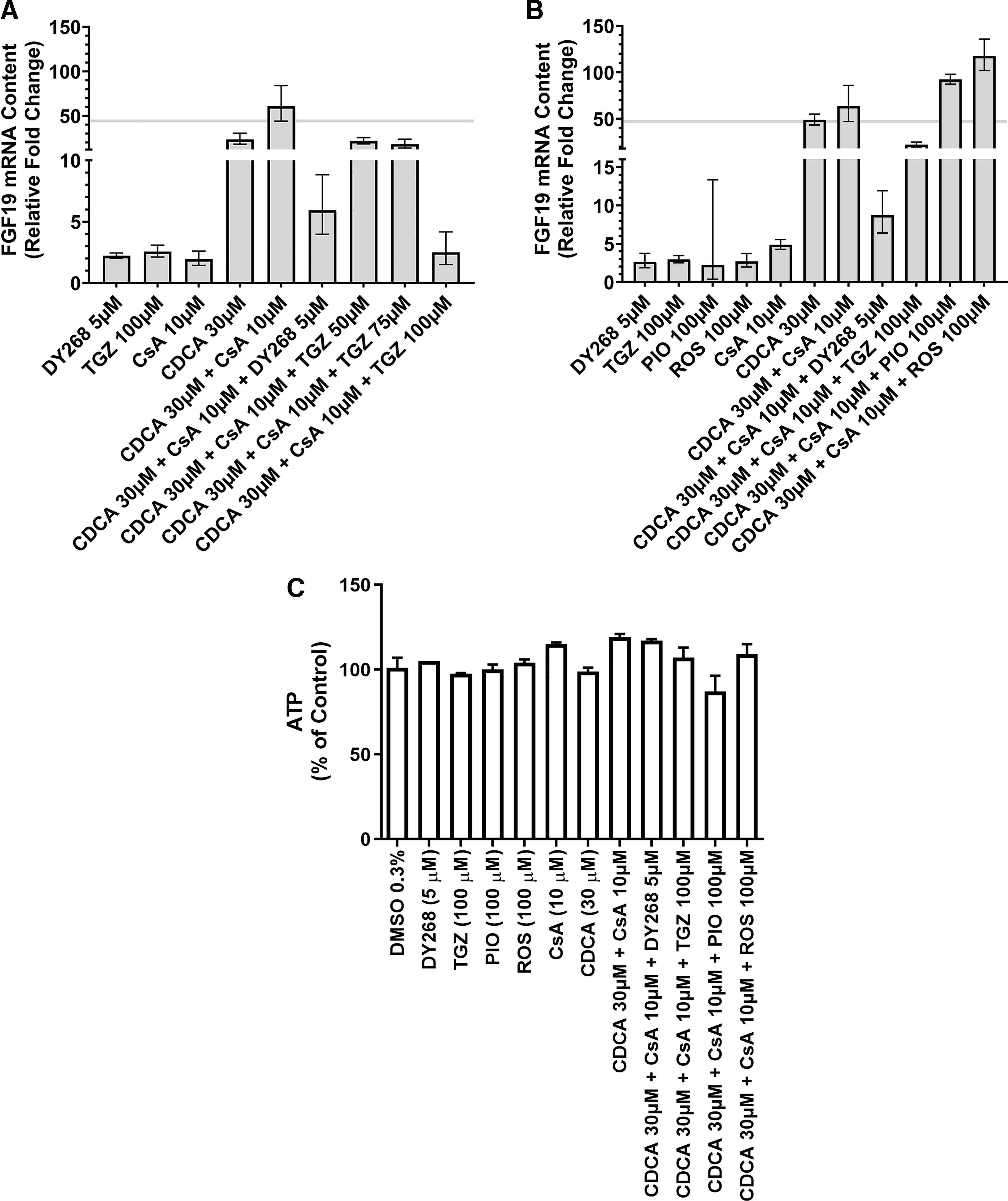
The expression of FGF19, a sentinel marker of FXR activation, was evaluated in SCHH following 24-hour treatment.
In a separate independent experiment re-demonstrating troglitazone FXR activation antagonism (Fig. 2B), neither pioglitazone nor rosiglitazone treatment at 100 µM reduced FGF19 induction in SCHH co-treated with CsA (10 μM) + CDCA (30 μM). These results suggested that pioglitazone and rosiglitazone lack FXR antagonism properties.
In a parallel cytotoxicity assessment, no evidence of cytotoxicity was observed in SCHH treated under the same conditions utilized to evaluate FXR antagonism (Fig. 2C). The lack of cytotoxicity indicated that the decreases of FGF19 mRNA content in SCHH exposed to troglitazone or DY268 in co-treatment with CsA + CDCA resulted from the inhibition of FXR activation by BA. These results also demonstrated that the co-treatment, namely CsA + CDCA, utilized to investigate FXR activation status was not cytotoxic to SCHH, suggesting that this co-treatment study design was adequate to assess FXR antagonism.
Mechanism-based assay to identify cholestatic hepatotoxicity safety hazard among thiazolidinediones
The cholestatic hepatotoxicity potential of the thiazolidinediones, including troglitazone, pioglitazone, and rosiglitazone, was evaluated using a BA mechanism-based assay, C-DILI Assay. SCHH were treated for 24 hours with the test article under either standard or BA sensitization medium conditions. BA sensitization medium was supplemented with an exogenous BA pool (250 µM) and FFA (1 mM). Cytotoxicity was measured by the leakage of LDH and by the depletion of ATP content (Fig. 3A, B).
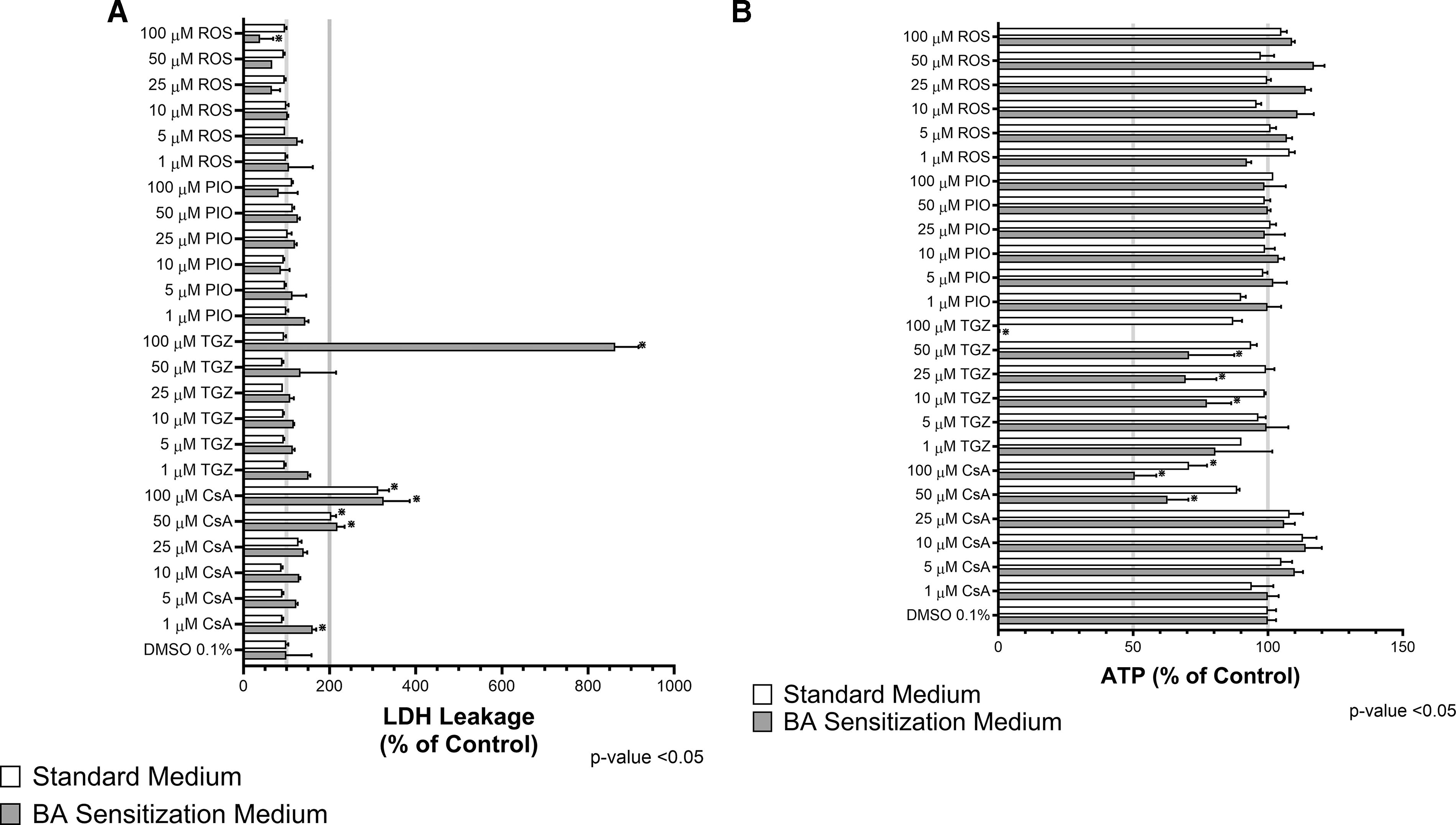
The BA-dependent hepatotoxicity potential of the thiazolidinediones, including troglitazone, pioglitazone, and rosiglitazone, was evaluated using the C-DILI™ Assay. SCHH were treated for 24 hours with the test article under either standard or sensitization medium conditions. Cytotoxicity was measured by leakage of
None of the thiazolidinediones at the concentrations evaluated significantly increased LDH secretion (Fig. 3A) or depleted ATP content (Fig. 3B) in SCHH under standard medium conditions. Under sensitization conditions, however, troglitazone (100 μM) was the only thiazolidinedione evaluated to significantly (p < 0.05) increase LDH secretion by >200% (Fig. 3A) and deplete ATP content (Fig. 3B). These results suggested that troglitazone cytotoxicity was BA-dependent.
Exposure to CsA, a well-known BSEP inhibitor, from 1 to 100 μM [maximum total plasma concentration (∼Cmax) to 125× Cmax; Table 2 resulted in significant (p < 0.05) LDH leakage and ATP loss under both standard and sensitization conditions at concentrations ≥50 µM (≥62.5× Cmax; Table 2). These results suggested that CsA cytotoxicity was independent of BA. Taken together, these results demonstrated the C-DILI Assay could discriminate between the BA-independent or -dependent hepatotoxicity potential of a test compound by employing two different media formulations with and without exogenous BAs.
Kinetics of cytotoxicity via drug-induced disruption of BA homeostasis
To better understand the kinetics of the cytotoxicity resulting from the drug-induced disruption of BA homeostasis, we assessed LDH leakage and ATP depletion of SCHH under standard and BA sensitization conditions following a 24-hour time course exposure to CsA (10 µM) or troglitazone (50–75 µM). No marked loss of ATP or LDH leakage was observed under standard conditions following the 24-hour time course with either CsA or troglitazone treatment, consistent with previous results (data not shown). However, a time-dependent increase in LDH leakage was observed in SCHH treated with troglitazone (75 µM) under BA sensitization conditions (Fig. 4A). Following 18 hours of exposure, LDH leakage significantly increased (p < 0.05) in SCHH treated with 75 µM (12× Cmax; Table 2) troglitazone, but not with 50 µM (8× Cmax; Table 2). No marked increase in LDH leakage was observed in SCHH treated with 10 µM CsA (13× Cmax; Table 2) under BA sensitization conditions following the 24-hour time course.
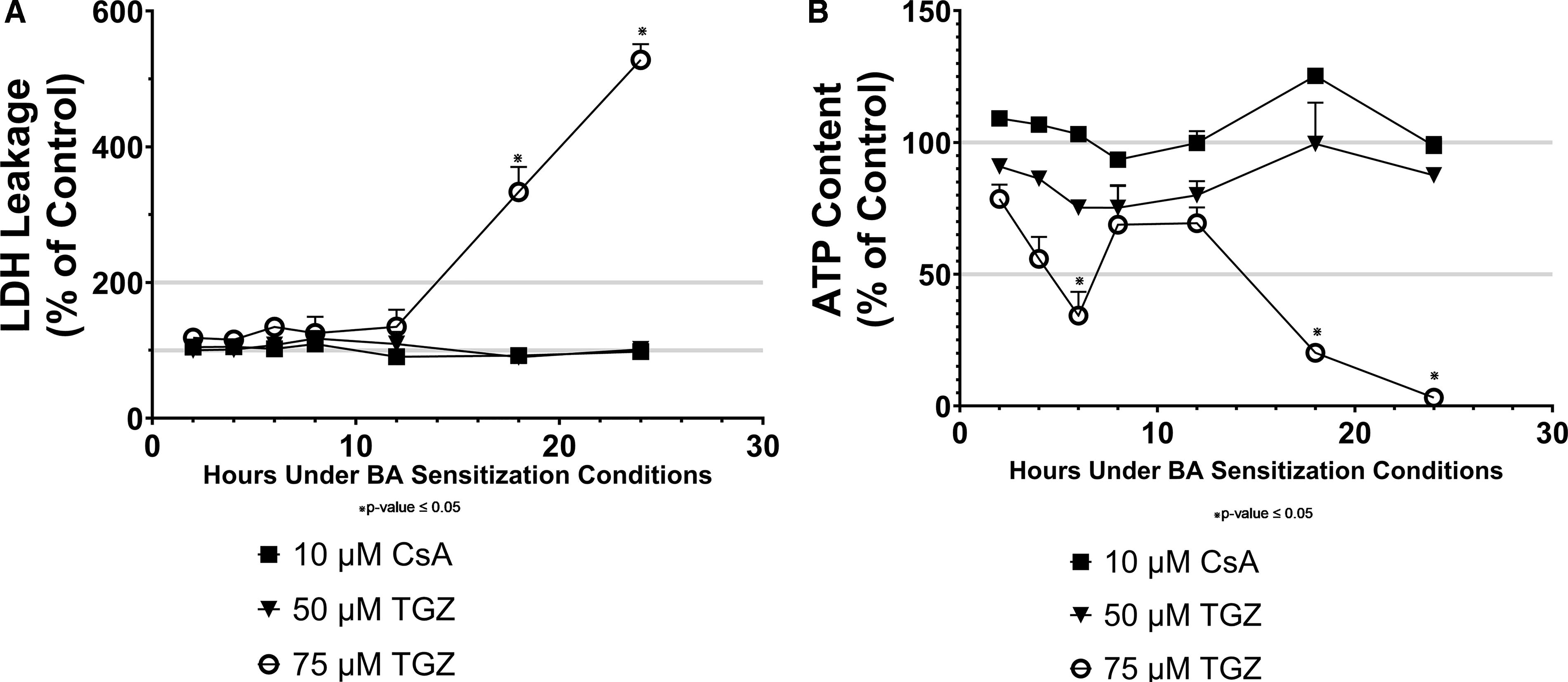
Interestingly, ATP content during the 24-hour time course under sensitization conditions was very dynamic and exhibited similar declines and recoveries across the treatments assessed, consistent with an adaptive or compensatory response (Fig. 4B). The most dramatic of these was demonstrated with a dose-dependent and time-dependent ATP loss in SCHH treated with troglitazone. ATP was reduced by 25% (p > 0.05) and 66% (p < 0.05) within the first 6 hours of exposure to 50 µM (∼8× Cmax; Table 2) or 75 µM (12× Cmax; Table 2) troglitazone, respectively. A noticeable rally in ATP content was observed (p < 0.05) during the 8–12-hour exposure period in SCHH treated with 75 µM (12× Cmax; Table 2) troglitazone (Fig. 4B). However, this recovery period was short-lived, with a significant ATP loss of ≥80% continuing during the 18–24-hour exposure period. The loss of ATP also coincided with the significant release of LDH previously described.
Initial screening of 45 drugs using BA mechanism-based cytotoxicity assay
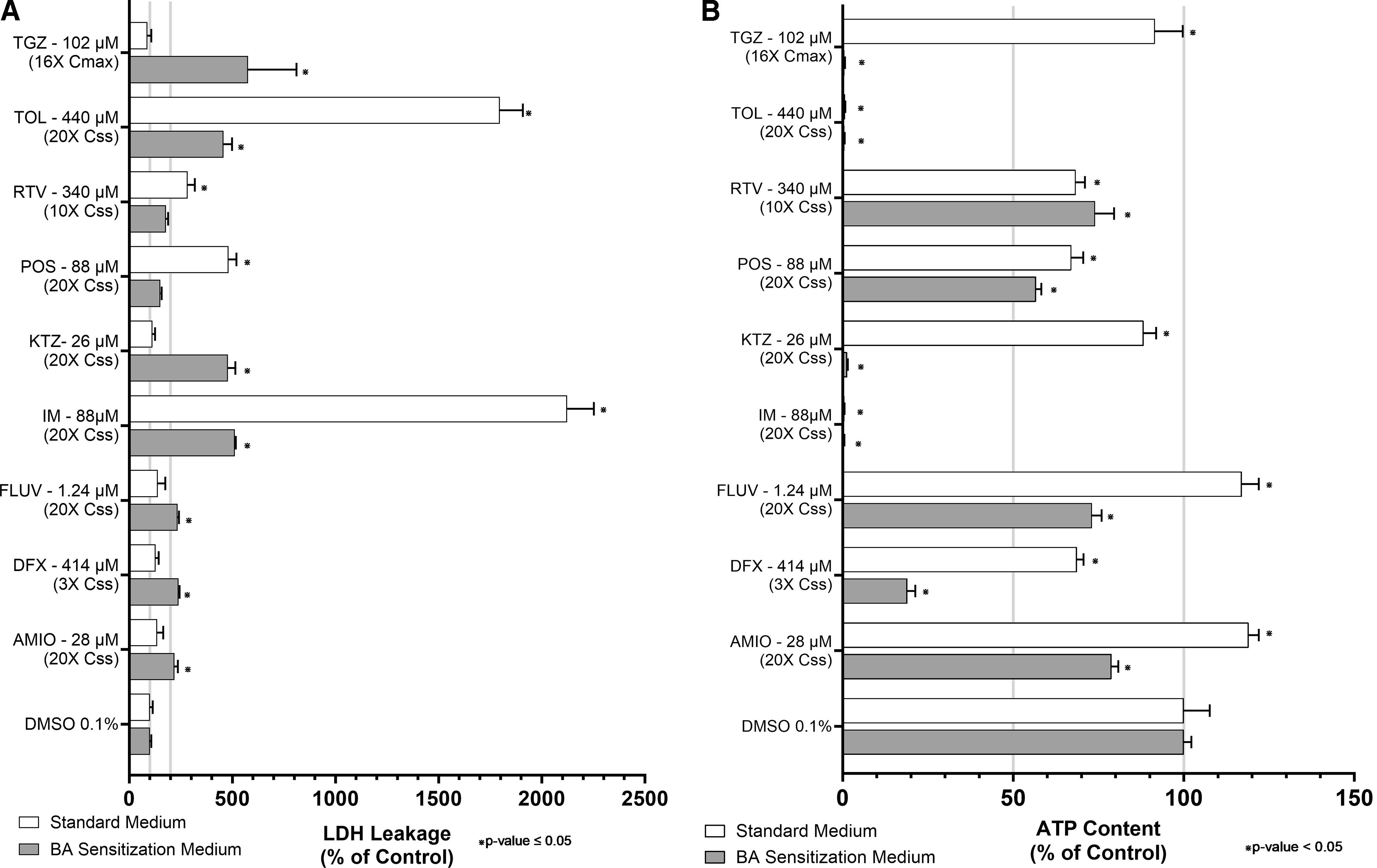
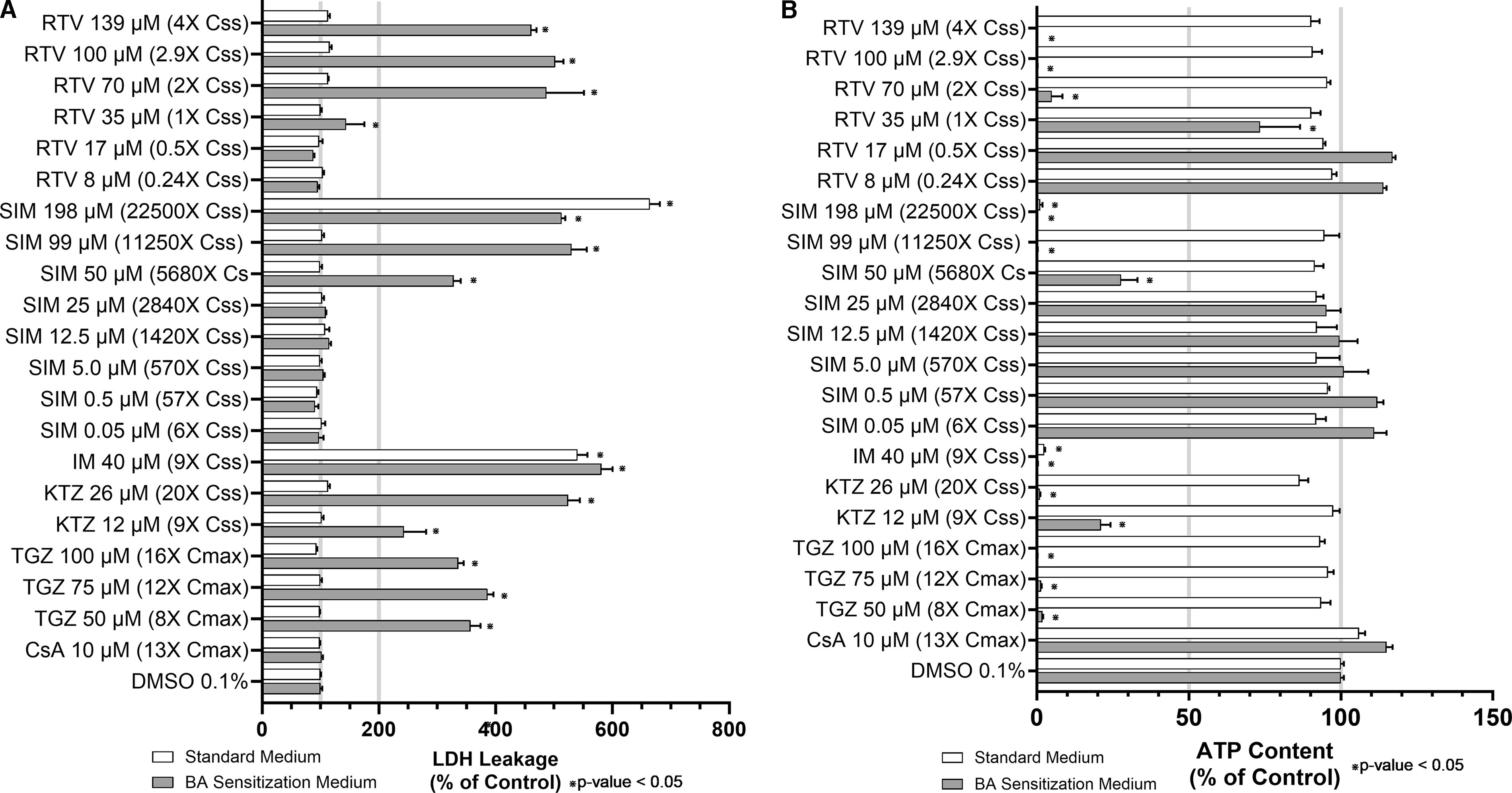
To assess the application of BA sensitization conditions to identify BA-induced hepatotoxicity safety hazards, we exposed SCHH to a diverse collection of 45 test compounds at 20× their systemic Cmax, steady-state plasma concentration (Css), or limit of solubility in standard or BA sensitization medium for 24 hours (Table 2). The 20× of their systemic concentrations was chosen to account for the higher portal vein concentrations observed following oral administration. All compounds evaluated have been reported to have varying degrees of BSEP inhibition potency (Table 3) determined in human BSEP vesicles studies. 5 Following exposure, LDH leakage and ATP content were measured to assess the cytotoxicity of each compound studied. Under standard medium, pronounced LDH leakage >200% (p ≤ 0.05) was observed in SCHH treated with imatinib (20× Css), posaconazole (20× Css), ritonavir (10× Css), and tolcapone (20× Css; Fig. 5A). Concomitant and significant (p ≤ 0.05) decreases in ATP content ranging from 33% to 99% were also observed in SCHH under the same conditions (Fig. 5B). These results suggested that a BA-independent mechanism of cytotoxicity may be involved for imatinib, posaconazole, ritonavir, and tolcapone; Table 4). This inference seems inconsistent with the well-established potency BSEP IC50 (Table 3) thresholds proposed by previous researchers.5,6
Reported BSEP Vesicle IC50 and FXR Antagonism Parameters of Marketed or Withdrawn Drugs
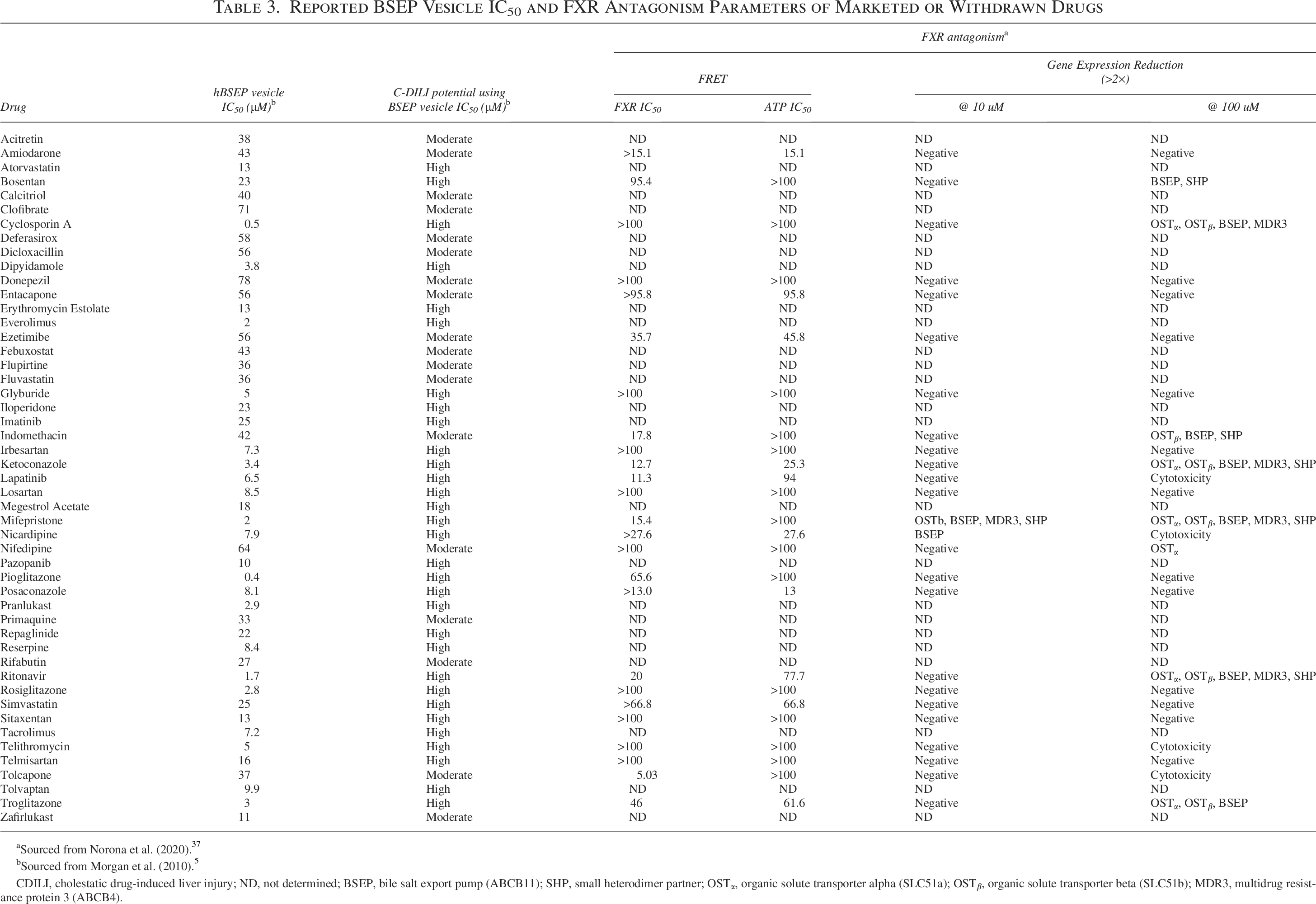
Sourced from Norona et al. (2020). 37
Sourced from Morgan et al. (2010). 5
CDILI, cholestatic drug-induced liver injury; ND, not determined; BSEP, bile salt export pump (ABCB11); SHP, small heterodimer partner; OSTα, organic solute transporter alpha (SLC51a); OSTβ, organic solute transporter beta (SLC51b); MDR3, multidrug resistance protein 3 (ABCB4).
The BA-dependent hepatotoxicity potential of 49 therapeutics with various BSEP inhibition potency and DILI incidence was evaluated using the C-DILI™ Assay. SCHH were treated for 24 hours with the test article under either standard or sensitization medium conditions. Cytotoxicity was measured by leakage of
C-DILI™ Assay BA Dependency Predictions of Marketed or Withdrawn Drugs

Figures 2A,B and 7.
Sourced from Morgan et al. (2010). 5
Sourced from Dawson et al. (2012). 6
Marked (p ≤ 0.05) LDH leakage ranging from 219% to 575% above the DMSO control (Fig. 5A) was observed in SCHH cultured under BA sensitization conditions and treated with amiodarone (20× Css), deferasirox (3× Css), fluvastatin (20× Css), imatinib (20× Css), ketoconazole (20× Css), tolcapone (20× Css), and troglitazone (16× Cmax). Simultaneous significant (p ≤ 0.05) decreases in ATP content ranging from 26% to >99% were also observed in SCHH under identical conditions (Fig. 5B). Apart from imatinib and tolcapone, cytotoxicity appeared to occur in SCHH in a BA-dependent manner with amiodarone, deferasirox, fluvastatin, ketoconazole, and troglitazone treatments. Again, as with BA-independent cytotoxicity, the observed BA-dependent cytotoxicity (Table 4) did not seem to correlate with the BSEP IC50 potencies (Table 3) thresholds proposed by previous researchers.5,6
No significant LDH leakage or ATP depletion (Supplementary Table S1) was observed in SCHH cultured under either standard or BA sensitization condition and treated with acitretin, atorvastatin, bosentan, calcitriol, clofibrate, dicloxacillin, dipyidamole, donepezil, entacapone, erythromycin, everolimus, ezetimibe, febuxostat, flupirtine, glyburide, iloperidone, indomethacin, irbesartan, lapatinib, losartan, megestrol, mifepristone, nicardipine, nifedipine, pazopanib, pranlukast, primaquine, repaglinide, reserpine, rifabutin, rosiglitazone, simvastatin, sitaxentan, tacrolimus, telithromycin, telmisartan, tolvaptan, or zafirlukast. Given that no evidence of cytotoxicity was observed under either culture condition, these results suggested that BA-dependency could not be determined (Table 4).
Confirmation of BA-dependent cytotoxicity
To confirm that the BA-dependent cytotoxicity mechanism was conserved across donors, three SCHH donors (DJJ, Hu8236, Hu8192) were exposed to amiodarone, deferasirox, fluvastatin, imatinib, ketoconazole, and troglitazone. Apart from imatinib, all test articles exhibited BA-dependent cytotoxicity in the previous assessment, as discussed above. Again, each test article was evaluated for 24 hours but at three different concentrations (Table 4).
Extensive dose-dependent increases (p < 0.05) in LDH leakage ranging from 112% to 1943% of the DMSO control were observed across all three SCHH donors cultured under BA sensitization conditions and exposed to troglitazone (8–16× Cmax; Fig. 6A). The concomitant significant (p < 0.05) dose-dependent depletion of ATP ranging from 8% to >99% was also observed across all three SCHH cultures treated with troglitazone under BA-sensitization conditions (Fig. 6B). In contrast, no significant LDH leakage or loss of ATP was observed across all three donors treated with troglitazone under standard conditions. These results were consistent with a troglitazone BA-dependent cytotoxicity mechanism, repeatedly replicated, and thus considered as a positive control BA-dependent cytotoxicity response for the assay (Table 4).
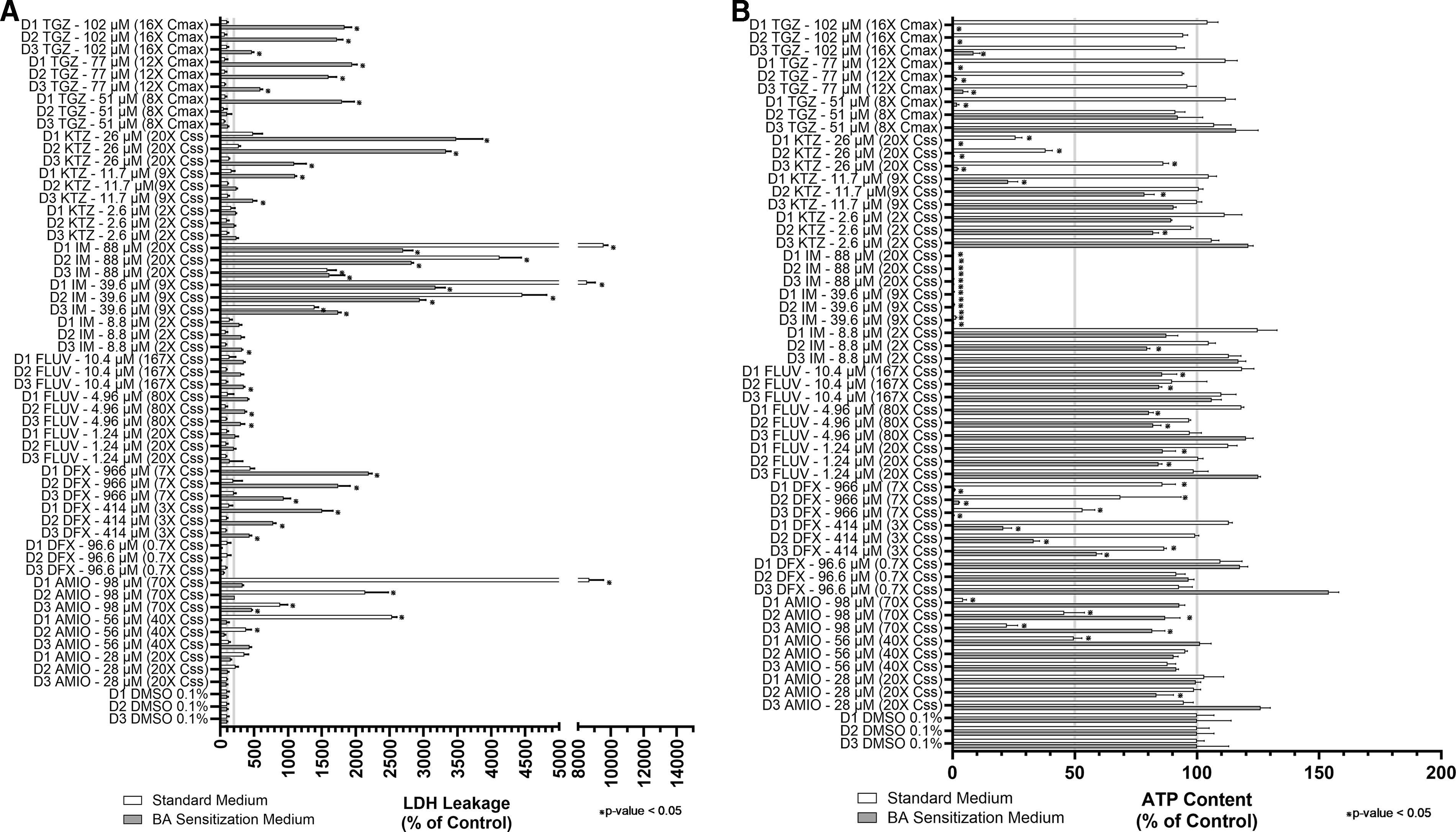
To confirm the bile acid dependency of cytotoxicity, three SCHH (Hu8192, Hu8236, DJJ) were exposed to amiodarone, deferasirox, fluvastatin, imatinib, ketoconazole, and troglitazone at three different concentrations (Table 4) under standard or BA sensitization conditions and evaluated for leakage of
Pronounced dose-dependent increases (p < 0.05) in LDH leakage ranging from 432% to 2191% of the DMSO control were observed across all three SCHH donors treated with deferasirox (0.7–7× Css; Fig. 6A) cultured under BA sensitization conditions. The coinciding significant (p < 0.05) loss of ATP ranging 41% to >99% was conserved under BA sensitization conditions across all three donors at 3× Css and 7× Css (Fig. 6B). Under standard conditions, LDH leakage >200% was observed in one (DJJ) of the three donors treated with deferasirox (7× Css), but this observation was not significantly different from vehicle control. In addition, ATP content remained >85% in DJJ under the same conditions. In general, these results were consistent with previous results demonstrating a BA-dependent mechanism of cytotoxicity at the concentrations assessed (Table 4).
Consistent with previous cytotoxicity results, marked dose-dependent LDH leakage (p < 0.05) was observed in SCHH treated with ketoconazole (2–20× Css) under BA sensitization conditions (Fig. 6A). Extensive LDH leakage (>1000%) was observed in all three donors treated with 20× Css ketoconazole under BA sensitization conditions, but at 9× Css, two (DJJ and Hu8192) of the three donors had marked LDH leakage (>450%). Under BA sensitization conditions, >99% of ATP was depleted in SCHH treated with 20× Css ketoconazole across all three donors, but only DJJ had significant loss (p < 0.05) of ATP at 9× Css (Fig. 6B). Under standard conditions, LDH leakage >200% was observed in DJJ and Hu8236 treated with ketoconazole (20× Css), but these observations were not statistically different from vehicle controls. Taken together, the results in two of the three donors suggested that ketoconazole exhibits a BA-dependent mechanism of cytotoxicity at concentrations ranging from 9 to 20× Css (Table 4).
Dose-dependent increases in LDH leakage >200% were observed in SCHH treated with fluvastatin (20–167× Css) under BA sensitization media conditions (Fig. 6A). LDH leakage was statistically significant (p < 0.05) in two (Hu8236 and Hu8192) of the three donors treated with 80× Css under these conditions. However, no significant dose-dependent ATP depletion was observed in the same cultures; ATP content remained >80% across all three SCHH donors at all fluvastatin concentrations assessed (Fig. 6B). Under standard media conditions, no significant LDH leakage and no significant loss of ATP were observed across any of the three SCHH donors treated at any fluvastatin concentration evaluated. The inconsistency between LDH leakage and ATP depletion under sensitization conditions suggested that fluvastatin lacks a BA-dependent mechanism of cytotoxicity (Table 4).
Clear dose-dependent significant (p < 0.05) increases in LDH leakage >200% were observed in SCHH treated with imatinib (2–20× Css) under standard or BA sensitization conditions (Fig. 6A). Significant (p < 0.05) simultaneous dose-dependent ATP depletion ranging from 5% to >99% was also observed across all three SCHH cultures treated with imatinib under the same culture conditions (Fig. 6B). Overall, these results suggested that imatinib cytotoxicity is generated by a BA-independent mechanism (Table 4).
Significant and >200% (p < 0.05) dose-dependent increases in LDH leakage were observed across SCHH treated with (40–70× Css) amiodarone under standard culture conditions (Fig. 6A). However, concomitant and significant (p < 0.05) dose-dependent ATP depletion ≥50% was observed in DJJ (40× Css and 70× Css), Hu8192 (70× Css), and Hu8236 (70× Css) in standard or both culture conditions (Fig. 6B). Taken together, these results suggested that amiodarone cytotoxicity is generated by a BA-independent mechanism (Table 4).
Evaluation of FXR antagonism
Using standard and BA sensitization culture conditions across three separate SCHH donors, we confirmed that deferasirox and ketoconazole have BA-dependent cytotoxicity mechanisms. Next, we investigated the potential of deferasirox and ketoconazole to the antagonism of FXR signaling in SCHH using FGF19 and OSTα/β gene expression as sentinel markers of FXR activation.8,10,18
Treatment with CDCA increased the FXR target genes, FGF19 and OSTβ mRNA content by 23-fold and 25-fold, respectively, over the DMSO control in SCHH, consistent with the literature (Fig. 7). 8 Co-treatment with CsA and CDCA resulted in a synergistic increase in FGF19 and OSTβ mRNA content of 61-fold and 77-fold greater than the DMSO control, respectively (Fig. 7).8,10,18 When compared with the gene expression responses in CsA and CDCA co-incubation, the response of FGF19 and OSTβ gene expression was reduced by ≥90% in SCHH co-treated with CsA, CDCA, and DY268, a potent FXR antagonist.8,14 Similarly, the response of FGF19 and OSTβ gene expression was decreased by ≥66%, ≥57%, and ≥52% in SCHH co-incubated with CsA, CDCA, and deferasirox; ketoconazole; or troglitazone, respectively (Fig. 7). These results confirm that deferasirox and ketoconazole disrupt BA-induced FXR activation at the same concentrations, resulting in BA-dependent toxicity in the C-DILI Assay. Our detection of FXR antagonism by ketoconazole in SCHH is consistent with the FXR fluorescence resonance energy transfer (FRET) reporter assay and gene expression results reported by Norona et al. (2020) (Table 4).
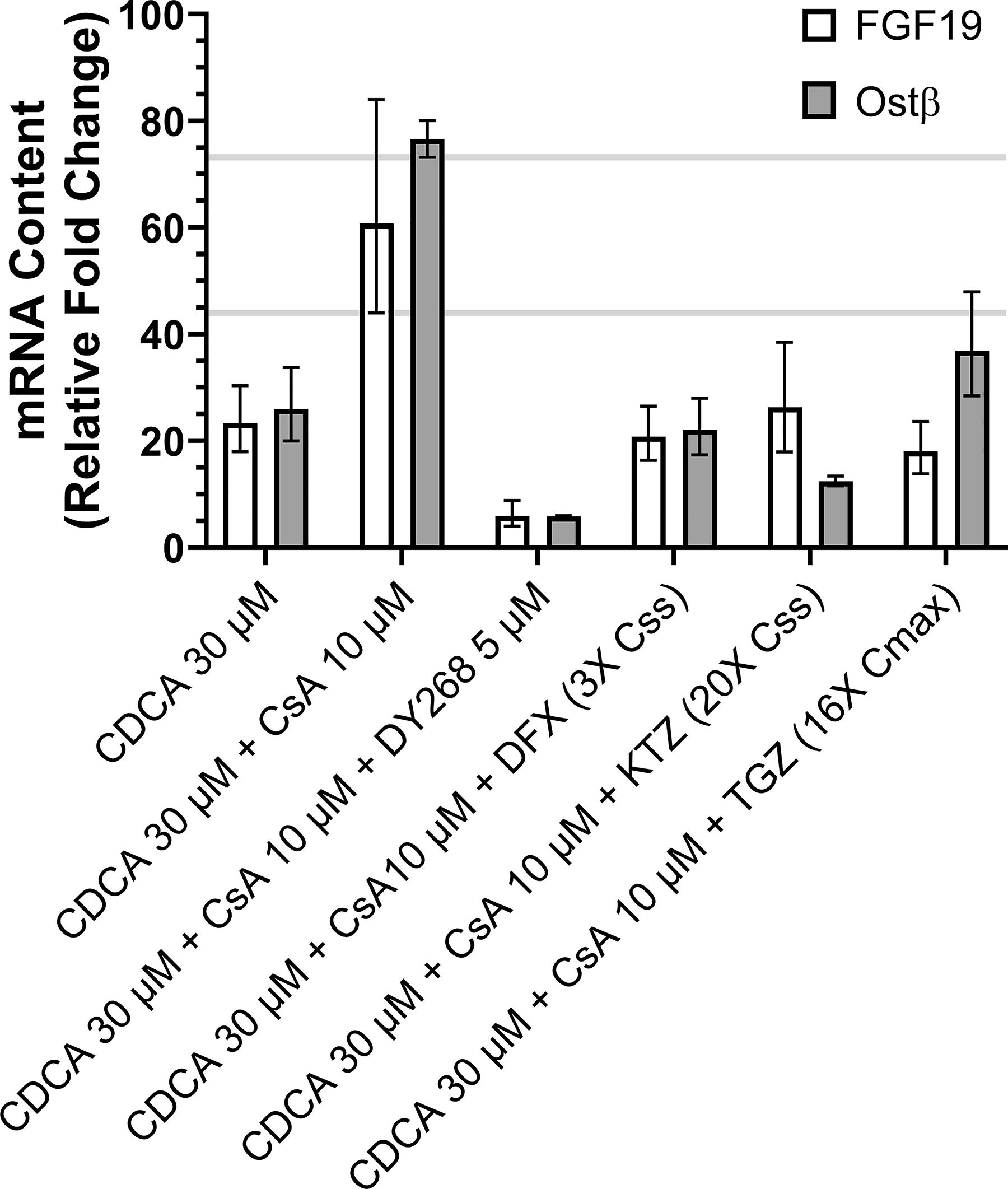
The expression of FGF19 and OSTβ, sentinel markers of FXR activation, was evaluated in SCHH following 24-hour treatment. The FXR agonist CDCA (30 μM) increased FGF19 and OSTβ mRNA content 23- and 26-fold, respectively, above the solvent control. A synergistic increase in FGF19 and OSTβ mRNA content of 61- and 77-fold, respectively, was observed in SCHH co-treated with CsA (10 μM) and CDCA (30 μM). DY268, a potent FXR antagonist, prevented the synergistic induction of FGF19 and OSTβ mRNA content. Troglitazone, ketoconazole, and deferasirox treatment also reduced the synergistic induction of FGF19 and OSTβ mRNA content, suggesting that FXR activation was inhibited. Values represent the RQ mean, error bars represent 95% confidence intervals of triplicates, and nonoverlapping CI indicates statistically significant differences among the groups. Gray lines represent the lowest RQ within 95% CI for gene expression results in SCHH treated with CDCA + CsA.
Reevaluation of mechanism of BA cytotoxicity
A small selection of compounds was chosen to re-assess the BA mechanism of cytotoxicity in a different SCHH donor, namely DJJ, under standard and BA sensitization culture conditions. The compounds chosen were initially identified as inconclusive (simvastatin) or BA-independent (ritonavir) in the donor Hu8192 in previous cytotoxicity assessments.
In our initial assessments of the antiretroviral protease inhibitor ritonavir and the lipid-lowering drug simvastatin, we observed a BA-independent and an inconclusive BA mechanism of cytotoxicity, respectively. Although choosing a single concentration is a great strategy for screening several compounds, a dose–response provides a more comprehensive evaluation. Therefore, we selected a broad range of concentrations to fully explore the BA cytotoxicity mechanism of ritonavir (0.24–4× Css) and simvastatin (6–22500× Css). Dose-dependent extensive (p ≤ 0.05) increases in LDH leakage and the accompanying decreases in ATP content were observed in SCHH treated with ritonavir cultured under sensitization conditions only (Fig. 8A, B, Table 4). Similarly, dose-dependent pronounced (p ≤ 0.05) increases in LDH leakage and the concomitant losses of ATP content were observed in SCHH treated with 5680–11,250× Css simvastatin cultured under sensitization conditions only. Nonlinear regression analysis of the ATP depletion curves for ritonavir and simvastatin exhibited left shifts in SCHH under BA sensitization when compared with standard conditions, resulting in the estimated TC50 parameters of 38.6 (R2 = 0.992) and 39.9 µM (R2 = 0.987) (Fig. 9A, B, Table 4), respectively. Simvastatin treatment at 22,500× Css resulted in marked (p < 0.05) LDH leakage and simultaneous ATP depletion in SCHH cultured under both standard and BA sensitization conditions. Overall, these results suggested that ritonavir and simvastatin induced cytotoxicity in SCHH in a BA-dependent manner across the concentrations examined (Table 4). The estimated ATP depletion TC50 for ritonavir and simvastatin within context to the therapeutic concentrations was 1.1× and >4000× Css, respectively, suggesting that this mechanism is more clinically relevant for ritonavir than for simvastatin.
Re-evaluation of the mechanism of BA cytotoxicity was performed using the C-DILI™ Assay using
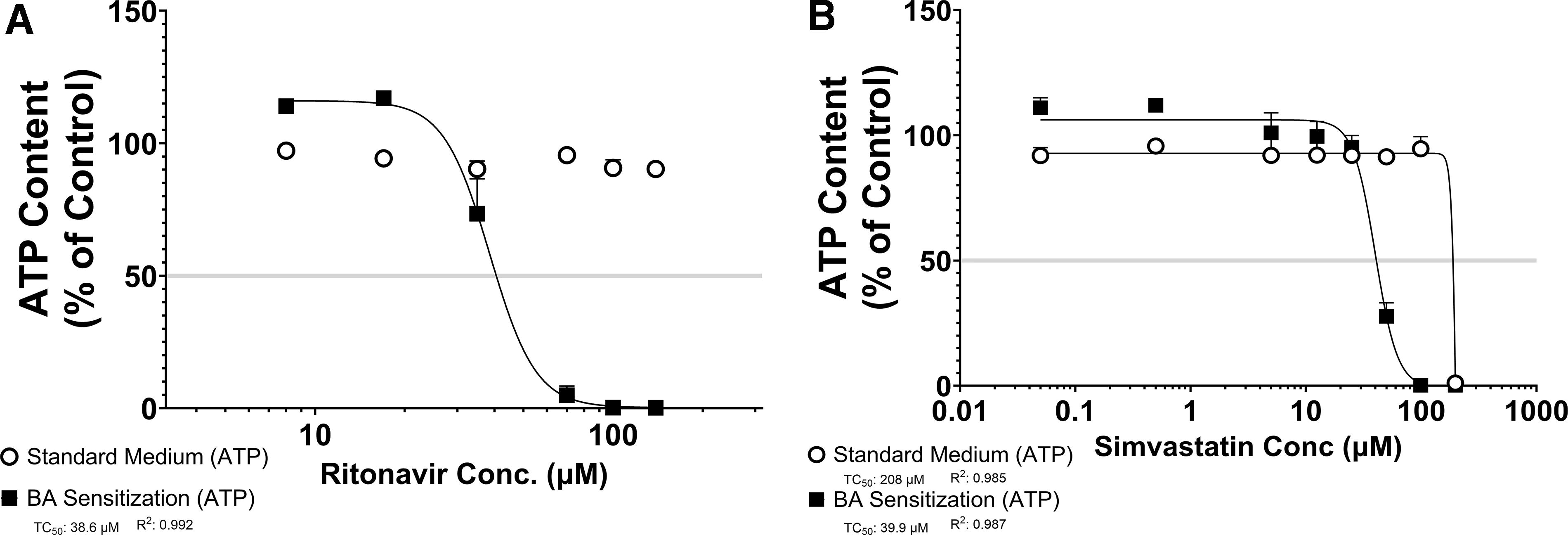
Nonlinear regression analysis of ATP depletion in standard or BA sensitization conditions in SCHH treated with increasing concentrations of
Screening of blinded drugs using cholestatic hepatotoxicity assay
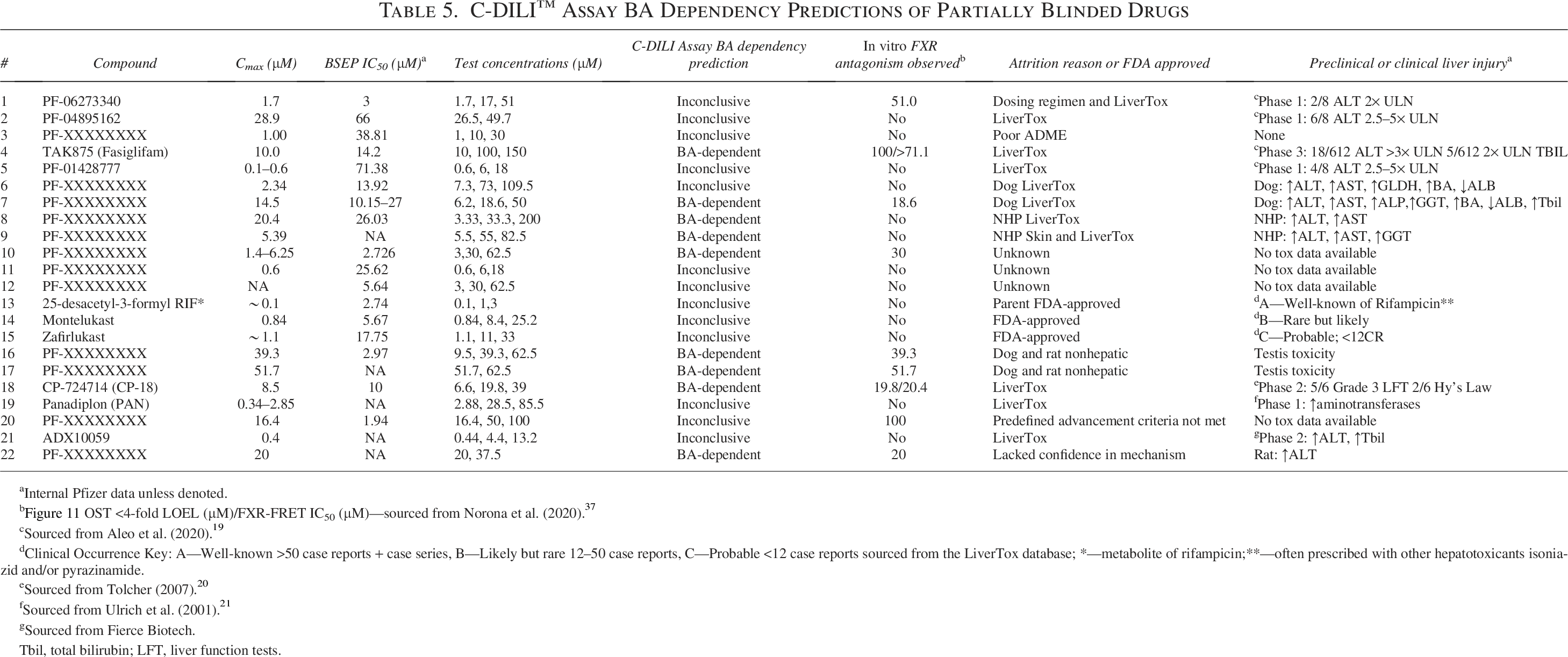
Pfizer provided 22 drugs, 19 failed and 3 approved, to BioIVT for assessment in a blinded manner (Table 5). Each compound was evaluated using 2–3 concentrations, with the lowest concentration at or near Cmax and with the highest concentrations chosen using solubility limitations.
C-DILI™ Assay BA Dependency Predictions of Partially Blinded Drugs
Internal Pfizer data unless denoted.
Sourced from Aleo et al. (2020). 19
Clinical Occurrence Key: A—Well-known >50 case reports + case series, B—Likely but rare 12–50 case reports, C—Probable <12 case reports sourced from the LiverTox database; *—metabolite of rifampicin;**—often prescribed with other hepatotoxicants isoniazid and/or pyrazinamide.
Sourced from Tolcher (2007). 20
Sourced from Ulrich et al. (2001). 21
Sourced from Fierce Biotech.
Tbil, total bilirubin; LFT, liver function tests.
CsA, a well-established BSEP inhibitor and lacking FXR antagonism, was utilized as a negative control and, as expected, failed to increase LDH leakage or decrease ATP content in comparison with the DMSO control under both media conditions (Fig. 10A, B). In contrast, troglitazone and imatinib treatment significantly increased LDH leakage (≥292% control) and depleted ATP (≥97% control) in SCHH. Troglitazone, a well-known BSEP inhibitor with FXR antagonism, markedly (p ≤ 0.05) increased LDH leakage and decreased ATP content in SCHH cultured under sensitization conditions only, representing a BA-dependent hepatotoxicity response. Imatinib treatment extensively (p ≤ 0.05) increased LDH leakage and reduced ATP content in SCHH under both culture conditions, representing a BA-independent cytotoxicity response.
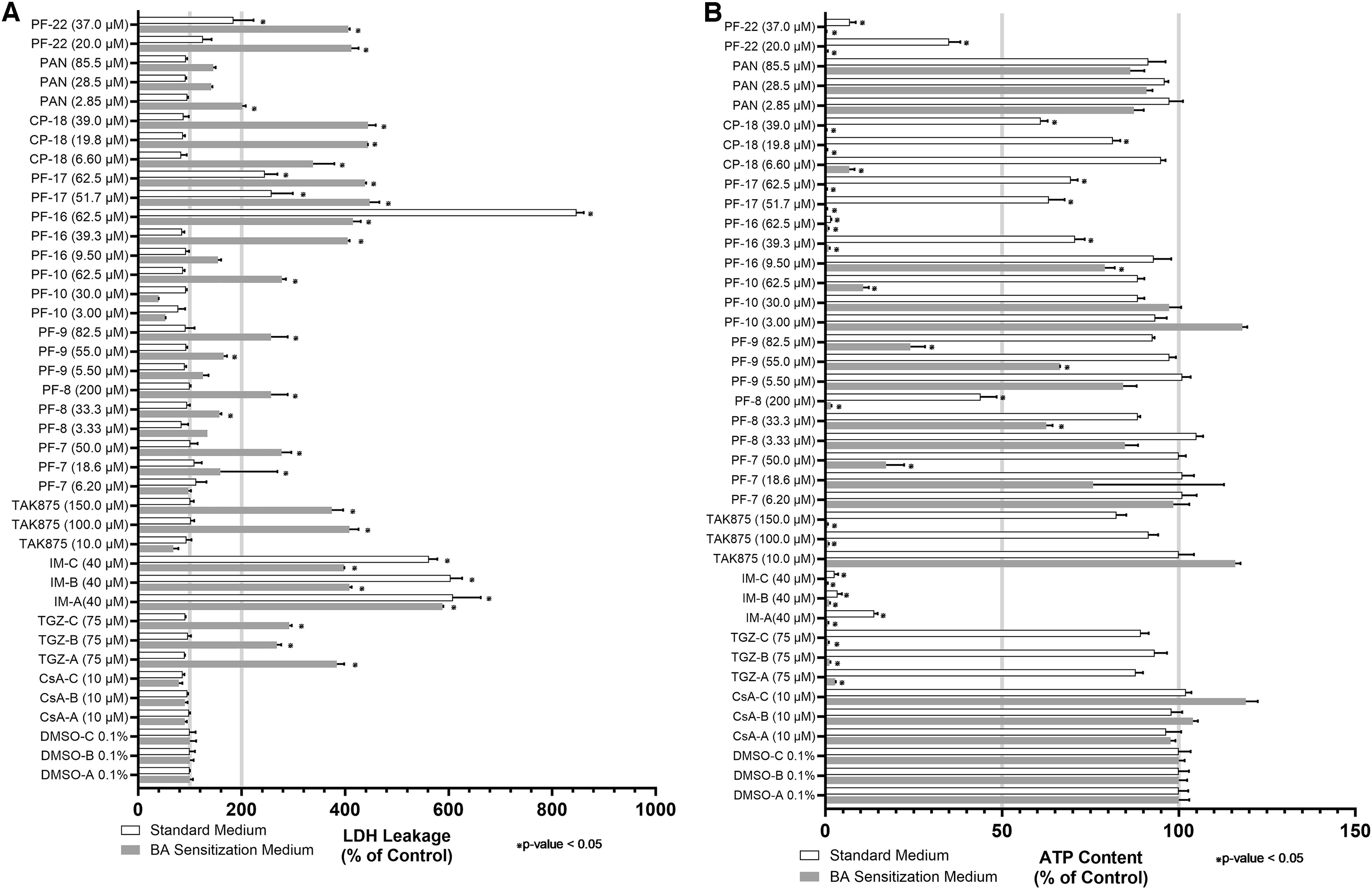
The BA-dependent hepatotoxicity potential of 22 therapeutics with varying BSEP inhibition potency and DILI incidence was evaluated using the C-DILI™ Assay. SCHH were treated for 24 hours with the test article under either standard or BA sensitization culture conditions. These assessments were performed across multiple plates in three separate independent experiments. Controls, including DMSO, CsA, TGZ, and IM, were included on each separate 96-well plate to ensure that the assay performed as anticipated and reproducibly. These independent controls were labeled -A, -B, -C. Cytotoxicity was measured by
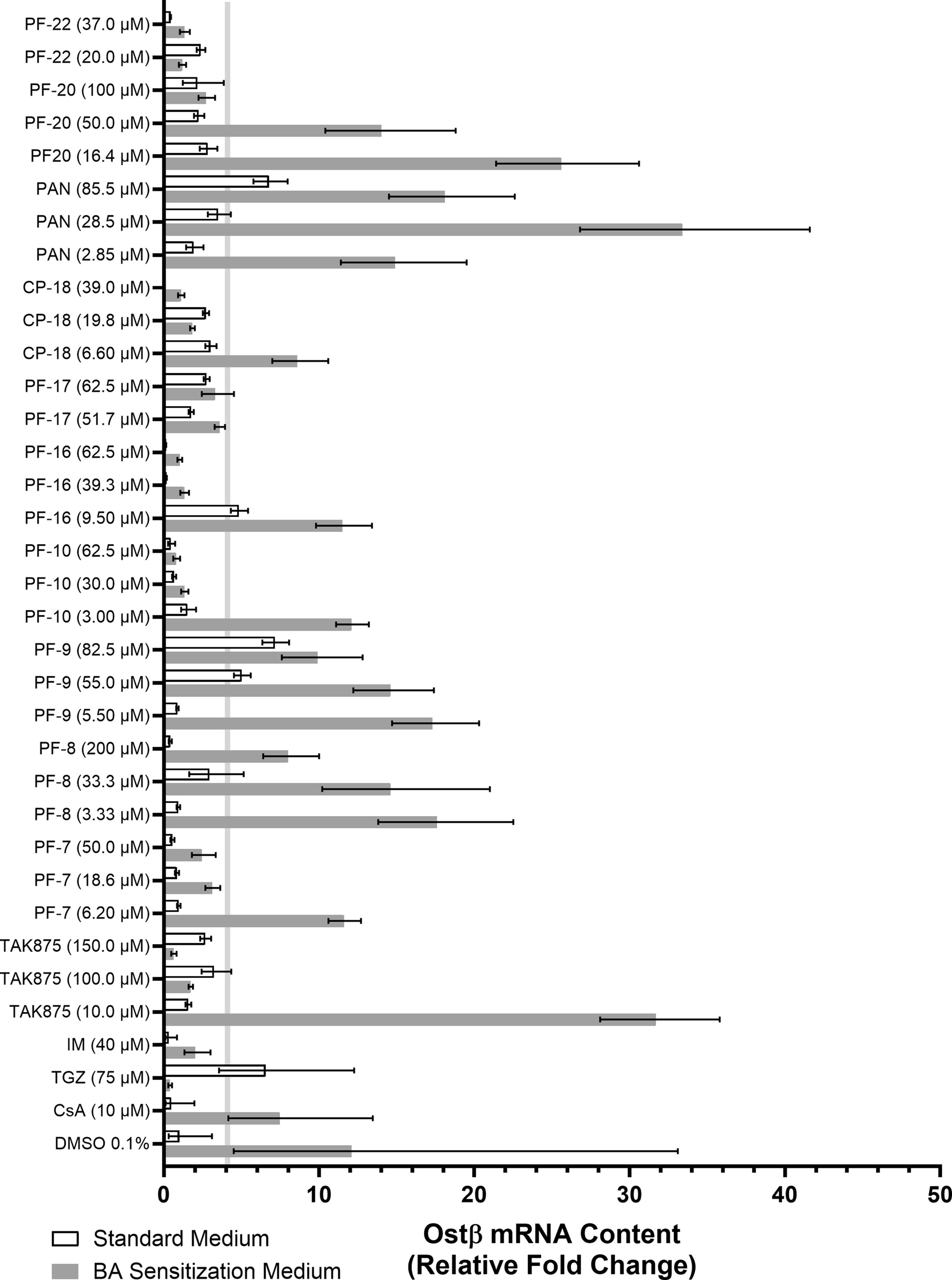
The expression of OSTβ, a sentinel marker of FXR activation, was evaluated in SCHH following 12-hour treatment with the test article under standard or BA sensitization culture conditions. The lower 95% confidence of OSTβ mRNA induction, 4.1-fold above the control and represented by the dotted line, in SCHH treated with CsA was utilized as a threshold to determine if induction, above the threshold, or repression, below the threshold, occurred due to treatment. Values represent the RQ mean, error bars represent 95% confidence intervals of triplicates, and nonoverlapping CI indicates statistically significant differences among the groups. Gray lines represent the lowest RQ within 95% CI for gene expression results in SCHH treated with CsA under BA sensitization conditions.
No significant dose-dependent increase in LDH leakage ≥150% of the control or loss of ATP content (≥36% of the control) was observed in SCHH under either standard or sensitization media following 24 hours of exposure to the test articles #1 (PF-06273340), #2 (PF-04895162), #3, #5 (PF-01428777), #6, #11, #12, #13 (25-desacetyl-3-formyl RIF), #14 (montelukast), #15 (zafirlukast), #19 (panadiplon), #20, and #21 (ADX10059) at any concentration evaluated (Table 5, Supplementary Table S2). The results for these selected drugs were inconclusive of a BA-independent or -dependent mechanism because no evidence of cytotoxicity was observed under either culture condition. However, the lack of a response in the presence of a BA pool at clinically relevant exposures may suggest that these compounds have low hepatotoxicity potential via a BA-dependent mechanism.
Extensive and significant (p ≤ 0.05) increases in LDH leakage >200% of the DMSO control with concomitant dose-dependent decreases in ATP content ≥80% of the DMSO control were observed in SCHH following 24 hours of exposure to the test articles #4 (TAK875), #7–10, #16, #18 (CP-724714), and #22 (PF-05746140) under the sensitization condition only (Fig. 10A, B; Table 5). The results suggested that these selected drugs induced cytotoxicity in SCHH in a BA-dependent manner across the concentrations examined. Interestingly, marked (p ≤ 0.05) LDH leakage >200% of the solvent control was observed in SCHH following 24 hours of exposure to #17 under both conditions. However, ATP depletion was ≤36% or ≥80% of the solvent control under standard or BA sensitization conditions, respectively. Because ATP depletion was more severe under BA sensitization conditions, it suggests that the cytotoxicity observed was BA-dependent (Table 5).
By employing a lowest observable effect level (LOEL) strategy for both endpoints, with the response thresholds of ≥200% for LDH leakage and ≥80% reduction in ATP content, the margin of safety (MOS = LOEL/Cmax) can be calculated to determine the result’s clinical relevance. The following order illustrates where this mechanism may have had greater clinical significance: 0.77× Cmax #18 (CP-724714) > 1× Cmax #22 ∼ 1× Cmax #16 ∼ 1× Cmax #17 > 3.4× Cmax #7 > 9.8× Cmax #8 > 10× Cmax #10 ∼ 10× Cmax #4 (TAK875) > 15× Cmax #9.
Evaluation of FXR antagonism of blinded compounds
To evaluate the effect of the blinded test articles on the activation status of FXR, the expression of OSTβ, an FXR-regulated efflux transporter, was assessed in SCHH cultured under standard or BA sensitization conditions following 12 hours of treatment. Twelve hours of exposure was utilized instead of 24 hours to minimize the impact of possible cytotoxicity on the OSTβ mRNA response, as determined by the cytotoxicity time-course studies conducted under BA sensitization conditions with troglitazone. Following exposure, total RNA was harvested, converted to cDNA, and quantitative RT-PCR analysis was performed using TaqMan (Table 5).
Following CsA treatment under sensitization conditions, OSTβ mRNA was markedly induced 7.5-fold above the control (Fig. 11). In contrast, the pronounced suppression of OSTβ induction responses of 97% (0.39-fold) was observed in SCHH cultured under sensitization conditions treated with troglitazone (Fig. 11). The lower 95% confidence of OSTβ mRNA induction, 4.1-fold above the control (Fig. 11), in SCHH treated with CsA was utilized as a threshold to determine if induction, above the threshold, or repression, below the threshold, occurred due to treatment. The results were consistent with previously reported data demonstrating that troglitazone, and not CsA, disrupts the BA activation of FXR under sensitization conditions.
Following exposure to increasing concentrations of the test articles #1 (PF-06273340), #2 (PF-04895162), #3, #5 (PF-01428777), #6, #8–9, #11, #12, #13 (25-desacetyl-3-formyl RIF), #14 (montelukast), #15 (zafirlukast), #19 (panadiplon), and #21 (ADX10059) under sensitization conditions, OSTβ mRNA was markedly induced ≥4.9-fold above the DMSO control (Table 5, Supplementary Table S3). These results were above the threshold and relatively consistent with the results observed in SCHH treated with the solvent control and CsA treatment under sensitization conditions. Taken together, these results suggested that these drugs have low potential to disrupt FXR activation.
Under sensitization conditions, the dose-dependent suppression of OSTβ induction response >70% was observed in SCHH following exposure to increasing concentrations of the test articles #4 (TAK875), #7, #10, #16, #18 (CP-724714), and #20 (Fig. 11, Table 5). Although the responses did not appear to be dose-dependent, OSTβ mRNA content under sensitization conditions was suppressed >70% below the DMSO control in SCHH treated with the test articles #17 and #22 (Fig. 11, Table 5). In general, these results demonstrated that the test articles #4 (TAK875), #7, #10, #16, #17, #18 (CP-724714), #20, and #22 disrupted the BA mediated activation of FXR.
Discussion
Although BA play many critical signaling roles in maintaining cellular bioenergetic homeostasis, they are toxic and can activate death receptors2–4,19–23 if their concentrations are not stringently controlled by feedback and feedforward mechanisms. 24
In the past two decades, BSEP inhibition has been accepted as a major DILI mechanism. Several studies provided evidence of a strong association of BSEP inhibition5–7 from drug exposure with the risk of clinical DILI. This association was supported by the identification of genetically defective BSEP alleles, a cause of liver injury in humans.25,26
Although it has been commonplace to utilize minimalist systems such as BSEP vesicles for preclinical safety DILI screening, it has been demonstrated that these simplistic systems are not always predictive of BA-induced hepatotoxicity because of drug exposure.26–29 The most likely explanation is the existence of a feedback mechanism controlled by the BA-sensing nuclear receptor, FXR. The concept of a compensatory mechanism for increased concentrations of BAs has been well-established in the literature.9,29–31 The “link” between BSEP inhibition, FXR activation, and compensatory mechanism failure was demonstrated in our previous work. 8
The International Transporter Consortium recommended the use of animal models32,33 as an alternative or in addition to BSEP inhibition screens. However, animal models often fail to identify liver injury due to BA homeostasis disruption for many reasons, including species differences regarding BA pool constituents, BA metabolism, BA regulation, or physiology (i.e., lack of gall bladder). 30 Given the complexities of identifying BA homeostasis disruption24,30 preclinically, there is a need to develop more comprehensive in vitro human translatable approaches to screen for BA homeostasis disruption and associated hepatotoxicity.
We used SCHH to screen 71 drugs with varying BSEP inhibition profiles and DILI incidence to evaluate the use of two hepatocyte culture media, both with and without a human BA pool supplement. This approach allows for the assessment of a drug candidate’s potential to cause BA-dependent (with the BA pool) or BA-independent (without the BA pool) cytotoxicity in a physiologically relevant and translatable system that maintains an intact BA feedback mechanism and functional drug-metabolizing enzymes. Susukida et al. 34 conducted a similar study using sandwich-cultured primary rat hepatocytes (SCRH) with an externally applied human BA pool to triage 38 drugs with diverse cholestatic DILI incidences, prior to the further analysis of 22 of these drugs in SCHH. This methodology presented translational challenges similar to those encountered in in vivo animal studies, particularly regarding the detection of BA homeostasis disruption, and was further complicated by combining a human BA pool with SCRH. In addition, the optimized human BA pool concentrations used in both SCRH (115×) and SCHH (150×) resulted in >20% cytotoxicity in the absence of drugs, which may have influenced their results. In contrast, our assay employed a BA pool concentration of 250 µM, which yielded minimal cytotoxicity. 8
Our assay strategy is aligned with the dynamic nature of portal venous BA concentrations in humans following postprandial elevations of 2–6× mean fasting concentrations of 14 µM in healthy individuals. 35 In some liver disease populations (i.e., primary biliary cirrhosis patients), the mean systemic plasma concentrations are reported to be 115–197 µM, with portal venous concentrations potentially 2–6× greater.35,36 Thus, it is imperative to use BA at low and high concentrations in the presence of a drug within a physiologically relevant in vitro human hepatic system to fully evaluate a drug’s potential to disrupt BA homeostasis.
To qualify this BA mechanism-based approach, we treated SCHH with increasing concentrations of CsA and thiazolidinediones, including troglitazone, pioglitazone, and rosiglitazone, under standard or BA sensitization media for 24 hours. The observed difference in the cytotoxic response in sensitization media in comparison with standard media suggested that troglitazone cytotoxicity was BA-dependent (Fig. 3A, B, Table 4). These results were consistent with the demonstrated BA biliary efflux inhibition (Fig. 1A–C) combined with FXR antagonism (Fig. 2A, B), consistent with our previous report. 8 Troglitazone FXR antagonism has also been confirmed by Norona et al. 37 at similar concentrations (Table 3). Neither pioglitazone nor rosiglitazone was demonstrated to possess both BA biliary efflux inhibition (Fig. 1B, C) and FXR antagonism (Fig. 2B), consistent with no cytotoxicity observed under BA sensitization conditions for either thiazolidinedione (Fig. 3A, B, Table 4). Although Norona et al. 37 reported FXR antagonism via FRET IC50 (Table 3) for pioglitazone, their results were not confirmed in SCHH, suggesting that the FRET result may be artifactual. The BA-dependent cytotoxicity mechanism requires both attributes as previously reported 8 and confirmed here. This concept is consistent with CsA BA-independent cytotoxicity (Fig. 3A, B, Table 4) demonstrated by dose-dependent cytotoxicity under both culture conditions. While Norona et al. 37 reported a signal of FXR antagonism with 100 µM CsA in SCHH, our results demonstrated that cytotoxicity is observed under both culture conditions at concentrations ≥63× Cmax (≥50 µM). Here and in past reports, we have demonstrated that CsA inhibits BA biliary efflux (Fig. 1B, C) but likely does not possess FXR antagonism at ≤31× Cmax (≤25 µM; Fig. 2A, B). 8 LiverTox DILI clinical injury (incidence) classifications of troglitazone, CsA, pioglitazone, and rosiglitazone appear to be consistent with our assay results (Tables 2 and 4).
After determining that our strategy could distinguish BA-dependent from BA-independent cytotoxicity, we explored the kinetics of BA-dependent cytotoxicity. Following 18 hours of exposure, troglitazone cytotoxicity was only significant under BA sensitization conditions in a time-dependent manner (Fig. 4A, B). ATP content exhibited declines and recoveries consistent with a successful or failed compensatory response during the time course. The timing of this response was consistent with a rapid BA adaptive response in SCHH described in our past work.8,10
Following optimization, 45 additional compounds were screened at a single concentration in SCHH under both culture conditions. The diverse collection of test compounds (Table 4) was evaluated at 20× their systemic Cmax, Css, or limit of solubility for 24 hours. The previously assessed CsA and thiazolidinediones were included as controls, and the assay response was consistent with previous assessments at the specified concentrations (Fig. 5A, B, Table 4). No cytotoxicity was observed under either culture condition for 37 compounds evaluated (Table 4 and Supplementary Table S1). According to the LiverTox database, 38 9 out of these 37 compounds (Table 2) have no evidence of clinical DILI; thus, these drugs would be anticipated to be negative under both media conditions. Interestingly, no cytotoxicity was observed for bosentan, sitaxentan, and telithromycin, which at first glance may seem inconsistent with their liver injury (incidence) as mixed (C), mixed (WD), 39 and hepatocellular (A), respectively (Table 2). It has been postulated that bosentan and sitaxentan, endothelin receptor antagonists (ERA), cause DILI by disrupting BA homeostasis. However, d8-TCA biliary excretion was inhibited by ≤20% (100 µM)40,41 with either ERA, and FXR antagonism IC50 was reported as >100 and 95.4 uM 37 for sitaxentan and bosentan, respectively. This evidence suggests that neither ERA compound possessed both attributes confirmed to disrupt BA homeostasis and is consistent with our presented results (Table 4 and Supplementary Table S1). In addition, clinical evidence indicates that hypersensitivity is the most likely mechanism of liver injury of telithromycin and ERAs. 38 Avoidance of macrolides or the use of corticosteroid therapy has proven effective to prevent or treat liver injury, respectively. 38
In the partially blinded screen, no evidence of cytotoxicity for #1 (PF-06273340), #2 (PF-04895162), #3, #5 (PF-01428777), #6, #11, #12, #13 (25-desacetyl-3-formyl RIF), #14 (montelukast), #15 (zafirlukast), #19 (panadiplon), #20, and #21 (ADX10059) was observed under either culture condition (Table 5 and Supplementary Table S2). These results appear to be inconsistent with the preclinical or clinical liver injury (Table 5) observed for #1 (PF-06273340), #2 (PF-04895162), #5 (PF-01428777), #6, #14 (montelukast), #15 (zafirlukast), #19 (panadiplon), and #21 (ADX10059). BSEP vesicle studies determined the pan-TKR inhibitor [#1 (PF-06273340)] and Kv7 channel opener [#2 (PF-04895162)] were strong and moderate BSEP inhibitors (Table 5), respectively. However, FXR antagonism was only detected with #1 (PF-06273340; LOEL: 51 µM) treatment (Fig. 11). Taken together, the evidence suggests that neither compound possessed sufficient BSEP inhibition and FXR antagonism at the clinically relevant concentrations assessed in a physiologically relevant hepatic model to demonstrate BA-dependent cytotoxicity. While it is possible for false negatives, the lack of a response in the presence of a BA pool may suggest that these compounds have low BA-dependent hepatotoxicity potential similar to pioglitazone or rosiglitazone. Caution should be applied to this inference if the studies were conducted without a dose–response; however, if a dose–response strategy was employed covering the relevant clinical exposures and included multiple donors, then such a deduction may be warranted. Alternatively, the model may lack the complexity required to recapitulate the injury (i.e., immune components); therefore, it still would be consistent with a BA-independent mechanism.
Amiodarone (20× Css), deferasirox (3× Css), fluvastatin (20× Css), or ketoconazole (20× Css) cytotoxicity was identified as BA-dependent (Fig. 5A, B, Table 4) in a single-concentration screening design. In a separate three-donor and solubility-limited three-dose response strategy, BA-dependent cytotoxicity was confirmed in all three donors for troglitazone (LOEL: 8× Cmax), defarasirox (LOEL: 3× Css), and ketoconazole (LOEL: 9× Css; Fig. 6A, B, Table 4). FXR antagonism was also confirmed for these compounds (Fig. 7) and consistent with Norona et al.’s 37 findings. Detection of BA-dependent liver injury at clinically relevant exposures ranging from 3 to 9× Css or Cmax seems to be consistent with the reported DILI clinical injury and incidence (Table 2) and falls below the 10×-fold exposure threshold recommended by the International Transporter Consortium. 32
With DJJ being the most sensitive donor, we reassessed the simvastatin and ritonavir BA cytotoxicity mechanisms over a broader dose–response. In this assessment, ritonavir and simvastatin exhibited dose-dependent cytotoxicity via a BA-dependent mechanism (Fig. 8A, B). The false negative in the initial screen for ritanovir and simavastatin was likely due to solubility issues and too low of a dose, respectively. The ATP depletion curves exhibited left shifts under BA sensitization, resulting in estimated TC50 parameters of 1.1× and >4000× Css, suggesting that this mechanism is more clinically relevant for ritonavir than for simvastatin, respectively (Fig. 9A,B, Table 4). Ritonavir’s estimated ATP TC50 in BA sensitization media was consistent with Norona et al.’s 37 (Table 3) reported FXR antagonism IC50 and consistent with BA biliary efflux inhibition in SCHH. 42
In the partially blinded screen, BA-dependent cytotoxicity (Fig. 10A, B; Table 5) was observed for #18 (CP-724714; 0.77× Cmax), #22 (1× Cmax), #16 (1× Cmax), #17 (1× Cmax), #7 (3.4× Cmax), #8 (9.8× Cmax), #10 (10× Cmax), #4 (TAK875; 10× Cmax), and #9 (15× Cmax). FXR antagonism was also confirmed for #4 (TAK875), #7, #10, #16, #18 (CP724714), and #20 (Fig. 11, Table 5). Confirmation of FXR antagonism for CP-724714, and not fasiglifam (#4: TAK875), was reported by Norona et al. 37 The reported CP-724714 FXR FRET IC50 of 20.4 µM was shockingly consistent with our SCHH LOEL of 19.8 µM (Table 5). In addition to FXR antagonism, both the HER2 tyrosine kinase (#18: CP-724714) and fasiglifam (#4: TAK875) were demonstrated to inhibit BA biliary efflux in SCH,43,44 consistent with the other examples of BA-dependent cytotoxicity presented. Both CP-724714 and fasiglifam (#4: TAK875) were both clinical failures due to liver injury. Similarly, preclinical liver injury was observed with #7, #8, #9, and #22 (Table 5). While the drugs #8 and #9 demonstrated BA-dependent cytotoxicity (Fig. 10A,B; Table 5), neither compound exhibited FXR antagonism (Fig. 11, Table 5), and the BSEP vesicle IC50 for #8 was determined to be 26 µM. Of course, there are BA-dependent alternative mechanisms, including a novel mechanism associated with tyrosine kinase inhibitors, recently demonstrated using our presented strategy. 45
During our multidonor re-evaluation, fluvastatin and amiodarone cytotoxicity mechanisms were changed from BA-dependent to BA-independent. Across the SCHH donors, amiodarone cytotoxicity at 20× Css was not repeated; however, ATP depletion and LDH leakage were significantly reduced in all three SCHH donors at 70× Css under standard conditions (Fig. 6A, B, Table 4). In all three SCHH donors treated with fluvastatin, ATP was not significantly reduced under either media condition at any concentration examined (Fig. 6A, B, Table 4). A BA-independent mechanism would be consistent with the postulated mechanisms of injury for amiodarone, phospholipidosis, and fluvastatin, hypersensitivity or reactive metabolite. 38
The cytotoxicity of imatinib, posaconazole, and tolcapone was identified as BA-independent during our single-concentration screen (Fig. 5A, B). In a separate three-donor and solubility-limited three-dose response strategy, we selected a single compound and confirmed imatinib’s BA-independent cytotoxicity (9× Css LOEL; Fig. 9A, B). A BA-independent mechanism would be consistent with the proposed mechanisms of injury, including immunological, sterol synthesis disruption, and toxic metabolite formation, respectively. 38
Conclusion
Identifying a drug candidate’s potential to disrupt BA homeostasis early in development is critical to improving clinical safety success. The use of BSEP inhibition and animal models in early drug development to reduce this liability risk has not always been effective.
SCHH have been demonstrated to properly recapitulate BA uptake and efflux (basolateral and biliary) while maintaining the BA feedback mechanisms.8,10,11 These mechanisms are vital to identify BA homeostasis disruption preclinically. Our results demonstrated the employment of two different hepatocyte culture medium, standard and BA sensitization, as a successful strategy to interrogate BA-dependency cytotoxicity across 71 different drugs assessed. This strategy has the potential to reduce false negatives in preclinical in vitro hepatotoxicity screens, thus reducing clinical DILI attrition. Our study demonstrated that FXR antagonism was typically a shared mechanism among BA-dependent cytotoxicity-positive drugs, but alternative disruption mechanisms do exist, and the method appeared to be sensitive to these too.
Our results demonstrated that this approach was successful, but false negatives and false positives are inherent to all assays. Utilization of dose–response and multiple donors is recommended to reduce the incidence of these errors. Lastly, increasing the exposure period may improve the approach’s ability to detect drugs that downregulate BA efflux pathways, such as mithramycin 46 or chlorpromazine. 47 Although these model compounds were not utilized in this assessment, their use along with other compounds with various potential BA homeostasis disruption mechanisms (i.e., tubule disruptors) should be included to further improve upon this strategy.
Footnotes
Acknowledgments
Mike Aleo selected the Pfizer blinded compounds and summarized the internal data on liver injury and the attrition reason. Matt Palmer and Kimberly Freeman performed B-CLEAR® Assays and initial C-DILI screening of the unblinded 49 therapeutics. Bob St. Claire performed LC-MS/MS on B-CLEAR® Assays. The authors would like to acknowledge Pfizer colleagues including Payal Rana, Chris Houle, and Anna Kopec for reviewing this article prior to journal submission. Your feedback improved the quality of this work and was greatly appreciated, thank you.
Author Disclosure Statement
J.P.J. is a co-inventor of the C-DILI™ Assay and was employed by BioIVT during 2014–2019 when this work was conducted and has no current monetary interest in BioIVT. J.P.J. was employed at Pfizer during article preparation and submission from 2019 to 2025. K.R.B. is employed by BioIVT and owns stock in the company and is a co-inventor of the C-DILI Assay.
Funding Information
No funding was received for this article.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.